

Abstract
Background
There is increasing evidence for the role of prenatal stress in shaping offspring DNA methylation and disease susceptibility. In the current study, we aimed to identify genes and pathways associated with pregnancy anxiety using a genome-wide DNA methylation approach.
Methods
We selected 22 versus 23 newborns from our Prenatal Early Life Stress (PELS) cohort, exposed to the lowest or highest degree of maternal pregnancy anxiety, respectively. Cord blood genome-wide DNA methylation was assayed using the HumanMethylation450 BeadChip (HM450, n = 45) and candidate gene methylation using EpiTYPER (n = 80). Cortisol levels were measured at 2, 4, and 12 months of age to test infant stress system (re)activity.
Results
Data showed ten differentially methylated regions (DMR) when comparing newborns exposed to low versus high pregnancy anxiety scores. We validated a top DMR in the GABA-B receptor subunit 1 gene (GABBR1) revealing the association with pregnancy anxiety particularly in male newborns (most significant CpG Pearson R = 0.517, p = 0.002; average methylation Pearson R = 0.332, p = 0.039). Cord blood GABBR1 methylation was associated with infant cortisol levels in response to a routine vaccination at 4 months old.
Conclusions
In conclusion, our results show that pregnancy anxiety is associated with differential DNA methylation patterns in newborns and that our candidate gene GABBR1 is associated with infant hypothalamic-pituitary-adrenal axis response to a stressor. Our findings reveal a potential role for GABBR1 methylation in association with stress and provide grounds for further research.
Electronic supplementary material
The online version of this article (10.1186/s13148-017-0408-5) contains supplementary material, which is available to authorized users.
Keywords: DNA methylation, Pregnancy anxiety, GABBR1, Prenatal stress, Gender differences, HPA axis
Background
Prenatal stress exposure can adversely influence infant development and alter susceptibility to obesity, metabolic disorders, and mental health outcomes [1]. Although the exact mechanisms of such links between the prenatal environment and postnatal (e.g., behavioral) phenotype are still unknown, many recent studies found evidence of a potential role for DNA methylation. Different forms and severity of maternal psychosocial stress were found to have an influence on fetal DNA methylation patterns [2]. Epigenetic studies investigating maternal well-being during pregnancy have largely made use of candidate gene approaches. For instance, gene-specific DNA methylation changes associated with prenatal stress were found in the glucocorticoid receptor gene (NR3C1) [3]. More specifically, studies showed associations of infant NR3C1 DNA methylation with maternal depressive symptoms [4–6], exposure to inter-partner violence [7], and war stress [8, 9]. A link of NR3C1 DNA methylation and altered infant stress reactivity of the hypothalamic-pituitary-adrenal (HPA) axis was found in the study of Oberlander et al. [4]. DNA methylation of other candidate genes with a role in HPA axis [6, 9] or immune system function [10] was associated with prenatal stress as well.
In addition to these candidate gene approaches, some epigenome-wide association studies (EWAS) have examined the potential role of prenatal stress. Genome-wide DNA methylation changes have been found in newborns from women with non-medicated depression or anxiety during pregnancy [11], while another study shows an association between the methyltransferase pathway and prenatal stress in two population-based cohorts [12]. Contrasting maternal perceived stress and objective measurements of hardship, Cao-Lei and colleagues found that only the latter was associated with DNA methylation at several CpG sites of children in the Project Ice Storm study [2]. Furthermore, a recent study by Serpeloni et al. described genome-wide DNA methylation changes associated with grandmaternal exposure to violence during pregnancy [13].
In previous publications examining our specific birth cohort, we have shown that pregnancy anxiety appeared to be an important factor associated with DNA methylation changes of the NR3C1 gene [14] and two imprinted genes involved in fetal development, i.e., insulin-like growth factor 2 (IGF2) and guanine nucleotide binding protein, alpha stimulating (GNAS) in cord blood [15]. These studies provide evidence that not only extreme forms of prenatal stress such as exposure to war or natural disasters [2, 8] but also maternal anxiety in a general healthy population, can alter DNA methylation in the offspring.
Following up on our candidate gene studies, we here performed an EWAS to identify the DNA methylation changes in newborns associated with maternal pregnancy-related anxiety. This hypothesis-generating approach provides the opportunity to identify novel candidate genes but also to study interactions using a biological pathway analysis. The magnitude of one of our EWAS discovery findings was subsequently investigated in an extended sample set using a different DNA methylation quantification technique, the Sequenom EpiTYPER platform. Finally, we examined the association of DNA methylation at our candidate region with infant HPA axis function.
Methods
Study population
Between October 2009 and December 2010, samples from 170 highly educated pregnant women at 6 to 12 weeks of gestation were recruited within the Prenatal Early Life Stress (PELS) study in the University Hospitals Leuven, Belgium. A detailed description of data and sample collection can be found elsewhere [14].
Briefly, each trimester, the pregnant women completed several questionnaires regarding mental health and general well-being. From 80 of these 170 participating mothers, we were able to collect a cord blood sample at birth, of which DNA was subsequently isolated. Of these 80 samples, 45 DNA samples were selected for genome-wide DNA methylation analysis based on maternal psychological measurements, while all 80 DNA samples were subsequently used for the candidate gene confirmation study. A schematic overview of the study design is represented in Additional file 1: Figure S1.
Psychological measurements
At each trimester of pregnancy, future mothers were asked to complete questionnaires assessing depression, anxiety, and mother-fetus relationship. The revised pregnancy-related anxiety questionnaire (PRAQ) was used to assess worries and anxiety specifically regarding the participating mother’s pregnancy [16]. The validated revised version, which we used for the current study, contains three subscales: (1) fear of integrity of the fetus, (2) concerns about one’s own appearance, and (3) fear of the delivery itself [17]. We previously reported a high internal reliability for the total scale (Cronbach’s α ≥ 0.95) and good internal reliability for the subscales (Cronbach’s α ≥ 0.73) of the revised questionnaire [14]. To select the samples for EWAS, we focused on the fear of integrity subscale of PRAQ, which appeared as most associated with DNA methylation in our candidate gene studies [14, 15]. This subscale provides a score ranging from zero to seven and measures maternal anxiety regarding the health of the unborn infant and fear of integrity of the newborn.
Child HPA axis measurements
In order to measure infant HPA axis stress response as well as baseline activity, cortisol was measured in response to a vaccination at 2 and 4 months old and during the day at 12 months old. In both vaccination studies and the day profile study, saliva samples for cortisol measurements were taken using BD Visitec eye sponges (Becton, Dickson and Company, Waltham, USA). To determine cortisol levels in saliva a High Sensitivity Salivary Cortisol Enzyme Immunoassay Kit (ELISA kit, Salimetrics, Europe) was used. This assay captures the full range of salivary cortisol levels (0.003 to 3.0 μg/dL) while using only 25 μL of saliva per test and it was designed by Salimetrics to be resilient to the effects of interference caused by collection techniques that affect pH.
Cortisol was measured in response to a physical stressor at 2 and 4 months of age. All visits were scheduled in the afternoon to control for circadian rhythm. Mothers were asked not to feed their infants from 1 hour before the test until after the test since this might compromise assay performance by lowering sample pH and influencing bacterial growth. The routine vaccination consisted of two intramuscular injections: one injection for Pneumococcus and one combined injection for diphtheria, whooping cough, tetanus, poliomyelitis, and Haemophilus influenza type B. Saliva for cortisol measurements was collected at arrival to the examination room and at 15, 30, 45, and 60 min following the injection. The time between arrival and vaccination included the time when the parent undressed the infant, when both waited for the doctor to arrive, and when the doctor was taking the health history and the physical examination. The use of this vaccination stressor has a number of advantages: (1) it is routinely performed on all infants and therefore is convenient; (2) it is standardized, and so equal treatment to all infants can be assumed (especially if all the data are collected at the same facility); (3) the onset of the application of the aversive stimulation can be precisely determined; and (4) the inoculation is aversive enough to evoke behavioral distress [18]. The maximum value following vaccination was noted and the area under the curve with respect to increase (AUCi), initial response (value at 15 min minus baseline), and recovery slope (value at 60 min minus 15 min) were calculated. Data for several of these variables were skewed (Shapiro-Wilk p value < 0.05) and were logit transformed to correct skewness. If measurements for one-time point were missing, AUCi, initial response and recovery slope were not calculated for this individual.
When infants were 12 months of age, mothers were instructed to collect saliva samples of their infant at awakening, 30 min, 4, and 12 h after awakening. Samples were sent back to our lab in a prepaid, addressed envelope, and processed to measure cortisol levels. The cortisol awakening response (CAR) was calculated and a repeated t test was carried out to verify a significant cortisol increase between awakening and 30 min.
Illumina Infinium HumanMethylation450 BeadChip Assay
Cord blood DNA samples were processed by the Barts and the London Genome Centre (London, UK), where bisulfite treatment was carried out using the Zymo EZ DNA methylation kit (Zymo Research) prior to running the Infinium HumanMethylation450 BeadChip (HM450) arrays (Illumina Inc., California, USA). In total, 48 samples were randomized over four chips based on PRAQ score, maternal smoking, maternal alcohol use, gender of the newborn, parity and gravidity, gestational age at birth, and maternal age. One sample was replicated on each chip to account for batch effects [19], resulting in a sample size of 45 unique samples analyzed on the HM450 array.
Data processing and quality control
Raw data generated by the iScan Illumina array were imported using GenomeStudio software (Illumina, Inc.) and the subsequent quality control and normalization were implemented using the wateRmelon package in R (available from the Bioconductor repository http://www.bioconductor.org) [20]. Data clean-up included removal of samples and CpG probes with insufficient data quality. In more detail, samples were removed if more than 5% of its sites had a poor detection p value (> 0.01), and probes with a detection p value of more than 0.01 in more than 1% of samples or a bead count of less than 3 in more than 5% of samples were removed from the analysis. Furthermore, cross-hybridizing probes, probes containing a common single nucleotide polymorphism (SNP) in the sequence or within 10 bp of the sequence, and probes on the X and Y chromosomes were removed [21]. The final analysis included 414,733 probes, and all samples passed the stringent quality control. A schematic overview of the analysis from data preprocessing until validation can be found in the Supplementary information (Additional file 1: Figure S1).
Genome-wide DNA methylation analysis
Differentially methylated regions (DMRs) were first explored using the R package DMRcate [22]. The DMRcate statistical model was corrected for gender, gestational age of the newborn at birth, batch (chip Sentrix ID), and cell type compositions as calculated with the Houseman algorithm [23]. Using the top 500 uniquely annotated DMRs from DMRcate, we performed a pathway analysis. Enrichment for Gene Ontology (GO) classes was conducted using the over-representation analysis (ORA) method in ErmineJ with a minimum gene set of 5, and a maximum of 100 [24].
DMR verification and validation using Sequenom EpiTYPER
In order to verify the top DMRs from the DMRcate analysis, we used a second region-specific method: the Python module comb-p [25]. Comb-p employs probe locations and p values from the differentially methylated CpG probe (DMP) analysis as input to identify differentially methylated regions. We, therefore, first calculated DMPs using a linear regression model with pregnancy anxiety as a categorical measurement (high versus low) corrected for gender, gestational age of the newborn, batch (chip Sentrix ID) effects, and cell type compositions, similar to the DMRcate model. A list of DMPs was further used for comb-p DMR analysis. Both comb-p and DMRcate have distinct underlying statistical methods to identify DMRs. DMRcate runs a model using a kernel smoothing function on logit-transformed beta values (“M values”), independently from the single CpG site significance. Comb-p, on the contrary, identifies DMRs based on the output of a probe-by-probe analysis, i.e., using the DMP analysis p values and locations of each CpG probe. Both methods calculate DMRs only accounting for their location in the genome, i.e., non-annotated. We combined their results to rank DMRs and further use this ranking to select candidate genes. To statistically test the overlap of unique, protein-coding genes between DMRcate and comb-p lists, the R package GeneOverlap (Bioconductor) was used [26].
Based on the HM450-identified DMR, an amplicon of 446 base pairs was designed using EpiDesigner. A schematic overview and the genomic sequence of the amplicon located at chr6:29594930–29595375 (hg19; forward primer: GGTTAGGGGGTTAGGTTTGTTAGTT; reverse primer: ACTCCCTCAAAAAATCAATATCTCC) can be found in Additional file 1: Figure S2. In silico analysis, using the MassArray package in Bioconductor [27] preceded laboratory experiments in order to assess (un)successful CpG units in advance [28]. EpiTYPER DNA methylation analysis was performed for a total of 80 cord blood samples (including 45 samples used for the HM450 analysis and 35 additional samples) of the PELS cohort as described [15]. In short, cord blood DNA (1 μg) was bisulfite treated using the MethylDetector kit with long cycling protocol (Active Motif, Carlsbad, CA, USA) [29]. Subsequently, amplicons were amplified in triplicate for each sample, followed by T-cleavage reactions and detection by mass spectrometry using the Sequenom MassARRAY (San Diego, CA, USA) protocol. The signal was subsequently translated by the EpiTYPER software resulting in DNA methylation percentages for each CpG unit. Values were excluded if the standard deviation between triplicates was more than 10%, and CpG units were excluded when having a success rate of less than 70%. A summary of CpG sites analyzed by EpiTYPER and their corresponding HM450 CpG probe identifiers is shown in the Supplementary data (Additional file 2). For this part of the study, all 80 samples of our PELS cohort were used and, therefore, PRAQ could be analyzed as a continuous variable. The association of DNA methylation at each CpG unit with PRAQ was analyzed using Pearson’s correlation tests. SPSS Statistics, version 23 (IBM Corp., Armonk, NY, USA) was used for these statistical analyses.
Results
Demographic data of the study cohort selected for EWAS
Cord blood DNA samples for the genome-wide methylation analysis were selected from the 80 mother-infant dyads of our PELS cohort. We aimed for the upper and lower quartiles of PRAQ-integrity scores and included 23 mothers with the highest and 22 mothers with the lowest scores.
All participating mothers were European, with a mean maternal age of 30.6 years and an average length of gestation of 277.2 days. An overview of selected demographic parameters and comparison between the high and low pregnancy anxiety group can be found in Table 1.
Table 1.
Study cohort for the genome-wide DNA methylation analysis with low versus high maternal pregnancy anxiety (PRAQ): fear of integrity scores
| Low pregnancy anxiety | High pregnancy anxiety | |||
|---|---|---|---|---|
| n = 22 | n = 23 | Test statistic | p value | |
| Pregnancy anxiety (PRAQ score) | 2.1 (0.4) | 5.2 (0.7) | t = − 18.906 | p < 0.0001 |
| Maternal age (years (SD)) | 30.0 (2.5) | 31.1 (4.0) | t = − 1.115 | 0.27 |
| Paternal age (years (SD)) | 32.0 (4.0) | 32.5 (4.6) | t = − 0.397 | 0.69 |
| Single parenthood, n (%) | 0 | 1 (4.3%) | Fisher’s exact | 1 |
| Method of birth, (n cesarean section (%)) | 2 (9.1%) | 8 (34.8%) | Fisher’s exact | 0.07 |
| Maternal BMI before pregnancy (kg/m2) | 23.3 (3.5) | 23.4 (4.3) | t = − 0.478 | 0.64 |
| Maternal smoking, n (%) | 2 (9.1) | 0 | Fisher’s exact | 0.23 |
| Maternal alcohol use, n (%) | 2 (9.1) | 4 (17.4) | Fisher’s exact | 0.67 |
| Gender of the baby, female n (%) | 11 (50) | 10 (43.5) | Chi squared = 0.192 | 0.77 |
| Gestational age (days (SD)) | 277.3 (7.2) | 277.1 (10.2) | t = 0.071 | 0.94 |
| Birth weight (g (SD)) | 3544.1 (426.1) | 3293.5 (447.2) | t = 1.923 | 0.06 |
Global DNA methylation is not influenced by pregnancy anxiety
Global DNA methylation difference between the low and the high prenatal anxiety group was assessed. We compared DNA methylation of all long interspersed nuclear element (LINE) repeats by analyzing preprocessed and normalized beta values at CpG probes in both LINE-1 and LINE-2 repeats extracted from the HM450 data as described previously [30]. This analysis involved 15,612 CpG probes at LINE repeats localized throughout the genome. The average DNA methylation percentage for these repetitive elements was 68.58 versus 68.61% for the high and low prenatal anxiety group, respectively.
Differentially methylated regions associated with pregnancy anxiety
Because a region of nearby CpG sites that are differentially methylated often provides stronger evidence for effective transcriptional regulation, we investigated the presence of DMRs using DMRcate. This method starts from preprocessed, normalized methylation values to analyze the HM450 data. DMRcate identified 901 DMRs with 3 or more probes that were differentially methylated between newborns exposed to high versus low pregnancy anxiety (top 50 DMRs presented in Additional file 3).
Epigenome-wide pathway analysis was performed in order to determine specific pathways, which are over-represented. For this purpose, we used ErmineJ on the top 500 uniquely annotated DMRs from DMRcate, enabling us to additionally assess multifunctionality of the resulting GO terms. The top enriched pathways are shown in Additional file 4, showing medium to high multifunctionality scores. Three of the top five GO terms relate to brain development, i.e., “pallium development” (p = 0.001), “hippocampus development” (p = 0.004), and “telencephalon development” (p = 0.004). Other top GO terms seem to be related to general cellular function and tight junctions with multifunctionality scores ranging from 0.002 to 0.932 (Additional file 4).
GABBR1 DNA methylation is positively associated with pregnancy anxiety in male newborns
We aimed at identifying a candidate gene to be further investigated in a larger sample set that includes the full PELS cohort (n = 80). For this purpose, we wanted to strengthen the DMR identification analysis by applying to our genome-wide data a second statistical method to identify DMRs, namely comb-p. The latter, in contrast to DMRcate, identifies DMRs based on the output of a probe-by-probe analysis, using the location and p values of DMPs. To this end, we first tested each CpG probe for differential methylation between neonates exposed to high or low pregnancy anxiety, while correcting for batch effects, gestational age, gender of the newborn, and cord blood cell type estimates (Additional file 5). In total, 17,180 CpG sites were differentially methylated with an unadjusted p value of < 0.05 (referred to as “DMP”) between the low and high pregnancy anxiety group. However, following the false discovery rate (FDR) correction for multiple testing none of the individual probes remained significant.
Subsequently, the comb-p tool was applied, returning ten DMRs (Table 2). Individual probes within the DMRs identified by comb-p are highlighted in a Manhattan plot (Fig. 1). The top ten DMRs according to the comb-p method are presented in Table 2 with indication of the rank order of the equivalent genomic region in the DMRcate results. There was a significant overlap between genes annotated to the DMRcate and comb-p lists, as assessed by GeneOverlap (p = 3.4E-7). The combined ranking of both comb-p and DMRcate DMR analyses identified homeobox C4 (HOXC4), palmitoyl-protein thioesterase 2 (PPT2) and GABA-B receptor subunit 1 gene (GABBR1) as top three DMRs of interest with a maximum beta fold change (FC) of 0.044 (minimum comb-p p = 1.8E-06, DMRcate p = 5.36E-14), 0.050 (minimum comb-p p = 4.5E-05, DMRcate p = 5.43E-18), and 0.053 (minimum comb-p p = 0.00015, DMRcate p = 1.08E-08), respectively. Contrary to HOXC4 and PPT2, GABBR1 DMR is located within a CpG island near active regulatory elements (Additional file 1: Figure S2) and was therefore selected for our validation study with Sequenom EpiTYPER. DNA methylation levels for each HM450 CpG probe in the GABBR1 DMR identified by DMRcate, including percent methylation difference between both groups (range 0.24 to 4) and effect sizes (range 0.170 to 0.825) can be found in Additional file 6.
Table 2.
Comb-p determined differentially methylated regions (DMR)
| Rank comb-p | Rank DMRcate | RefGene name | Location (GRCh37/hg19 assembly) | Minimum p value | Number of probes | Stouffer-Liptak-Kechris p value | Šidák corrected | CpG island |
|---|---|---|---|---|---|---|---|---|
| 1 | 624 | TP53INP1 | chr8:95962083–95962464 | 0 | 6 | 0 | 0 | yes |
| 2 | NA | PRSS50 | chr3:46759334–46759699 | 0 | 8 | 0 | 0 | yes |
| 3 | 3 | HOXC4 | chr12:54446278–54446577 | 0 | 6 | 0 | 0 | no |
| 4 | 169 | PKP3 | chr11:396685–397078 | 0 | 3 | 0 | 0 | yes |
| 5 | NA | ZNF764 | chr16:30572738–30573014 | 0 | 5 | 0 | 0 | yes |
| 6 | 2 | PPT2 | chr6:32120954–32121421 | 0 | 14 | 0 | 0 | no |
| 7 | 21 | GABBR1 | chr6:29595001–29595316 | 0 | 7 | 2.70E-07 | 0.07 | yes |
| 8 | 358 | PITPNM3 | chr17:6358362–6358600 | 0 | 4 | 2.88E-07 | 0.1 | yes |
| 9 | NA | ESRRG | chr1:217306589–-217306764 | 0.01 | 3 | 1.01E-06 | 0.39 | no |
| 10 | NA | HS3ST2 | chr16:22959593–22959820 | 0 | 2 | 0 | 1 | yes |
Both uncorrected (Stouffer-Liptak-Kechris) p values and p values corrected for multiple testing (Sidak) are shown, as well as the rank order of the DMR obtained using the DMRcate method
Fig. 1.
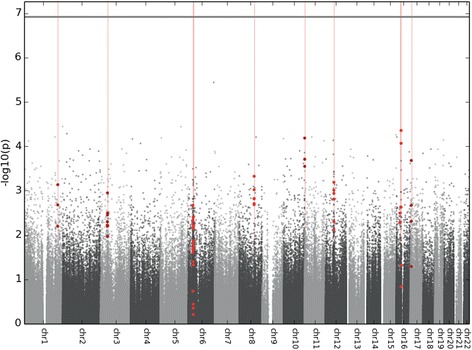
Manhattan plot showing the −log10 p values for all CpG probes and their genomic locations included in the genome-wide methylation analysis (n = 414,733). The false discovery rate p value threshold is represented by a horizontal line. Vertical lines and overlaid bold dots correspond to differentially methylated regions (DMRs) identified by comb-p, as represented in Table 2 [25]
An overview of the GABBR1 gene and the region analyzed by Sequenom EpiTYPER platform can be found in Additional file 1: Figure S2. A summary of CpG units analyzed or excluded for technical reasons is shown in Additional file 2 and correlations between EpiTYPER and HM450 probes are shown in Additional file 7. The 17 CpG sites present in the GABBR1 DMR corresponded to 14 CpG units, of which 4 could not be analyzed due to technical reasons inherent to the MassArray and EpiTYPER methodology (e.g., low mass, fragment overlap) and one had a low success rate (CpG_4). Correlations between pregnancy anxiety and DNA methylation of the GABBR1 DMR were not significant in the total sample set (mean GABBR1 DMR methylation: Pearson R = 0.100, p = 0.379). However, as DNA methylation at several GABBR1 CpG units was associated with the pregnancy anxiety x gender interaction term (CpG_8 p = 0.045, CpG_9.10 p = 0.014, CpG_14.15 p = 0.029, and a trending effect at CpG_3 p = 0.06), data were stratified by gender. When analyzing the EpiTYPER data for this GABBR1 separately for male and female newborns, we observed a positive correlation between fetal GABBR1 DNA methylation and pregnancy anxiety in male newborns on average across the amplicon (Pearson R = 0.332, p = 0.039), at CpG_6, CpG_8, CpG_9.10, CpG_14.15, and a trend was found (p < 0.1) at CpG_1.2, CpG_16, and CpG_17 (Table 3). Only single CpG units CpG_8 (Pearson R = 0.517, p = 0.002) and CpG_14.15 (Pearson R = 0.462, p = 0.003) remained significant following Bonferroni correction for multiple tests (at α = 0.0056) (Fig. 2).
Table 3.
Pearson correlations of pregnancy anxiety (PRAQ) integrity scores with cord blood GABBR1 DNA methylation (average and separate CpG units) generated by EpiTYPER, for boys (n = 39) and girls (n = 41) separately
| GABBR1 DNA methylation | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CpG_1.2 | CpG_3 | CpG_6 | CpG_8 | CpG_9.10 | CpG_11 | CpG_14.15 | CpG_16 | CpG_17 | Average GABBR1 | ||
| Boys (n = 39) | Pearson R | 0.302 | 0.184 | 0.37 | 0.517a | 0.37 | 0.156 | 0.462a | 0.295 | 0.299 | 0.33 |
| p value | 0.061 | 0.306 | 0.043 | 0.002 | 0.023 | 0.395 | 0.003 | 0.095 | 0.086 | 0.039 | |
| Girls (n = 41) | Pearson R | − 0.029 | − 0.288 | 0.032 | 0.035 | − 0.192 | − 0.128 | − 0.040 | − 0.078 | − 0.106 | − 0.049 |
| p value | 0.860 | 0.093 | 0.858 | 0.844 | 0.278 | 0.451 | 0.824 | 0.655 | 0.531 | 0.761 | |
aSignificant after correction for tests at nine CpG sites (α = 0.0056)
Fig. 2.
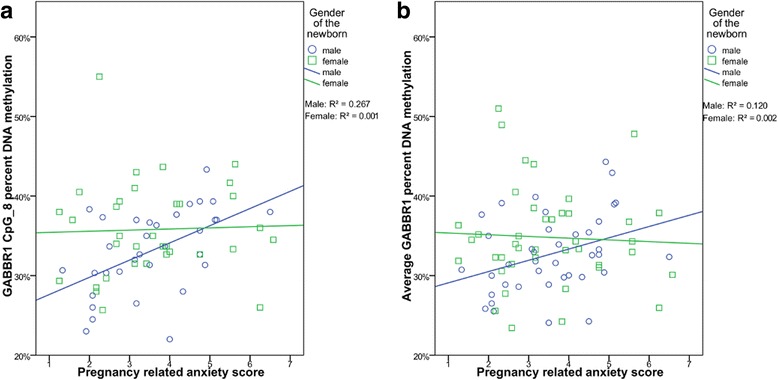
Correlations for male (blue; n = 39) and female (green; n = 41) newborns between pregnancy anxiety (PRAQ) integrity scores and GABA-B receptor subunit 1 gene (GABBR1) DNA methylation at a the most significant CpG site (CpG_8) and b on average across the DMR
GABBR1 DNA methylation significantly correlates with HPA axis function
Using EpiTYPER data (n = 80), we aimed to assess whether the DNA methylation changes in the GABBR1 DMR are also functionally relevant. For this, we examined how cord blood GABBR1 DNA methylation relates to cortisol levels during a stress response at 2 and 4 months and to the cortisol awakening response at 12 months of age. Information on the raw data of these cortisol measurements can be found in Additional file 1: Figure S3. At 2 and 4 months, infants were exposed to a physical stressor, more specifically a routine vaccination injection. Each time, five saliva samples were taken to measure the cortisol levels and calculate the elicited stress response. A paired t test confirmed a significant cortisol increase between T0 and 15 min at 2 months (p = 0.002) and 4 months (p < 0.001). At 2 months, one CpG unit was associated with the initial cortisol response to the stressor. At 4 months, several CpG units appeared to be associated with the AUCi, initial response and recovery slope (Table 4). However, following multiple testing corrections for 27 tests at each study moment (α = 0.0019), only the following associations remained. At 4 months, DNA methylation at CpG_1.2 was significantly associated with the initial response (Pearson R = 0.392, p = 0.001). This same CpG unit was significantly associated with the recovery slope at 4 months (Pearson R = 0.400, p = 0.001; Table 4).
Table 4.
Pearson correlations of cord blood GABBR1 DNA methylation (average and separate CpG units) with infant HPA axis cortisol measurements in response to routine vaccination at 2 and 4 months of age
| GABBR1 DNA methylation | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Child HPA axis stress response | CpG_1.2 | CpG_3 | CpG_6 | CpG_8 | CpG_9.10 | CpG_11 | CpG_14.15 | CpG_16 | CpG_17 | Average GABBR1 | ||
| 2 M (n = 20) | AUCi | Pearson R | 0.372 | − 0.476 | 0.029 | − 0.452 | 0.109 | 0.062 | 0.375 | 0.238 | 0.218 | − 0.152 |
| p value | 0.141 | 0.053 | 0.924 | 0.091 | 0.678 | 0.826 | 0.153 | 0.375 | 0.418 | 0.547 | ||
| Initial response | Pearson R | 0.141 | − 0.47 | 0.092 | − 0.270 | 0.120 | − 0.004 | 0.448 | 0.125 | 0.172 | − 0.098 | |
| p value | 0.565 | 0.043 | 0.745 | 0.295 | 0.624 | 0.989 | 0.062 | 0.621 | 0.494 | 0.680 | ||
| Recovery slope | Pearson R | 0.141 | − 0.283 | 0.063 | − 0.162 | 0.118 | − 0.103 | 0.342 | − 0.012 | 0.083 | − 0.086 | |
| p value | 0.576 | 0.255 | 0.830 | 0.550 | 0.641 | 0.716 | 0.179 | 0.964 | 0.751 | 0.727 | ||
| 4 M (n = 67) | AUCi | Pearson R | 0.260 | 0.239 | 0.241 | 0.200 | 0.084 | 0.096 | 0.080 | 0.186 | 0.082 | 0.168 |
| p value | 0.041 | 0.079 | 0.091 | 0.150 | 0.538 | 0.487 | 0.553 | 0.177 | 0.547 | 0.189 | ||
| Initial response | Pearson R | 0.392a | 0.215 | 0.35 | 0.3 | 0.166 | 0.184 | 0.077 | 0.160 | 0.136 | 0.202 | |
| p value | 0.001 | 0.102 | 0.009 | 0.023 | 0.210 | 0.166 | 0.556 | 0.230 | 0.301 | 0.100 | ||
| Recovery slope | Pearson R | 0.400a | 0.219 | 0.38 | 0.35 | 0.209 | 0.27 | 0.130 | 0.192 | 0.182 | 0.225 | |
| p value | 0.001 | 0.102 | 0.005 | 0.010 | 0.115 | 0.047 | 0.327 | 0.159 | 0.173 | 0.071 | ||
aSignificant at the α level of 0.0019, corrected for 27 tests (3 cortisol variables and 9 CpG sites)
At 12 months, salivary cortisol levels of infants were measured to calculate cortisol awakening responses. However, performing a paired t test indicated that there was no significant increase in cortisol between awakening and 30 min (mean difference = − 0.009, p = 0.647). This is also apparent from Additional file 5: Figure S3B and S3C. Since there was no significant awakening response found, association analyses with GABBR1 DNA methylation levels were not carried out.
Discussion
In our PELS cohort, we found that prenatal exposure to maternal pregnancy-related anxiety is associated with region-specific DNA methylation changes across the genome. Previously, we have shown that pregnancy anxiety is linked to fetal DNA methylation in the glucocorticoid receptor gene (NR3C1), which is a key player in the hypothalamic-pituitary-adrenal axis, and to DNA methylation of imprinted genes IGF2 and GNASXL important in fetal growth and development [14, 15]. This led us to hypothesize that PRAQ measurements, more specifically the “fear of integrity” subscale, could indeed have more widespread associations with fetal DNA methylation.
DNA methylation of repetitive sequences such as LINE elements have been associated with growth trajectories and birthweight [31]. However, in our study, global fetal DNA methylation was not different in the high versus low prenatal anxiety group and these groups did not cluster together.
Pathway and GO analysis using ErmineJ [24] results of the current study should be interpreted carefully, taking into account the multifunctionality scores of each GO term. High multifunctionality scores indicate that many genes involved have several different biological functions, complicating interpretation of these results. Our findings showed that the top-ranked DMRs annotated to genes identified in our study were enriched in pathways related to brain development, general cellular functions, and tight junctions.
We based the selection of the GABBR1 gene for further validation on the merged analysis from both DMRcate and comb-p, two methods that use different strategies to identify DMRs using HM450 data. Although absolute DNA methylation differences appeared small, Cohen’s d effect sizes in the region were medium to large. Additionally, small absolute differences have previously been found to be relevant and correlated to biologically meaningful measurements [32, 33]. Interestingly, the EpiTYPER validation study of the GABBR1 DMR on the full PELS cohort confirmed a gender-specific association of pregnancy anxiety with GABBR1 DNA methylation. More specifically, in male neonates, CpG_8 and CpG_14.15 were significantly associated with pregnancy anxiety following multiple testing corrections, with a large effect size (Pearson correlation coefficients R = 0.517 and 0.462, respectively). None of the GABBR1 DMR CpG units were significantly associated with pregnancy anxiety in female newborns. A possible explanation for the gender differences might be that the exposure to an adverse environment is processed in a different way by both genders. Already during fetal development differences in vulnerability to a range of diseases, including psychiatric disorders, arise between males and females [34] while susceptibility as well as presentation and therapeutic outcomes appear to be gender-specific for certain psychiatric disorders later in life [35]. Evidence from rodent and human research also indicates that the influence of prenatal environment on neurodevelopment is modulated by sex [36, 37]. Finally, gender-specific DNA methylation trajectories were found during fetal neurodevelopment [38], suggesting that gender is an important factor to take into account in analyses similar to the current study.
The identified candidate gene GABBR1 encodes a G-protein coupled receptor subunit GABA-B1, which heterodimerizes with the GABA-B2 subunit to form the GABA-B receptor. Gamma-aminobutyric acid (GABA) can reduce neuronal excitability in the central nervous system by binding these GABA-B or the GABA-A receptors. GABA signaling has a crucial, yet complex, role in the neuroendocrine adaptation and neuronal plasticity in response to stress, mainly in the paraventricular nucleus of the hypothalamus [39]. GABAergic neurotransmission has been implicated in many psychiatric disorders, including anxiety, depression, and schizophrenia [40]. Compelling recent research reveals GABBR1 methylation changes in HM450 data of a high-risk population of youths exposed to childhood maltreatment and further suggests that the identified markers may provide evidence for a molecular link between early life stress and mental health [41]. DNA methylation of the GABBR1 gene has further been implicated in obsessive-compulsive disorder (OCD) [42] as well as schizophrenia [43]. In female OCD patients, GABBR1 DNA methylation was found to be different from healthy controls and associated with symptom severity at baseline, effect of treatment, and responder status. In rats, NR3C1 DNA methylation changes have been found in the hippocampal tissue of rat pups exposed to poor maternal care, which eventually displayed poor maternal care themselves and were more anxious in adult life. Furthermore, maternal care and early life stress in rats were linked to the GABAergic synapse and altered GABA-A receptor expression [44]. In the current study, we identified GABBR1 DNA methylation as affected by prenatal anxiety exposure in a birth cohort. Interestingly, using data from a previous study, one of our co-authors (DvdH) found that the hippocampal tissue of female rat pups exposed to prenatal stress showed a 36% decrease in Gabbr1 gene expression. In general, gene expression in these pups showed an involvement of GABAergic neurotransmission [45]. We hypothesize that the GABAergic network, in particular GABBR1, could play an important role in prenatal stress and that regulation via GABBR1 DNA methylation may potentially influence HPA axis response.
Furthermore, we showed that fetal GABBR1 DNA methylation at CpG_1.2 was associated with HPA axis reactivity in response to a routine vaccination at 4 months old. The observation that we find significant associations of GABBR1 DNA methylation with stress response at 4 months but not at 2 months is likely due to the smaller sample size at 2 months of age, when only 20 out of 80 mother-child dyads participated. The lack of significant associations with infant cortisol day profiles at 12 months could be due to the fact that we could not observe an awakening response. Most likely, an increase of cortisol between awakening and 30 min could not be observed because infants were already lying awake for some time before the mothers could obtain the first cortisol sample of the day.
When comparing previously published EpiTYPER DNA methylation data of NR3C1-1F, IGF2, and GNASXL regions to the current HM450 data, we noticed that most analyzed CpG sites were not present on the HM450 chip. In the IGF2-DMR0 region, no probes were present. The GNASXL and NR3C1-1F regions were represented by one and five probes, respectively, which were not significant.
Some limitations to our current study should be acknowledged. First, the sample size of our genome-wide study is relatively small and therefore no CpG sites survived multiple testing corrections at the probe-by-probe (DMP) analysis level, which is often the case in similar studies where expected effect sizes are small [46, 47]. Instead, we focused on the validation of our findings by examining nearby CpG sites in a DMR analysis, additionally performing a validation study of our candidate gene region using EpiTYPER. Moreover, this current study was intended as a hypothesis-generating study in which we identified and validated GABBR1 as a gene of interest, which was consistent with findings in prenatally stressed rodent brain tissue and associated with infant phenotype, i.e., HPA axis response. Second, we observe a cortisol peak at 15 min post-vaccination, as found by Lewis and Thomas [18]. However, as we have measured cortisol at 15, 30, 45, and 60 min following vaccination, we are presumably not measuring the highest cortisol response peak which is at 20–25 min post-stressor, by consensus [48, 49]. Yet, it is reasonable to assume that the AUC and slope values do reflect the inter-individual variability in cortisol response. Also, the fact that we could not observe an awakening response at 12 months is a major limitation for this part of the study. A delay in sampling at awakening and diurnal sampling during only 1 day likely limit the validity of this measurement. Third, we examine DNA methylation patterns in umbilical cord blood samples in response to pregnancy anxiety. There has been much debate on whether peripheral blood samples can be used as a surrogate tissue to study brain-related phenotypes. Although DNA methylation indeed shows tissue-specific differences [50], several published studies report valuable DNA methylation differences associated with disease found in peripheral tissues [51–53]. Moreover, data from the online Blood Brain DNA Methylation Comparison Tool [54] suggested that DNA methylation of the HM450 probes in the identified GABBR1 DMR was correlated between peripheral blood and prefrontal cortex samples (most significant probe: cg05812266, R = 0.425, p = 0.00012). Furthermore, when analyzing genome-wide DNA methylation data from peripheral blood samples, it is important to take blood cell type composition into account, as these can vary between individuals and may confound associations of exposure [55]. A number of recent methodological studies question the validity of applying the Houseman algorithm [23] to cord blood samples for blood cell type correction [56–58]. However, the method has been extensively used in genome-wide DNA methylation studies using DNA from umbilical cord blood [59–66]. The Houseman algorithm estimations remain an approximate calculation, although they are a good alternative to correct for the potential influence of cell type composition in the absence of actual cell type measurements. The results presented in the current study should be interpreted with caution, taking these limitations into account.
Conclusions
In conclusion, we showed that pregnancy anxiety is associated with fetal DNA methylation changes, identifying GABBR1 as one of the top candidate genes associated with pregnancy anxiety in male newborns. We further provide evidence for a potential link of GABBR1 DNA methylation with infant HPA axis reactivity to a stressor. Future longitudinal studies performed on larger cohorts are needed to verify and further explore these findings.
Additional files
Schematic overview of the study design from data processing to validation of our main findings. Figure S2. Overview of the GABBR1 gene based on UCSC genome browser. Figure S3. Salivary cortisol measurements of infants at 2, 4 and 12 months old. (DOCX 1379 kb)
GABBR1 CpG sites analyzed by EpiTYPER and corresponding HM450 CpG probe identifiers. (XLSX 9 kb)
Top 50 DMRcate results. (XLSX 17 kb)
ErmineJ Gene Ontology (GO) pathways analysis results based on the top 500 annotated differentially annotated regions. (XLSX 14 kb)
Top 100 ranked CpG probes associated with prenatal anxiety. (XLSX 20 kb)
Overview of CpG probes in the GABBR1 region identified by DMRcate. (XLSX 14 kb)
Correlations between HM450 CpG probes and their respective CpG units measured by Sequenom EpiTYPER (n = 41). (XLSX 12 kb)
Acknowledgements
Not applicable.
Funding
This project was supported by the Fund for Scientific Research Flanders (FWO; grant number ELG-C5778-G.0A69.13). BI is a post-doctoral fellow of the Fund for Scientific Research Flanders (12M2715N). SC is a Senior Clinical Researcher supported by the Fund for Scientific Research Flanders (1800411 N). Furthermore, this work was supported by the Deutsche Forschungsgemeinschaft (DFG) Sonderforschungsbereich Transregio (SFB TRR) 58/A5 to DvdH. The PELS study is a 3-centers European study, supported by national funding agencies participating in the Eurocores Program EuroSTRESS of the European Union.
Availability of data and materials
Data generated by the HumanMethylation450 BeadChip array discussed in this publication are available in NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) through the accession number GSE104376.
Abbreviations
- AUCi
Area under the curve with respect to increase
- CAR
Cortisol awakening response
- DMP
Differentially methylated CpG probe
- DMR
Differentially methylated regions
- EWAS
Epigenome-wide association study
- FDR
False discovery rate
- GABBR1
GABA-B receptor subunit 1 gene
- GNAS
Guanine nucleotide binding protein, alpha stimulating
- GO
Gene Ontology
- HM450
HumanMethylation450 BeadChip
- HOXC4
Homeobox C4
- HPA
Hypothalamic-pituitary-adrenal
- IGF2
Insulin-like growth factor 2
- LINE
Long interspersed nuclear element
- NR3C1
Glucocorticoid receptor gene
- PELS
Prenatal Early Life Stress
- PPT2
Palmitoyl-protein thioesterase 2
- PRAQ
Pregnancy-related anxiety questionnaire
- SNP
Single nucleotide polymorphism
Authors’ contributions
SC, KF, BI, EP, EBV, and TH contributed to study conception and design. EBV and EP analyzed HM450 DNA methylation data. EBV and BI carried out EpiTYPER methodology and analyzed data. TH recruited the study sample and collected sample material. DL supervised technical aspects of the study and revised the manuscript. KA clinically examined infants in the cortisol response study and helped with interpretation of cortisol data. EBV, BI, and EP drafted the manuscript. TH, DvdH, DL, KF, and SC critically reviewed and edited the manuscript. The final manuscript was read and approved by all authors.
Ethics approval and consent to participate
We obtained informed consent from all participating mothers, and the study was approved by the ethical committee of the University Hospitals of Leuven, Belgium (S51757).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s13148-017-0408-5) contains supplementary material, which is available to authorized users.
Contributor Information
Elise Beau Vangeel, Phone: +32 16 340548, Email: Elise.Vangeel@kuleuven.be.
Ehsan Pishva, Email: E.Pishva@exeter.ac.uk.
Titia Hompes, Email: titia.hompes@upckuleuven.be.
Daniel van den Hove, Email: d.vandenhove@maastrichtuniversity.nl.
Diether Lambrechts, Email: diether.lambrechts@vib-kuleuven.be.
Karel Allegaert, Email: karel.allegaert@uzleuven.be.
Kathleen Freson, Email: kathleen.freson@kuleuven.be.
Benedetta Izzi, Email: izzi.epigenomica@gmail.com.
Stephan Claes, Email: Stephan.Claes@upckuleuven.be.
References
- 1.Entringer S, Buss C, Wadhwa PD. Prenatal stress, development, health and disease risk: a psychobiological perspective. Psychoneuroendocrinology. 2015;62:366–375. doi: 10.1016/j.psyneuen.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao-Lei L, Massart R, Suderman MJ, Machnes Z, Elgbeili G, Laplante DP, Szyf M, King S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS One. 2014;9:e107653. doi: 10.1371/journal.pone.0107653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Palma-Gudiel H, Cordova-Palomera A, Eixarch E, Deuschle M, Fananas L. Maternal psychosocial stress during pregnancy alters the epigenetic signature of the glucocorticoid receptor gene promoter in their offspring: a meta-analysis. Epigenetics. 2015;10:893–902. doi: 10.1080/15592294.2015.1088630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3:97–106. doi: 10.4161/epi.3.2.6034. [DOI] [PubMed] [Google Scholar]
- 5.Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ. The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics. 2013;8:1321–1329. doi: 10.4161/epi.26634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braithwaite EC, Kundakovic M, Ramchandani PG, Murphy SE, Champagne FA. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics. 2015;10:408–417. doi: 10.1080/15592294.2015.1039221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, Elbert T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiatry. 2011;1:e21. doi: 10.1038/tp.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulligan CJ, D’Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics. 2012;7:853–857. doi: 10.4161/epi.21180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kertes DA, Kamin HS, Hughes DA, Rodney NC, Bhatt S, Mulligan CJ. Prenatal maternal stress predicts methylation of genes regulating the hypothalamic-pituitary-adrenocortical system in mothers and newborns in the Democratic Republic of Congo. Child Dev. 2016;87:61–72. doi: 10.1111/cdev.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao-Lei L, Veru F, Elgbeili G, Szyf M, Laplante DP, King S. DNA methylation mediates the effect of exposure to prenatal maternal stress on cytokine production in children at age 13(1/2) years: Project Ice Storm. Clin Epigenetics. 2016;8:54. doi: 10.1186/s13148-016-0219-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Non AL, Binder AM, Kubzansky LD, Michels KB. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics. 2014;9:964–972. doi: 10.4161/epi.28853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rijlaarsdam J, Pappa I, Walton E, Bakermans-Kranenburg MJ, Mileva-Seitz VR, Rippe RC, Roza SJ, Jaddoe VW, Verhulst FC, Felix JF, et al. An epigenome-wide association meta-analysis of prenatal maternal stress in neonates: a model approach for replication. Epigenetics. 2016;11:140–149. doi: 10.1080/15592294.2016.1145329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serpeloni F, Radtke K, de Assis SG, Henning F, Natt D, Elbert T. Grandmaternal stress during pregnancy and DNA methylation of the third generation: an epigenome-wide association study. Transl Psychiatry. 2017;7:e1202. doi: 10.1038/tp.2017.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A, Schops G, Dom M, Van Bree R, Freson K, et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatr Res. 2013;47:880–891. doi: 10.1016/j.jpsychires.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Vangeel EB, Izzi B, Hompes T, Vansteelandt K, Lambrechts D, Freson K, Claes S. DNA methylation in imprinted genes IGF2 and GNASXL is associated with prenatal maternal stress. Genes Brain Behav. 2015;14:573–582. doi: 10.1111/gbb.12249. [DOI] [PubMed] [Google Scholar]
- 16.Van den Bergh BR, Mulder EJ, Mennes M, Glover V. Antenatal maternal anxiety and stress and the neurobehavioural development of the fetus and child: links and possible mechanisms. A review. Neurosci Biobehav Rev. 2005;29:237–258. doi: 10.1016/j.neubiorev.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 17.Huizink AC, Mulder EJ, Robles de Medina PG, Visser GH, Buitelaar JK. Is pregnancy anxiety a distinctive syndrome? Early Hum Dev. 2004;79:81–91. doi: 10.1016/j.earlhumdev.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Lewis M, Thomas D. Cortisol release in infants in response to inoculation. Child Dev. 1990;61:50–59. doi: 10.2307/1131046. [DOI] [PubMed] [Google Scholar]
- 19.Buhule OD, Minster RL, Hawley NL, Medvedovic M, Sun G, Viali S, Deka R, McGarvey ST, Weeks DE. Stratified randomization controls better for batch effects in 450K methylation analysis: a cautionary tale. Front Genet. 2014;5:354. doi: 10.3389/fgene.2014.00354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pidsley R, Wong CCY, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013;14:293. doi: 10.1186/1471-2164-14-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–209. doi: 10.4161/epi.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peters TJ, Buckley MJ, Statham AL, Pidsley R, Samaras K, V Lord R, Clark SJ, Molloy PL. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, Wiencke JK, Kelsey KT. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillis J, Mistry M, Pavlidis P. Gene function analysis in complex data sets using ErmineJ. Nat Protoc. 2010;5:1148–1159. doi: 10.1038/nprot.2010.78. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen BS, Schwartz DA, Yang IV, Kechris KJ. Comb-p: software for combining, analyzing, grouping and correcting spatially correlated P-values. Bioinformatics. 2012;28:2986–2988. doi: 10.1093/bioinformatics/bts545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen L. GeneOverlap: Test and visualize gene overlaps., R package version 1.12.0 edition; 2013.
- 27.Thompson RF, Greally JM. MassArray: Analytical Tools for MassArray Data. R package version 1.28.0 edition; 2009.
- 28.Suchiman HE, Slieker RC, Kremer D, Slagboom PE, Heijmans BT, Tobi EW. Design, measurement and processing of region-specific DNA methylation assays: the mass spectrometry-based method EpiTYPER. Front Genet. 2015;6:287. doi: 10.3389/fgene.2015.00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Izzi B, Binder AM, Michels KB. Pyrosequencing evaluation of widely available bisulfite conversion methods: considerations for application. Med Epigenet. 2014;2:28–36. doi: 10.1159/000358882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rochtus A, Winand R, Laenen G, Vangeel E, Izzi B, Wittevrongel C, Moreau Y, Verpoorten C, Jansen K, Van Geet C, Freson K. Methylome analysis for spina bifida shows SOX18 hypomethylation as a risk factor with evidence for a complex (epi)genetic interplay to affect neural tube development. Clin Epigenetics. 2016;8:108. doi: 10.1186/s13148-016-0272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Michels KB, Harris HR, Barault L. Birthweight, maternal weight trajectories and global DNA methylation of LINE-1 repetitive elements. PLoS One. 2011;6:e25254. doi: 10.1371/journal.pone.0025254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Palma-Gudiel H, Cordova-Palomera A, Leza JC, Fananas L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev. 2015;55:520–535. doi: 10.1016/j.neubiorev.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 33.Breton CV, Marsit CJ, Faustman E, Nadeau K, Goodrich JM, Dolinoy DC, Herbstman J, Holland N, LaSalle JM, Schmidt R, et al. Small-magnitude effect sizes in epigenetic end points are important in children’s environmental health studies: The Children’s Environmental Health and Disease Prevention Research Center's Epigenetics Working Group. Environ Health Perspect. 2017;125:511–526. doi: 10.1289/EHP595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bale TL, Epperson CN. Sex differences and stress across the lifespan. Nat Neurosci. 2015;18:1413–1420. doi: 10.1038/nn.4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bale TL, Baram TZ, Brown AS, Goldstein JM, Insel TR, McCarthy MM, Nemeroff CB, Reyes TM, Simerly RB, Susser ES, Nestler EJ. Early life programming and neurodevelopmental disorders. Biol Psychiatry. 2010;68:314–319. doi: 10.1016/j.biopsych.2010.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress. 2011;14:348–356. doi: 10.3109/10253890.2011.586447. [DOI] [PubMed] [Google Scholar]
- 37.Weinstock M. Gender differences in the effects of prenatal stress on brain development and behaviour. Neurochem Res. 2007;32:1730–1740. doi: 10.1007/s11064-007-9339-4. [DOI] [PubMed] [Google Scholar]
- 38.Spiers H, Hannon E, Schalkwyk LC, Smith R, Wong CC, O'Donovan MC, Bray NJ, Mill J. Methylomic trajectories across human fetal brain development. Genome Res. 2015;25:338–352. doi: 10.1101/gr.180273.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bains JS, Wamsteeker Cusulin JI, Inoue W. Stress-related synaptic plasticity in the hypothalamus. Nat Rev Neurosci. 2015;16:377–388. doi: 10.1038/nrn3881. [DOI] [PubMed] [Google Scholar]
- 40.Hines RM, Davies PA, Moss SJ, Maguire J. Functional regulation of GABAA receptors in nervous system pathologies. Curr Opin Neurobiol. 2012;22:552–558. doi: 10.1016/j.conb.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cecil CA, Smith RG, Walton E, Mill J, McCrory EJ, Viding E. Epigenetic signatures of childhood abuse and neglect: implications for psychiatric vulnerability. J Psychiatr Res. 2016;83:184–194. doi: 10.1016/j.jpsychires.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 42.Nissen JB, Hansen CS, Starnawska A, Mattheisen M, Borglum AD, Buttenschon HN, Hollegaard M. DNA methylation at the neonatal state and at the time of diagnosis: preliminary support for an association with the estrogen receptor 1, gamma-aminobutyric acid B receptor 1, and myelin oligodendrocyte glycoprotein in female adolescent patients with OCD. Front Psychiatry. 2016;7:35. doi: 10.3389/fpsyt.2016.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gumerov V, Hegyi H. MicroRNA-derived network analysis of differentially methylated genes in schizophrenia, implicating GABA receptor B1 [GABBR1] and protein kinase B [AKT1] Biol Direct. 2015;10:59. doi: 10.1186/s13062-015-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Diorio J, Meaney MJ. Maternal programming of defensive responses through sustained effects on gene expression. J Psychiatry Neurosci. 2007;32:275–284. [PMC free article] [PubMed] [Google Scholar]
- 45.Van den Hove DL, Kenis G, Brass A, Opstelten R, Rutten BP, Bruschettini M, Blanco CE, Lesch KP, Steinbusch HW, Prickaerts J. Vulnerability versus resilience to prenatal stress in male and female rats; implications from gene expression profiles in the hippocampus and frontal cortex. Eur Neuropsychopharmacol. 2013;23:1226–1246. doi: 10.1016/j.euroneuro.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 46.Dempster EL, Wong CC, Lester KJ, Burrage J, Gregory AM, Mill J, Eley TC. Genome-wide methylomic analysis of monozygotic twins discordant for adolescent depression. Biol Psychiatry. 2014;76:977–983. doi: 10.1016/j.biopsych.2014.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, Plomin R, Mill J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morelius E, He HG, Shorey S. Salivary cortisol reactivity in preterm infants in neonatal intensive care: an integrative review. Int J Environ Res Public Health. 2016;13. [DOI] [PMC free article] [PubMed]
- 49.Gunnar MR. Studies of the human infant’s adrenocortical response to potentially stressful events. New Dir Child Dev. 1989;45:3–18. [DOI] [PubMed]
- 50.Davies MN, Volta M, Pidsley R, Lunnon K, Dixit A, Lovestone S, Coarfa C, Harris RA, Milosavljevic A, Troakes C, et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012;13:R43. doi: 10.1186/gb-2012-13-6-r43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fisher HL, Murphy TM, Arseneault L, Caspi A, Moffitt TE, Viana J, Hannon E, Pidsley R, Burrage J, Dempster EL, et al. Methylomic analysis of monozygotic twins discordant for childhood psychotic symptoms. Epigenetics. 2015;10:1014–1023. doi: 10.1080/15592294.2015.1099797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. J Hum Genet. 2013;58:434–438. doi: 10.1038/jhg.2013.65. [DOI] [PubMed] [Google Scholar]
- 53.Masliah E, Dumaop W, Galasko D, Desplats P. Distinctive patterns of DNA methylation associated with Parkinson disease: identification of concordant epigenetic changes in brain and peripheral blood leukocytes. Epigenetics. 2013;8:1030–1038. doi: 10.4161/epi.25865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hannon E, Lunnon K, Schalkwyk L, Mill J. Interindividual methylomic variation across blood, cortex, and cerebellum: implications for epigenetic studies of neurological and neuropsychiatric phenotypes. Epigenetics. 2015;10:1024–1032. doi: 10.1080/15592294.2015.1100786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, Houseman EA, Izzi B, Kelsey KT, Meissner A, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10:949–955. doi: 10.1038/nmeth.2632. [DOI] [PubMed] [Google Scholar]
- 56.Yousefi P, Huen K, Quach H, Motwani G, Hubbard A, Eskenazi B, Holland N. Estimation of blood cellular heterogeneity in newborns and children for epigenome-wide association studies. Environ Mol Mutagen. 2015;56:751–758. doi: 10.1002/em.21966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bakulski KM, Feinberg JI, Andrews SV, Yang J, Brown S, LMcKenney S, Witter F, Walston J, Feinberg AP, Fallin MD. DNA methylation of cord blood cell types: applications for mixed cell birth studies. Epigenetics. 2016;11:354–362. doi: 10.1080/15592294.2016.1161875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cardenas A, Allard C, Doyon M, Houseman EA, Bakulski KM, Perron P, Bouchard L, Hivert MF. Validation of a DNA methylation reference panel for the estimation of nucleated cells types in cord blood. Epigenetics. 2016;11:773–779. doi: 10.1080/15592294.2016.1233091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Simpkin AJ, Suderman M, Gaunt TR, Lyttleton O, McArdle WL, Ring SM, Tilling K, Davey Smith G, Relton CL. Longitudinal analysis of DNA methylation associated with birth weight and gestational age. Hum Mol Genet. 2015;24:3752–3763. doi: 10.1093/hmg/ddv119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bakulski KM, Lee H, Feinberg JI, Wells EM, Brown S, Herbstman JB, Witter FR, Halden RU, Caldwell K, Mortensen ME, et al. Prenatal mercury concentration is associated with changes in DNA methylation at TCEANC2 in newborns. Int J Epidemiol. 2015;44:1249–1262. doi: 10.1093/ije/dyv032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Engel SM, Joubert BR, Wu MC, Olshan AF, Haberg SE, Ueland PM, Nystad W, Nilsen RM, Vollset SE, Peddada SD, London SJ. Neonatal genome-wide methylation patterns in relation to birth weight in the Norwegian mother and child cohort. Am J Epidemiol. 2014;179:834–842. doi: 10.1093/aje/kwt433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Finer S, Mathews C, Lowe R, Smart M, Hillman S, Foo L, Sinha A, Williams D, Rakyan VK, Hitman GA. Maternal gestational diabetes is associated with genome-wide DNA methylation variation in placenta and cord blood of exposed offspring. Hum Mol Genet. 2015;24:3021–3029. doi: 10.1093/hmg/ddv013. [DOI] [PubMed] [Google Scholar]
- 63.Joubert BR, Haberg SE, Bell DA, Nilsen RM, Vollset SE, Midttun O, Ueland PM, Wu MC, Nystad W, Peddada SD, London SJ. Maternal smoking and DNA methylation in newborns: in utero effect or epigenetic inheritance? Cancer Epidemiol Biomark Prev. 2014;23:1007–1017. doi: 10.1158/1055-9965.EPI-13-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kile ML, Houseman EA, Baccarelli AA, Quamruzzaman Q, Rahman M, Mostofa G, Cardenas A, Wright RO, Christiani DC. Effect of prenatal arsenic exposure on DNA methylation and leukocyte subpopulations in cord blood. Epigenetics. 2014;9:774–782. doi: 10.4161/epi.28153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Koestler DC, Avissar-Whiting M, Houseman EA, Karagas MR, Marsit CJ. Differential DNA methylation in umbilical cord blood of infants exposed to low levels of arsenic in utero. Environ Health Perspect. 2013;121:971–977. doi: 10.1289/ehp.1205925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharp GC, Lawlor DA, Richmond RC, Fraser A, Simpkin A, Suderman M, Shihab HA, Lyttleton O, McArdle W, Ring SM, et al. Maternal pre-pregnancy BMI and gestational weight gain, offspring DNA methylation and later offspring adiposity: findings from the Avon Longitudinal Study of Parents and Children. Int J Epidemiol. 2015;44:1288–1304. doi: 10.1093/ije/dyv042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Schematic overview of the study design from data processing to validation of our main findings. Figure S2. Overview of the GABBR1 gene based on UCSC genome browser. Figure S3. Salivary cortisol measurements of infants at 2, 4 and 12 months old. (DOCX 1379 kb)
GABBR1 CpG sites analyzed by EpiTYPER and corresponding HM450 CpG probe identifiers. (XLSX 9 kb)
Top 50 DMRcate results. (XLSX 17 kb)
ErmineJ Gene Ontology (GO) pathways analysis results based on the top 500 annotated differentially annotated regions. (XLSX 14 kb)
Top 100 ranked CpG probes associated with prenatal anxiety. (XLSX 20 kb)
Overview of CpG probes in the GABBR1 region identified by DMRcate. (XLSX 14 kb)
Correlations between HM450 CpG probes and their respective CpG units measured by Sequenom EpiTYPER (n = 41). (XLSX 12 kb)
Data Availability Statement
Data generated by the HumanMethylation450 BeadChip array discussed in this publication are available in NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) through the accession number GSE104376.