

ABSTRACT
Deficiency of interleukin (IL)-36 receptor antagonist (DITRA) syndrome is a rare autosomal recessive disease caused by mutations in IL36RN. IL-36R is a cell surface receptor and a member of the IL1R family that is involved in inflammatory responses triggered in skin and other epithelial tissues. Accumulating evidence suggests that IL-36R signaling may play a role in the pathogenesis of psoriasis. Therapeutic intervention of IL-36R signaling offers an innovative treatment paradigm for targeting epithelial cell-mediated inflammatory diseases such as the life-threatening psoriasis variant called generalized pustular psoriasis (GPP). We report the discovery and characterization of MAB92, a potent, high affinity anti-human IL-36 receptor antagonistic antibody that blocks human IL-36 ligand (α, β and γ)-mediated signaling. In vitro treatment with MAB92 directly inhibits human IL-36R-mediated signaling and inflammatory cytokine production in primary human keratinocytes and dermal fibroblasts. MAB92 shows exquisite species specificity toward human IL-36R and does not cross react to murine IL-36R. To enable in vivo pharmacology studies, we developed a mouse cross-reactive antibody, MAB04, which exhibits overlapping binding and pharmacological activity as MAB92. Epitope mapping indicates that MAB92 and MAB04 bind primarily to domain-2 of the human and mouse IL-36R proteins, respectively. Treatment with MAB04 abrogates imiquimod and IL-36-mediated skin inflammation in the mouse, further supporting an important role for IL-36R signaling in epithelial cell-mediated inflammation.
KEYWORDS: Generalized Pustular Psoriasis, IL-36R, IL1RL2, IL1R family, antagonist antibody
Abbreviations
- AUC
analytical ultracentrifugation
- CH
constant heavy region
- CL/F
clearance/ fraction absorbed (bioavailability)
- DITRA
deficiency of interleukin 36-receptor antagonist
- GAHA
goat anti-human IgG gamma antibody
- GPP
generalized pustular psoriasis
- IL-8
interleukin-8
- IL-36
Interleukin-36
- PBS
phosphate-buffered saline
- RU
resonance units
- SPR
surface plasmon resonance
- VH
variable heavy region
- Vκ
variable kappa
Introduction
Interleukin (IL)-36 cytokines are members of the IL-1 superfamily that signal through the heterodimeric IL-36 receptor (IL-36R), leading to activation of nuclear factor- κB (NF- κB) and mitogen-activated protein kinases (MAPKs).1-6 There are three IL-36 agonistic cytokines, IL-36α, IL-36β, and IL-36γ, and an antagonistic cytokine, IL-36Ra (Fig. 1).3,7 Members of the IL-36 family are located on chromosome 2 in close proximity to other members of the IL1 superfamily.8 The IL-36 signaling pathway shares some similarities with the IL-1 signaling in that both pathways activate NF-κB and MAPKs, and both the IL-1R and IL-36R heterodimers share the β chain of the IL-1RacP molecule that is required for the trimer signaling complex.9,10 In psoriatic tissues, IL-36 ligands are expressed at higher levels compared with IL-1 β after IL-17 stimulation.11 More recently, investigation into the signaling mechanism of IL-36R has demonstrated differences between IL-1R and IL-36R after ligand binding.12 In the absence of cognate agonists, IL-36R is endocytosed by clathrin and recycled to the plasma membrane. In the presence of IL-36 ligands, IL-36R increased accumulation in LAMP1+ lysosomes. In contrast, IL-1R is endocytosed via caveolae and is degraded after reaching the endosome. Both IL-1R and IL-36 can interact with Tollip; however, while this interaction targets IL-1R for degradation, it stabilizes IL-36R. Thus, IL-1R and IL-36R have distinct mechanisms for endocytosis and degradation that differentially regulate signaling and function. These differences support the concept that IL-36 signaling may have a unique pathogenic contribution to inflammatory diseases.
Figure 1.
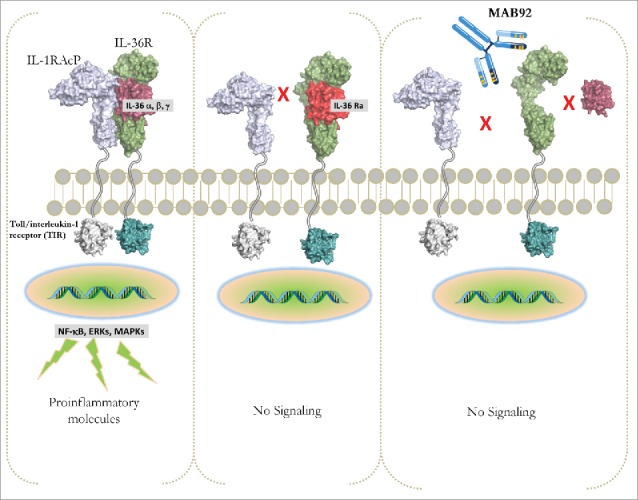
IL-36R signaling. Binding of the proteolytically maturated agonistic IL-36 ligands (α, β and γ) to the IL-36R results in the recruitment of the co-receptor (IL-1RAcP) and lead to activation of the NFκB, MAPK and ERK. Intracellular signaling activates gene transcription of proinflammatory mediators. IL-36Ra competes with IL-36 for IL-36R binding, thereby acting as an IL-36 antagonist. IL-36 can directly or indirectly stimulate various cellular responses and is involved in models of psoriasis and in generalized pustular psoriasis in humans. Binding of MAB92 to huIL-36R would antagonize the IL-36R signaling.
Members of the IL-36 family were first observed to be most highly expressed in squamous epithelial tissues, and in particular overexpressed in skin of psoriatic lesions.5,13 The link between IL-36R and epithelial-mediated diseases was confirmed in humans based on loss of function mutations in IL-36Ra that lead to generalized pustular psoriasis (GPP).4,14,15 Elevated IL-36α mRNA and protein expression was reported also in chronic kidney disease.16 IL-36R signaling may contribute to inflammation and tissue homeostasis in rheumatoid arthritis, psoriatic arthritis,17 asthma, chronic obstructive pulmonary disease18 and inflammatory bowel diseases.19-21 Initial support for a role of IL-36R signaling in driving epithelial-mediated inflammation stemmed from findings demonstrating that transgenic mice overexpressing IL-36α in keratinocytes exhibit inflammatory skin lesions with some features common to human psoriasis.13 This phenotype was more severe when transgenic mice were crossed with IL-36Ra-deficient mice, supporting a regulatory function of IL-36Ra in vivo. The inflammatory skin condition in the keratinocyte-specific IL-36α transgenic is even more similar to human psoriasis if the mice are treated with 12-O-tetradecanoylphorbol 13-acetate, resembling the human disease histologically, molecularly, and in its response to therapeutics. Moreover, human psoriatic lesional skin transplanted onto immunodeficient mice is normalized when the mice are treated with anti-IL-36R antibody,5 demonstrating that the IL-36 axis is required to maintain the lesional phenotype in human psoriatic skin. GPP is a rare, life-threatening variant of psoriasis that is characterized by widespread eruption of sterile, subcorneal pustules and epidermal scaling coupled with systemic symptoms. A homozygous loss-of-function mutation in IL-36RN, L27P, was first identified in GPP patients.4 Recently, different mutations were found by exome sequencing on unrelated individuals with GPP in Caucasians and Asian populations.14,22
Taken together, these data indicate that IL-36R ligands, including IL-36α, IL-36β, and IL-36γ, exert proinflammatory effects in vitro and in vivo, and that IL-36Ra act as a natural antagonist, similar to but distinct from the well-characterized IL-1/IL-1Ra system. There is evidence that IL-36R ligands are involved in several disease conditions, and there is a need for new therapeutic agents targeting this pathway, in particular for use in the treatment of inflammatory diseases such as GPP and other epithelial mediated inflammatory diseases.23-37
Here, we report the identification and characterization of a high affinity humanized antagonistic antibody (MAB92) that targets human IL-36R. MAB92 exhibits exquisite species specificity as it is highly specific for the human IL-36R. To enable in vivo studies aimed at enhancing the understanding of IL-36R's role in modulation of inflammatory pathways, we identified an antagonistic antibody (MAB04) targeting mouse IL-36R as a surrogate that shares key molecular attributes with MAB92.
Results
Immunization, screening and epitope binning to identify functional lead mouse anti-human IL-36R monoclonal antibody
High affinity anti-IL-36R antibodies were identified by immunizing mice with recombinant human IL-36R (huIL-36R). The selection of mice for fusion was driven by the assessment of serum titers. In all, we performed 4 fusions and screened >50,000 fusion-product supernatants that led to the identification of ∼7100 clones specific for binding to huIL-36R. The 150 hits were subsequently sub-cloned by limited-dilution method. The hybridoma supernatants were purified by affinity chromatography (protein-A) and purified mouse monoclonal antibodies (mAbs) were used for subsequent screening. Eight functionally potent mAbs (IC90< 5 nM, Table 1) were identified by screening for the blockade of NFκB activation induced by IL-36 ligands in the NCI/ADR-RES ovarian epithelial cell line that express endogenous functional IL-36R. Target engagement and selectivity were determined by counter-screening against IL-1β-induced NFκB in the same cells and the use of an NCI/ADR-RES IL-36R knockout (KO) cell line, respectively. All of these clones had high affinity binding as determined by surface plasmon resonance (SPR) in the pM range (Table 1). The variable genes were isolated by standard PCR-based methodology as described in detail in the Materials and Methods section. The antibodies were binned by cross-competition using SPR. Clone mMAB92 ranked as the top hit based on the profiling criteria, including binding, function and sequence quality, and was thus chosen for humanization.
Table 1.
Assessment of biophysical properties.
| Mouse anti-huIL-36R lead antibodies |
ka (M−1s−1) |
kd (s−1) |
KD (pM) |
NCI/ADR-RES IC90 (nM) |
Human primary dermal fibroblast IC90 (nM) |
| mMAB92 | 1.36 × 106 | 3.28 × 10−5 | 24 | 0.9 | 1.9 |
| mMAB93 | 1.1 × 106 | 1.56 × 10−5 | 14 | 0.9 | n.d. |
| mMAB94 | 1.53 × 106 | 2.38 × 10−5 | 16 | 1.3 | n.d. |
| mMAB95 | 9.3 × 105 | 5.2 × 10−5 | 55 | 3.8 | n.d. |
n.d.: not determined
Humanization
A mouse-human chimeric antibody (Chi.MAB92) was generated by sub-cloning the mouse variable region of mMAB92 with human constant domains (CH1–3 domain of IgG1). Chi.MAB92 was re-profiled (for binding and function, data not shown) to ensure that its properties remained unchanged, thus confirming seamless v-gene recovery from the hybridoma clone. Humanization of the mMAB92 antibody essentially followed the protocol described by Singh et al.38 Briefly, we selected the most homologous human germ-line antibody sequences, IGKV3–20*01, KJ2 for the light chain and IGHV1–46*03, HJ4 for the heavy chain. To evaluate the framework residue requirements for maintaining mMAB92 functionality, a library of variants was generated that mostly focused on residues flanking the complementarity-determining regions (CDRs), in which each residue being considered was altered between the human and murine residue.39 The variable heavy (VH) chain of mMAB92 (murine) fused with human constant region 1 (CH1) was used as the heavy chain in expressing the light chain variants (humanized) as antigen-binding fragments (Fabs) using a M13-phage secretion system. The variable kappa (Vκ) chain was sequence optimized in the context of the parental VH chain to lower the possibility of functional or epitope drift. The variants were evaluated by an ELISA for huIL-36R binding to sufficiently cover the diversity in the library. A chimeric mMAB92 Fab with murine variable regions grafted to human constant regions was produced to serve as a positive control for screening and measuring the maintenance of affinity and potency.
Approximately 195 candidate hits with a binding affinity for huIL-36R greater than or equal to the Chi. MAB92 Fab were sequenced and ranked to identify those with the lowest number of retained mouse residues in the framework regions and the most favorable Epivax (in silico immunogenicity prediction) scores.40 The top three optimized leads were subcloned into pTT5 vector system IgG1 expression cassettes, using standard PCR restriction enzyme-based cloning techniques. The Fc domain of IgG1 was engineered to reduce effector functions by two mutations (L234A/L2345A)38. The sequence optimized IgGs was expressed transiently in CHO-E cells.41 Based on the binding affinity and potency data, MAB92 was selected for further profiling. Table 2 shows the KD for MAB92 binding to hIL-36R. In addition, MAB92 did not show any detectable binding to human IL-1R at concentrations up to 1.0 μM (Suppl.Fig. S1).
Table 2.
Assessment of biophysical properties.
| Parameter |
MAB92 |
| Binding affinity by surface plasmon resonance (hIL-36R) | KD = 20 pM |
| Aggregation after 2-step purification process (% monomer) | 98 |
| pI as determined by isoelectric focusing | >8 |
| Thermal stability (DSC- Fab Tm)) | 76.1°C |
| Deamidation within the Fab (pH 9.0 @ 37°C for 1 week) | <5% |
| Whole blood stability (48 h at 37°C) | <2-fold |
| Valence at pH 5.0 | +21.7 |
| Solubility (mg/ml) | 127 |
Assessment of functional potency
MAB92 was tested for blockade of the agonistic IL-36 ligand (α,β,γ)-induced activation of NFκB and IL-8 (Fig. 2A-C) production in primary human intestinal myofibroblasts, dermal fibroblasts and human keratinocytes. The values calculated from the dose response curves (at a concentration of ligand representing the EC80 of binding) demonstrate potent functional blockade (results for the primary cells and 2 relevant human cell lines are shown in Table 3).
Figure 2.
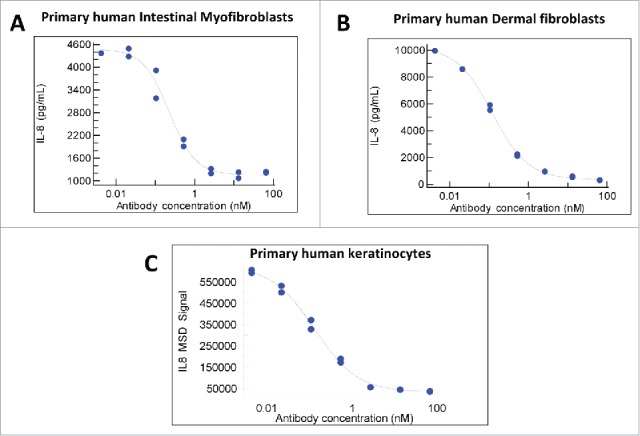
Functional potency. Blockade of IL-36 ligand induced IL-8 secretion by MAB92. The agonistic IL-36 ligand (α,β,γ)-induced activation of NFκB and IL-8 production in primary human intestinal myofibroblasts (a), dermal fibroblasts (b) and human keratinocytes (c) was effectively neutralized by MAB92.
Table 3.
Assessment of functional potency for MAB92 in primary cells.
| |
Agonist Ligand |
IC90 nM (SD) |
| Human NCI/ADR-RES ovarian epithelial cancer cell line | IL-36 α | 1.8 (0.3) |
| IL-36 β | Two.4 (1.1) | |
| IL-36 γ | One.5 (0.6) | |
| Human primary intestinal myofibroblasts | IL-36 α | 3.2 (1.6) |
| IL-36 β | Two.8 (1.1) | |
| IL-36 γ | Three.7 (3.5) | |
| Human primary keratinocytes | IL-36 α | 2.2 (0.1) |
| IL-36 β | One.9 (0.5) | |
| IL-36 γ | 0.7 (0.1) | |
| Human primary dermal fibroblasts | IL-36 α | 3.8 (1.9) |
| IL-36 β | Nine.0 (0.9) | |
| IL-36 γ | Sixteen (7.7) | |
| Cynomolgus monkey primary dermal fibroblasts | IL-36 α | |
| IL-36 β | >3000 | |
| IL-36 γ |
Assessment of biophysical properties
As a measure of specificity, MAB92 was analyzed for binding to human IL-36R in the presence of 50% human serum to assess the potential for interference due to non-specific protein binding. The ratio of the on-rate in serum relative to buffer was <2 for MAB92, demonstrating that the antibody did not associate with serum proteins, and was specific for its target (Table 2). The stability of MAB92 was also assessed in whole blood (for 2 days at 37°C). No significant change in binding to huIL-36R was observed. In addition to assessing the purity and homogeneity using standard methods such as SDS-PAGE and analytical size-exclusion chromatography, we examined homogeneity using analytical ultracentrifugation (AUC) for MAB92 (Table 2). AUC is a solution technique requiring no separation media or membranes that can lead to the loss of sample components due to protein/gel matrix interactions. The valence of MAB92 at pH 5.0 was measured as an assessment of its solubility and the likelihood for low viscosity at high concentrations. The valence values are shown in Table 2. A solubility concentration test was also performed using 60 mM sodium citrate, 115 mM NaCl, pH 6.0 buffer to assess the potential challenges of concentrating the antibody and propensity for aggregation in concentrated form. MAB92 was concentrated to >100 mg/ml and assessed for monomer percentage by sedimentation velocity. MAB92 remained >96% monomers at this concentration without additional formulation (Table 2). The thermal stability of MAB92 was measured by differential scanning calorimetry (DSC). MAB92 showed the thermal unfolding profile that is typical for mAbs. The level of deamidation within the Fab region was significantly lower when assessed using an accelerated stability test condition (Table 2). In summary, MAB92 showed favorable biophysical attributes.
Epitope Mapping
To map the binding site on human IL-36R, epitope mapping was performed by hydrogen-deuterium exchange mass spectrometry. Clone mMAB92 was incubated with an equimolar concentration of IL-36R in a deuterated buffer. IL-36R protein alone served as the control. Extent of protection was inferred by measuring the differences in hydrogen/deuterium exchange between IL-36R alone and IL-36R in complex with the antibody. The protection map (Fig. 3A-B) and further refinements of the analysis showed significant protection of regions identified as residues: 9 to 14 (MKNEIL), 96 to 110 (EKHWCDTSIGGLPNL), 113 to 119 (YKQILHL), 149 to 154 (IKGERF) and 177 to 186 (QAILTHSGKQ) on IL-36R. This indicated that the lead antibody primarily bound to within domain-2 of the IL-36R. Substantial differences in sequence homology between human and cynomolgus monkey IL-36R were noted within the lead antibody binding epitope.
Figure 3.

Epitope mapping. (a) The binding epitope for the parental mouse anti-human IL-36R antibody and for the rat anti-mouse IL-36R antibody was mapped by hydrogen-deuterium exchange mass spectrometry. Both antibodies show overlapping binding preference to domain-2 on IL-36R. (b) A homology model of IL-36R extracellular domain, highlighting the overlapping binding epitope for mMAB92 and MAB04 on domain-2.
Exquisite specificity of MAB92 for human IL-36R: evaluation of cross-species binding for MAB92 to enable preclinical safety assessment
MAB92 does not show functional cross-reactivity with the cynomolgus IL-36 receptor under physiologic conditions. MAB92 was ∼1000 fold weaker in NFκB assays in primary cynomolgus dermal fibroblasts with IC90 of > 3000 nM, compared with primary human dermal fibroblasts. Using SPR, no detectable binding was observed when up to 15 μM of the Fab from MAB92 was incubated with the immobilized cynomolgus IL-36R, consistent with the poor functional activity observed in primary cynomolgus cell-based assays. Subsequently, we expressed and purified IL-36R from commonly used toxicology species such as rhesus, marmoset, mouse, rat, minipig and hamster. MAB92 did not bind recombinant IL-36R from these species up to 1 μM (data not shown).
Design and development of a surrogate antibody (MAB04, anti-mouse IL-36R) for in vivo studies
We developed MAB04, an anti-mouse IL-36R antibody by immunization in the rat. The Fc domain of MAB04 was also engineered to reduce effector function by two mutations (D265A, N297A).42,43 The engineered surrogate antibody (MAB04) was profiled in several in vitro/ in vivo studies and results are summarized in Table 3 (also see Fig. S2, Table 4). The rat-mouse chimeric IgG2a, MAB04, binds to mouse IL-36R and was profiled for use as a surrogate antibody for in vivo pharmacology studies and nonclinical drug safety studies in mice. In C57BL/6 mice, MAB04 was tested for its pharmacokinetic (PK) characteristics with a study design including three dose groups (0.3, 1.5 and 10 mg/kg intraperitoneal (i.p.)) to assess potential target-mediated drug disposition (TMDD) impact and saturability, and clearance/fraction absorbed (CL/F) across a dose range potentially covering the human therapeutic dose. Blood samples collected over one and two weeks for the 0.3 mg/kg and two higher dose groups, respectively, were analyzed using a validated ELISA bioanalytical method to determine drug concentrations. At the presumed TMDD-saturating dose of 10 mg/kg i.p., MAB04 CL/F in the mouse was 3.1 ± 0.4 mL/d/kg (Fig. 4). This value is comparable to that of clearance observed for a 1.5 mg/kg dose of the non-cross-reactive human lead, MAB92, in the monkey (4.5 ± 1.2 mL/d/kg). MAB04 clearance in mouse was dose-dependent with values for CL/F of 37.7, 11.2 and 3.1 for the 0.3, 1.5 and 10 mg/kg doses, respectively, suggestive of TMDD impact on clearance.
Table 4.
In vitro/ in vivo potency profiles for surrogate anti-mouse IL-36R antibody (MAB04).
| Parameter |
MAB04 |
| Unique binding epitope (IL-36R) | Domain-2 |
| Binding affinity (KD) (mouse IL-36RECD-Fc, Fab binding) | 238 pM |
| Functional binding avidity (KDapp) (mouse IL-36RECD-Fc, IgG) | 146 pM |
| Serum shift (mouse) (fold change in on-rate) | <2-fold |
| In vitro selectivity (SPR) (mouse IL-1R1) | No binding |
Figure 4.
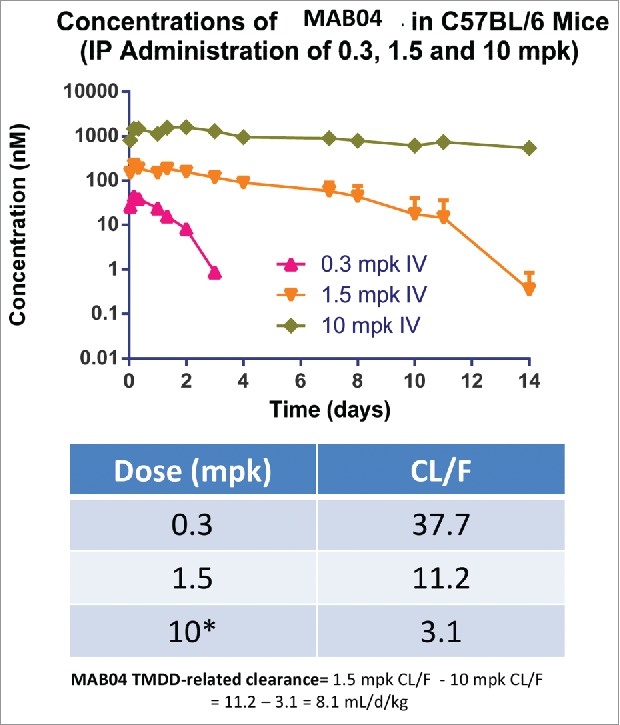
Pharmacokinetics profile of MAB04 in mice following a single intraperitoneal dose of 0.3, 1.5 or 10 mg/kg. MAB04 clearance in mouse was dose-dependent with values for CL/F of 37.7, 11.2 and 3.1 for the 0.3, 1.5 and 10 mg/kg doses, respectively suggestive of TMDD impact on clearance.
MAB04 inhibits skin inflammation in mice
To demonstrate the ability of MAB04 to inhibit IL-36R activity in vivo, we developed a model of IL-36-induced skin inflammation in mice. IL-36 cytokines (0.01 μg each of IL-36 α,β,γ) were injected intradermally into the mouse ear once daily for 2 consecutive days. Twenty-four hours following the second injection, ear thickness was measured and tissue harvested for protein extraction and RNA isolation. A single administration of MAB04 (250 μg i.p.) 24 hours before the first IL-36 injection was able to inhibit IL-36 induced ear swelling by 77% (Fig. 5A). Lipocalin (LCN2) in the ear tissue homogenate was measured as an indication of neutrophil infiltration. In addition to inhibiting IL-36-induced ear swelling, MAB04 also reduced LCN2 expression by 71% (Fig. 5B), suggesting a reduction in cellular inflammation as well as edema.
Figure 5.

(a) MAB04 inhibits IL-36 induced skin swelling in mice (b) MAB04 inhibits IL-36 induced production of Lipocalin2. Intradermal L-36 injection into ear of C57B/6 mice induced increases in ear thickness (swelling) and Lipocalin2 (LCN2) which was significantly inhibited by a single 250 µg i.p. injection of MAB04 before challenge. Mean +/− SEM. N = 8/group.
To further examine the role of IL-36R in skin inflammation, MAB04 was tested in imiquimod-induced skin inflammation model in mice. Imiquimod (Aldara cream, 5%), was applied daily to the ear of mice for 7 days. MAB04 or isotype control (250 μg i.p.) was administered day 0, 3 and 6. On day 7 ear thickness measurements were obtained and tissue harvested for protein isolation. Consistent with previous reports using IL-36R−/− KO mice,44 MAB04 reduced imiquimod-induced swelling by 63% compared with isotype control (Fig. 6A). Additionally, IL-36R blockade resulted in 68% reduction in IL17 production in skin homogenates (Fig. 6B).
Figure 6.
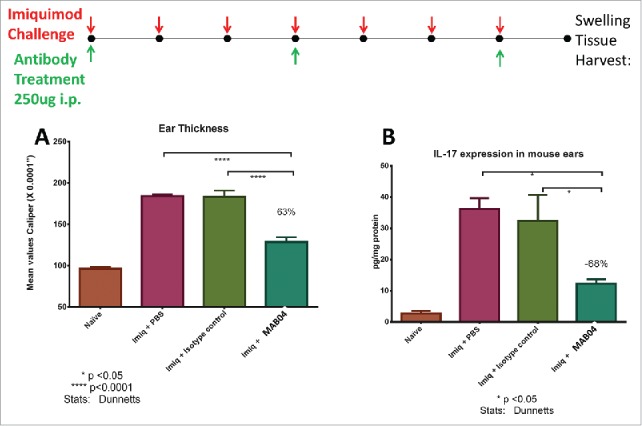
(a) MAB04 inhibits imiquimod induced skin swelling and (b) IL-17 production in skin of mice. Imiquimod cream (Aldara™) applied daily to mouse ears daily for 7 d induced robust ear swelling and IL-17 which was blocked by treatment with MAB04. Antibody was delivered i.p. every 3 d beginning 1 hour before initial imiquimod challenge. Values are Mean +/− SEM, n = 8/group.
Discussion
DITRA is a rare autosomal recessive disease caused by mutations in IL36RN.4, 14 Therapeutic intervention of IL-36R signaling offers an innovative treatment paradigm for GPP. Marketed therapeutics such as anakinra (IL-1RA), adalimumab (anti-tumor necrosis factor) and secukinumab (anti-IL-17A) are being currently explored as treatment options for GPP.45-47 Neutralizing of the IL-36 signaling by an antibody targeting IL-36R could in principle be achieved by interfering with the binding of IL-36 agonistic ligands or preventing the recruitment of IL1RAcP to IL-36/IL-36R complex. MAB92 was humanized from the mouse antibody (mMAB92), characterized in several in vitro pharmacological assays and profiled for CMC characteristics. MAB92 binds to recombinant and cell surface expressed human IL-36R with high affinity as demonstrated by SPR (affinity of 223 pM) and flow cytometry analysis. The functional activity of MAB92 is enhanced by avidity when binding to the IL-36 receptor expressed on the surface (EC90 = 3.6 ± 1.7 nM) of BaF/3 cells transfected with hIL-36R and IL1RAcP. Avidity is achieved through the bivalent nature of the antibody and the ability to simultaneously bind multiple receptors on the cell surface. In functional NF κB activation assays and cytokine (IL8) release assays in both the human NCI/ADR-RES cell line and in human primary intestinal myofibroblasts, human primary dermal fibroblasts, and human primary keratinocytes, MAB92 meets our criteria for IC90 < 5 nM for all 3 IL-36 ligands. MAB92 did not inhibit IL-1β-induced NFκB activation or cytokine release in the human NCI/ADR-RES cells or human primary cells. MAB92 was also assessed (in vitro) in complement-mediated cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) assays. As expected, the lead mAb did not elicit CDC or ADCC activities (data not shown).
To gain insights onto the mechanism of action, we sought to map the epitope for mAb binding to huIL-36R. The results from the epitope mapping analysis by HX-MS indicated that the primary binding site for the lead antibody is on domain-2 of human IL-36R. Domain-2 in the IL1R family has been shown to be pivotal for ligand binding. As shown in Fig. 3A-B, the sequence within the epitope is quite divergent among IL-36R from a variety of species. Notably, human and cynomolgus IL-36R share only ∼75% sequence homology within the epitope (see also Fig. S3). Nonetheless, the precise molecular determinants for narrow species cross-reactivity of MAB92 would be evident from the crystal structure of MAB92 in complex with IL-36R. A similar example of another therapeutic antibody (canakinumab, anti-IL-1β) was recently reported to be not cynomolgus/rhesus cross-reactive despite high homology (∼96%).48 As MAB92 showed insufficient (IC90 > 3 μM) pharmacological profile in blocking the functional activity of cynomolgus IL-36R, we identified, MAB04 as a surrogate for in vivo pharmacology studies and preclinical toxicology assessment in mice. Mouse mAbs reactive with mouse antigens are often needed for early preclinical proof-of concept studies in development programs. Our choice for MAB04 as surrogate stems from our observations on analogous binding and pharmacological profile of MAB04 and MAB92. MAB04 binds to mouse IL-36R (Table 4) with a marginally reduced binding affinity compared with MAB92s binding to human IL-36R (Table 2). The functional potency of MAB04 toward mouse IL-36R is similar to MAB92 in several primary human cell-based assays. The epitope for MAB04 on the mouse IL-36R overlaps with the epitope for MAB92 on human IL-36R as they both primarily bind to domain-2.
The IL-36 receptor family of cytokines has been extensively linked to psoriasis and this has been strengthened by human genetic mutations in the IL-36 pathway linked to skin inflammatory diseases.4,14,15 Mouse models of skin inflammation have demonstrated that IL-36R signaling is sufficient to induce skin inflammation5 and required for a full cascade of inflammatory processes in vivo.44 To further characterize MAB04 and support its use as a surrogate for MAB92, we performed several in vivo skin inflammation models where IL-36R has been shown to be instrumental. First, using direct injection of IL-36 cytokines into mouse skin, we were able to show that a single dose of MAB04 was able to block IL-36-induced skin inflammation. Both the swelling response as well as cellular inflammation resulting from IL-36 injection was significantly reduced by MAB04 (Fig. 6A, B).
To extend these observations and support the use of MAB04 to study IL-36R pharmacology, we evaluated the surrogate antibody in an imiquimod-induced skin inflammation model, relevant to human disease based on a report demonstrating exacerbation of psoriasis in patients treated with imiquimod.24 The importance of IL-36 pathway in the imiquimod mouse model has been supported by studies using IL-36R−/− KO mice where a significant reduction of imiquimod-induced inflammation was observed compared with wild type controls.44 In the same studies, the investigators show that the lack of IL-36R resulted in reduced numbers of IL17+ cells in the skin following imiquimod challenge. As the IL-23 pathway including IL-17 is known to be critical for human psoriasis, the link between IL-36 and IL-23 further supports the potential relevance of IL-36R signaling in human disease. Our in vivo data with MAB04 demonstrating both blockade of the swelling response as well as reduction of IL-17 protein in skin tissue of imiquimod-challenged mice confirms the role for IL-36R pathway in skin inflammation and supports the use MAB04 to interrogate the role of IL-36 pathway in epithelial-mediated inflammatory diseases. In conclusion, given the predominant expression of IL-36 family members in epithelial tissues (such as skin), a strong rationale exists for the development of anti-IL-36R therapy for the management of inflammatory pathways in psoriasis.
Materials and methods
Expression and Purification of recombinant human IL-36R
TransIT PRO system (Mirus Bio LLC) was used to transfect suspension HEK293F cells with full-length IL-36R (C-terminally fused with poly-Histidine tag). Transfected cells were incubated for 4 days. Media was harvested and applied to a Ni-NTA His-Bind Superflow column (Novagen) following the manufacturer's conditions. IL-36R eluted from the Ni column was further purified by size-exclusion chromatography.
Immunization, screening and identification of mMAB92 and mMAB04
Mice were immunized with various forms of recombinant IL-36R. Hybridomas were generated via fusions with PAI myeloma cells using standard methods. MabSelect SuRe resin (GE Healthcare) was used for antibody purification from cultured hybridoma supernatants. Binding of purified mouse anti-human IL-36R or rat anti-mouse IL-36R IgGs from three different immunization campaigns was performed on an Octet QK (ForteBio, Menlo Park, CA) equipped with streptavidin (SA) biosensor tips (ForteBio) to rank order hits. Variable gene sequences were recovered from hybridomas using standard methods and chimeric antibodies were generated to verify the characteristics of the recovered antibodies.
Expression and Purification of MAB92 and MAB04
CHO-3E7 (CHO-E) cells are maintained in an actively dividing state in FreeStyle CHO (FS-CHO) medium supplemented with 8 mM Glutamax at 37°C, 5% CO2, and 150 rpm shake speed. CHO-E cells are transfected at 2 × 10E6 cells/mL in FS-CHO supplemented with 2 mM glutamine. For 1 L transfection, 1 mg light chain DNA and 0.5 mg heavy chain DNA are diluted in 100 mL of OptiPro SFM and sterile filtered through a 0.2 µm filter. 1.5 mL of Mirus TransIT Pro transfection reagent is added to the diluted DNA mixture and allowed to incubate for 15–30 minutes at room temperature. After the incubation period, the complex is added to the prepared CHO-E cells, and the shake flask returned to the 37 °C, 5% CO2 shaker at 150 rpm. Four hours post transfection, 10 mL of Gibco Anti-Clumping Agent and 10 ml of Pen/Strep is added to the transfected cells. 24 hours post-transfection, 150 mL of CHO CD Efficient Feed B is added to the transfection and the temperature is shifted to 32 °C. The transfected culture is maintained for 6—12 days. Harvest is done by centrifuging at 4700 rpm, 4 °C for 40 minutes, followed by sterile filtration through a 0.2 µm filter. The clarified cell culture supernatant is sampled for titer by ForteBio/Pall Octet Red 96 instrument with Protein A biosensors; supernatant samples and reagents are equilibrated to room temperature before the experiment. The biosensors are pre-soaked in conditioned medium and the sample plate is incubated at 30 ˚C for 10 minutes before data acquisition. The experiment is performed at 200 rpm and data acquired for 120 seconds. The raw data are imported into the analysis software and compared with an appropriate standard curve of matching IgG isotype and medium.
Sample is captured from the harvested cell culture fluid (HCCF) by recombinant Protein-A affinity chromatography using MabSelect SuRe resin from GE healthcare. The column was pre-equilibrated with DPBS buffer (8.05 mM sodium phosphate, 137 mM NaCl, 1.47 mM potassium phosphate, 2.6 mM potassium chloride, pH 7.2). The column was washed with 5 column volumes (CVs) of DPBS followed by 5 CVs of DPBS plus 0.5 M NaCl to remove DNA, endotoxin, cell culture media components, host cell proteins and aggregates. The column was then re-equilibrated in Wash Buffer 1 for 2 CVs. Protein was eluted from the column using 30 mM sodium acetate pH 3.5. Fractions were pooled based on the UV absorbance at 280 nm, where fractions showing >15 mAu were pooled. Pooled samples were then neutralized using 1% by volume 3 M sodium acetate pH 9.0 to bring the final formulation to 60 mM sodium acetate pH 5.0. Neutralized protein is sterile filtered into a 50 mL Falcon tube by 0.22 µm Millipore Steriflip using vacuum filtration system.
As a polishing step, the affinity-chromatography purified sample was further purified by Poros 50 HS column on an ÄKTA system at 4 °C. The antibody was bound to the column that was pre-equilibrated in 60 mM sodium acetate, pH 5.0 and washed in the same buffer. The second wash was performed with 20 mM citrate, pH 6.0. The majority of the negatively charged process-related impurities, such as residual DNA, endotoxin, and some host cell proteins, were removed during loading and washing steps. The antibody was then eluted gradiently using 20 mM citrate supplemented with 1 M NaCl, pH 6.0 in 20 CV, and the eluted sample was adjusted to the final ionic strength of 150 mM NaCl. Samples were pooled based on purity in an SDS-PAGE analysis. The protein was sterile filtered into a 50 mL Falcon tube by 0.22 µm Millipore Steriflip using vacuum filtration system. The concentration was measured by UV280 by nanodrop 8000 spectrophotometer. The endotoxin level in the purified antibody was determined by LAL cartridges, Charles River Laboratories.
Humanization and Sequence optimization of mMAB92
Humanization was performed using M-13-based Fab expression system methods described previously.38,39 Libraries for the light chain and heavy chain were designed individually, wherein selected framework positions would contain the mouse amino acid residue or the amino acid from the chosen human germline template and selected CDR positions would undergo saturation mutagenesis using the NNK codon or custom mutagenesis using unique mixtures of codons. A Fab library of variants was screened in a binding ELISA to determine which variants maintain binding potency equivalent to or greater than the chimera. Favored residues at the library positions were determined by >80% prevalence within the confirmed hits. Sequences were analyzed for percent human in the framework regions and for potential immunogenicity using the Treg adjusted scores from the EpiVax Epimatrix in silico immunogenicity prediction program.49
Analytical Ultracentrifugation
All experiments were conducted on a Beckman XLI analytical ultracentrifuge (Beckman Coulter, Inc., Fullerton, CA). All sedimentation velocity experiments were conducted at 40,000 rpm and 20 °C. Experiments were conducted in a pH 6.0 buffer containing 20 mM citrate and 115 mM NaCl. Data were collected at 280 nm and were analyzed using continuous c(S) model in SedFit version 12.1c.50
Differential scanning calorimetry
Thermal unfolding and aggregation of sequence-optimized anti-human IL-36R IgGs at a concentration of 1 mg/ml in 20 mM sodium citrate, 115 mM sodium chloride, pH 6.0 buffer were monitored from 25 °C to 110 °C at a scan rate of 60 °C/hr via an automated capillary DSC (MicroCal, LLC, Northampton, Massachusetts). The data were analyzed using Origin 7.0 software (Origin-Lab, Northampton, Massachusetts). All thermograms were baseline corrected and fitted using the two-state model in Origin to obtain apparent midpoint temperatures (Tm) of unfolding.
Solubility
The sequence-optimized antibodies were concentrated from 10 mg/ml to 120 mg/ml using Amicon ultra centrifugal filter 15 mL with MWCO of 50 kDa (EMD Millipore, Billerica, MA). After concentration, the solution concentrations were determined by UV/VIS Nanodrop 8000 (Thermo Scientific, Wilmington, DE) at A280 nm of a solution diluted to < 40 mg/ml. The homogeneity of the concentrated antibody samples was analyzed via AUC following the method described above.
Serum interference
The interaction of each of the antibodies with human IL-36R in 1 x kinetic buffer and human serum (Sigma, St. Louis, MO) was performed on an Octet QK (ForteBio) instrument equipped with streptavidin (SA) biosensor tips (ForteBio). The sensors captured with biotinylated human IL-36R were dipped in human serum to establish a baseline for the binding in serum before the interaction of MAB92 with human IL-36R. The response of the binding sensorgrams in 1x kinetic buffer and human serum at different association time points (60 sec, 120 sec, and 240 sec) were compared with determine if the antibodies bound to off-target molecules in human serum.
Molecular affinity of human IL-36R binding to MAB92
ProteOn XPR36 (Bio Rad, Hercules, CA) was used to measure the kinetics and affinity of human IL-36R binding to the sequence optimized antibodies. Goat anti-human IgG gamma Fc specific (GAHA) (Invitrogen, Grand Island, NY, Catalog# 31125) was immobilized to the dextran matrix of a GLM chip (Bio Rad, Hercules, CA) along 6 horizontal channels using an amine coupling kit (Bio Rad, Hercules, CA) at a surface density between 8000 RU and 10000 RU according to the manufacturer's instructions. MAB92 was captured to the GAHA surface along 5 vertical channels at a surface density of ∼200 RU. The last vertical channel was used as a column reference to remove bulk shift. The binding kinetics of human IL-36R to each antibody were determined by global fitting of duplicate injections of human IL-36R at five dilutions (10, 5.0, 2.5, 1.25, 0.625, and 0 nM). The collected binding sensorgrams of human IL-36R at five concentrations with duplicates were double referenced using inactive channel / inter-spot reference and extraction buffer reference. The referenced sensorgrams were fit into 1:1 Langmuir binding model to determine association rate (ka), dissociation rate (kd), and dissociation constant (KD). Selectivity for MAB92 binding to IL-1R1 was determined in a similar experimental set-up. Duplicate injections of four dilutions (1, 0.5, 0.25, 0.125 μM) of recombinant human IL-1R1 (R&D systems, catalog# 269–1R-100) were used as analyte. A human anti-IL-1R1 mAb was used as a positive control.51
Epitope mapping: Identification of peptides and deuterium exchange experiments
Protein alone or protein/antibody complex were mixed with an excess volume of buffer (50 mM phosphate-buffered saline (PBS) in H2O) or deuterated buffer (50 mM PBS in D2O). After 100 seconds at room temperature, urea/TCEP was added and mixed. After 1 minute, pepsin or protease XIII P was added and the entire volume was immediately transferred to a 96-well plate, at 4 °C. After 5 minutes, 50 μl of this solution was injected onto a Phenomenex Jupiter C5 column The peptides were eluted from the column with a gradient of Mobile Phase A (water/acetonitrile/formic acid, 99/1/0.1) and Mobile Phase B (acetonitrile/water/formic acid, 95/5/0.1). Time = 0 min (3%B), Time = 2.2 min (3%B), Time = 10.1 min (90%B), Time = 12.0 min (90%B), Time = 12.1 min (3%B). Three replicates were done for both the control (IL-36R alone) and experiment (IL-36R / antibody complex). The effluent was directed into a Thermo Orbitrap Velos mass spectrometer and electrospray ionization was used for online monitoring of the chromatographic effluent. For peptide identification experiments, data was acquired over 12 minutes (3 minute delay start time), in full-scan mode at 30,000 resolution followed by seven ion trap data dependent scans (CID). Peptides were identified using fragmentation data and the program Proteome Discoverer (Thermo Scientific). For deuterium-exchange experiments, data was acquired over 12 minutes (3 minute delay start time), in full-scan mode at 60,000 resolution. Data was analyzed using the program PepMap (Thermo Scientific).
Pharmacokinetic analysis
Serum concentrations of MAB04 were determined using a validated, antigen-capture ELISA assay. Briefly, recombinant mouse IL-1 Rrp2/IL-1 R6 (R&D Systems, Catalog# 2354-RP-100), was immobilized on Nunc MaxiSorp 96-well plates (Affimetrix eBioscience, San Diego, CA). Plates were washed and then blocked with 5% bovine serum albumin (BSA) with 0.05% Tween, w/v of SeraCare in PBS. Matrix reference standards, quality control and test samples were then diluted to a final concentration of 10% C57BL6 serum and transferred to the blocked plate. Plates were washed before addition of goat anti-mouse IgG2a-horseradish peroxidase-conjugated (Southern Biotech, Catalog# 1030–05) at a concentration of 0.05 µg/ml. Plates were washed again then BioFx (SurModics, Eden Prairie, MN) TMBW substrate was added. Plates were allowed to develop for ∼2 minutes at room temperature before addition of BioFx liquid stop solution for TMB substrate (0.2 M H2SO4) and were then read using a SpectraMax (Molecular Devices, Sunnyvale, CA) M5 Plate Reader at OD 450 nm. Concentrations were derived by plotting standard curve concentrations versus 450 OD signal intensity in a log-log curve fit using Softmax Pro software (Molecular Devices, Sunnyvale, CA). Non-compartmental pharmacokinetic analysis was performed WinNonlin (v. 5.3, Pharsight Corporation, Mountain View, CA, USA). Areas under the serum concentration-time curve to the last quantifiable time point (AUC0-t) were calculated using the linear trapezoidal method and were extrapolated to time infinity (AUCinf) using log-linear regression of the terminal portion of the individual curves. The elimination rate constant (kel) was determined by least-squares regression of the log-transformed concentration data using the terminal phase, identified by inspection between days 1 and 14 and terminal half-life was equal to ln2/ kel.
IL-36 induced mouse skin inflammation
Female C57B/6 mice from Charles River Laboratories were used for all in vivo studies and all procedures were approved by Boehringer Ingelheim's Institutional Animal Care and Use Committee. Mouse IL-36 cytokines were purchased from R&D Systems and prepared in sterile filtered 0.1% BSA in PBS. Truncated forms of IL-36α (Cat#6995-IL), IL-36 β (Cat# 6834-IL) and IL-36 gamma (Cat#6835-IL) were combined and diluted to a final concentration of 1.5 µg/ml each and 20 µl was injected into ears intradermally into anesthetized mice on two consecutive days. Antibody treatments were administered in 200 µl volume intraperitoneally 24 hours before the first intradermal injection. 24 hours after the second skin injection, ear thickness measurements were collected using an engineer's micrometer. Ear tissue were collected and snap frozen in liquid nitrogen and homogenized in 1 ml of Tris-buffered saline with protease inhibitor. Samples were homogenized with fast-prep matrix tubes (MP-Biomedical) and centrifuged at 14,000 rpm at 4°C. Supernatants were stored at −20°C until assayed for lipocalin-2 with R&D Systems kit (Cat# MLCN20) according to manufacture protocol.
Imiquimod induced skin inflammation
20 µl volume imiquimod (Aldara 5%) in a glass syringe was applied to the dorsal side of both ears of isoflurane-anesthetized mice once daily for 7 challenges. 24 hours after the last challenge, ear-thickness was measured and mice were killed. Tissues were collected and snap frozen homogenized as described above. R&D Systems mouse IL-17 ELISA (Cat#M1700) was used according to manufacturer's protocol.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge Julie Ritchie, John Miglietta, Kathleen Haverty, Lucaz Chlewicki, Cynthia Kenny, David Hayes, Spurgeon Hogan, Haiguang Xiao, Elizabeth Greene, Philip Gorman, Helen Wu, Gale Hansen, Meredith Liberto, Maria Myzithras, Hua Li, Michelle Lewis for technical support.
Reference
- 1.Towne JE, Sims JE. IL-36 in psoriasis. Curr Opin Pharmacol 2012; 12:486-90; PMID:22398321; https://doi.org/ 10.1016/j.coph.2012.02.009 [DOI] [PubMed] [Google Scholar]
- 2.Cowen EW, Goldbach-Mansky R. DIRA, DITRA, and new insights into pathways of skin inflammation: what's in a name? Arch Dermatol 2012; 148:381-4; PMID:22431779; https://doi.org/ 10.1001/archdermatol.2011.3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Towne JE, Renshaw BR, Douangpanya J, Lipsky BP, Shen M, Gabel CA, Sims JE. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36alpha, IL-36beta, and IL-36 gamma) or antagonist (IL-36Ra) activity. J Biol Chem 2011; 286:42594-602; PMID:21965679; https://doi.org/ 10.1074/jbc.M111.267922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marrakchi S, Guigue P, Renshaw BR, Puel A, Pei XY, Fraitag S, Zribi J, Bal E, Cluzeau C, Chrabieh M, et al.. Interleukin-36-receptor antagonist deficiency and generalized pustular psoriasis. N Engl J Med 2011; 365:620-8; PMID:21848462; https://doi.org/ 10.1056/NEJMoa1013068 [DOI] [PubMed] [Google Scholar]
- 5.Blumberg H, Dinh H, Dean C Jr., Trueblood ES, Bailey K, Shows D, Bhagavathula N, Aslam MN, Varani J, Towne JE, et al.. IL-1RL2 and its ligands contribute to the cytokine network in psoriasis. J Immunol 2010; 185:4354-62; PMID:20833839; https://doi.org/11574262 10.4049/jimmunol.1000313 [DOI] [PubMed] [Google Scholar]
References
- 6.Sims JE, Nicklin MJ, Bazan JF, Barton JL, Busfield SJ, Ford JE, Kastelein RA, Kumar S, Lin H, Mulero JJ, et al.. A new nomenclature for IL-1-family genes. Trends Immunol 2001; 22:536-7; PMID:11574262; https://doi.org/ 10.1016/S1471-4906(01)02040-3 [DOI] [PubMed] [Google Scholar]
- 7.Afonina IS, Muller C, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015; 42:991-1004; PMID:26084020; https://doi.org/ 10.1016/j.immuni.2015.06.003 [DOI] [PubMed] [Google Scholar]
- 8.Born TL, Smith DE, Garka KE, Renshaw BR, Bertles JS, Sims JE. Identification and characterization of two members of a novel class of the interleukin-1 receptor (IL-1R) family. Delineation of a new class of IL-1R-related proteins based on signaling. J Biol Chem 2000; 275:29946-54; PMID:10882729; https://doi.org/ 10.1074/jbc.M004077200 [DOI] [PubMed] [Google Scholar]
- 9.Towne JE, Garka KE, Renshaw BR, Virca GD, Sims JE. Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-kappaB and MAPKs. J Biol Chem 2004; 279:13677-88; PMID:14734551; https://doi.org/ 10.1074/jbc.M400117200 [DOI] [PubMed] [Google Scholar]
- 10.Thomas C, Bazan JF, Garcia KC. Structure of the activating IL-1 receptor signaling complex. Nat Struct Mol Biol 2012; 19:455-7; PMID:22426547; https://doi.org/ 10.1038/nsmb.2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muhr P, Zeitvogel J, Heitland I, Werfel T, Wittmann M. Expression of interleukin (IL)-1 family members upon stimulation with IL-17 differs in keratinocytes derived from patients with psoriasis and healthy donors. Br J Dermatol 2011; 165:189-93; PMID:21410667; https://doi.org/ 10.1111/j.1365-2133.2011.10302.x [DOI] [PubMed] [Google Scholar]
- 12.Saha SS, Singh D, Raymond EL, Ganesan R, Caviness G, Grimaldi C, Woska JR Jr, Mennerich D, Brown SE, Mbow ML, et al.. Signal transduction and intracellular trafficking by the interleukin 36 receptor. J Biolo Chem 2015; 290(39):23997-4006; PMID:26269592; https://doi.org/17908936 10.1074/jbc.M115.653378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blumberg H, Dinh H, Trueblood ES, Pretorius J, Kugler D, Weng N, Kanaly ST, Towne JE, Willis CR, Kuechle MK, et al.. Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J Exp Med 2007; 204:2603-14; PMID:17908936; https://doi.org/ 10.1084/jem.20070157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO, Burden AD, et al.. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J hum Genet 2011; 89:432-7; PMID:21839423; https://doi.org/22428995 10.1016/j.ajhg.2011.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugiura K, Takeichi T, Kono M, Ogawa Y, Shimoyama Y, Muro Y, Akiyama M. A novel IL36RN/IL1F5 homozygous nonsense mutation, p.Arg10X, in a Japanese patient with adult-onset generalized pustular psoriasis. Br J Dermatol 2012; 167:699-701; PMID:22428995; https://doi.org/ 10.1111/j.1365-2133.2012.10953.x [DOI] [PubMed] [Google Scholar]
- 16.Shaik Y, Sabatino G, Maccauro G, Varvara G, Murmura G, Saggini A, Rosati M, Conti F, Cianchetti E, Caraffa A, et al.. IL-36 receptor antagonist with special emphasis on IL-38. Int J Immunopathol Pharmacol 2013; 26:27-36; PMID:23527706; https://doi.org/ 10.1177/039463201302600103 [DOI] [PubMed] [Google Scholar]
- 17.Frey S, Derer A, Messbacher ME, Baeten DL, Bugatti S, Montecucco C, Schett G, Hueber AJ. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann Rheum Dis 2013; 72:1569-74; PMID:23268368; https://doi.org/ 10.1136/annrheumdis-2012-202264 [DOI] [PubMed] [Google Scholar]
- 18.Chen H, Wang Y, Bai C, Wang X. Alterations of plasma inflammatory biomarkers in the healthy and chronic obstructive pulmonary disease patients with or without acute exacerbation. J Proteomics 2012; 75:2835-43; PMID:22343073; https://doi.org/ 10.1016/j.jprot.2012.01.027 [DOI] [PubMed] [Google Scholar]
- 19.Medina-Contreras O, Harusato A, Nishio H, Flannigan KL, Ngo V, Leoni G, Neumann PA, Geem D, Lili LN, Ramadas RA, et al.. Cutting Edge: IL-36 Receptor Promotes Resolution of Intestinal Damage. J Immunol 2016; 196:34-8; PMID:26590314; https://doi.org/26783184 10.4049/jimmunol.1501312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scheibe K, Backert I, Wirtz S, Hueber A, Schett G, Vieth M, Probst HC, Bopp T, Neurath MF, Neufert C. IL-36R signalling activates intestinal epithelial cells and fibroblasts and promotes mucosal healing in vivo. Gut 2016; 66(5):823-38; PMID:26783184; https://doi.org/ 10.1136/gutjnl-2015-310374 [DOI] [PubMed] [Google Scholar]
- 21.Russell SE, Horan RM, Stefanska AM, Carey A, Leon G, Aguilera M, Statovci D, Moran T, Fallon PG, Shanahan F, et al.. IL-36alpha expression is elevated in ulcerative colitis and promotes colonic inflammation. Mucosal Immunol 2016; 9:1193-204; PMID:26813344; https://doi.org/ 10.1038/mi.2015.134 [DOI] [PubMed] [Google Scholar]
- 22.Wang TS, Chiu HY, Hong JB, Chan CC, Lin SJ, Tsai TF. Correlation of IL36RN mutation with different clinical features of pustular psoriasis in Chinese patients. Arch Dermatol Res 2016; 308:55-63; PMID:26589685; https://doi.org/ 10.1007/s00403-015-1611-x [DOI] [PubMed] [Google Scholar]
- 23.Sugiura K, Oiso N, Iinuma S, Matsuda H, Minami-Hori M, Ishida-Yamamoto A, Kawada A, Iizuka H, Akiyama M. IL36RN mutations underlie impetigo herpetiformis. J Invest Dermatol 2014; 134:2472-4; PMID:24717243; https://doi.org/ 10.1038/jid.2014.177 [DOI] [PubMed] [Google Scholar]
- 24.Sugiura K. The genetic background of generalized pustular psoriasis: IL36RN mutations and CARD14 gain-of-function variants. J Dermatol Sci 2014; 74:187-92; PMID:24656634; https://doi.org/ 10.1016/j.jdermsci.2014.02.006 [DOI] [PubMed] [Google Scholar]
- 25.Shu D, Jin HZ. Mutation analysis of IL36RN in a Chinese Daur family with generalized pustular psoriasis. Eur J Dermatol: EJD 2014; 24:415-6; PMID:24979538; https://doi.org/ 10.1684/ejd.2014.2382 [DOI] [PubMed] [Google Scholar]
- 26.Renert-Yuval Y, Horev L, Babay S, Tams S, Ramot Y, Zlotogorski A, Molho-Pessach V. IL36RN mutation causing generalized pustular psoriasis in a Palestinian patient. Int J Dermatol 2014; 53:866-8; PMID:24898045; https://doi.org/ 10.1111/ijd.12525 [DOI] [PubMed] [Google Scholar]
- 27.Li X, Chen M, Fu X, Zhang Q, Wang Z, Yu G, Yu Y, Qin P, Wu W, Pan F, et al.. Mutation analysis of the IL36RN gene in Chinese patients with generalized pustular psoriasis with/without psoriasis vulgaris. J Dermatol Sci 2014; 76:132-8; PMID:25212972; https://doi.org/ 10.1016/j.jdermsci.2014.08.007 [DOI] [PubMed] [Google Scholar]
- 28.Hayashi M, Nakayama T, Hirota T, Saeki H, Nobeyama Y, Ito T, Umezawa Y, Fukuchi O, Yanaba K, Kikuchi S, et al.. Novel IL36RN gene mutation revealed by analysis of 8 Japanese patients with generalized pustular psoriasis. J Dermatol Sci 2014; 76:267-9; PMID:25468355; https://doi.org/ 10.1016/j.jdermsci.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 29.Carapito R, Isidor B, Guerouaz N, Untrau M, Radosavljevic M, Launay E, Cassagnau E, Frenard C, Aubert H, Romefort B, et al.. Homozygous IL36RN mutation and NSD1 duplication in a patient with severe pustular psoriasis and symptoms unrelated to deficiency of interleukin-36 receptor antagonist. Br J Dermatol 2014; 172(1):302-5; PMID:25039711; http://dx.doi.org/ 10.1111/bjd.13261 [DOI] [PubMed] [Google Scholar]
- 30.Berki DM, Mahil SK, Burden AD, Trembath RC, Smith CH, Capon F, Barker JN. Loss of IL36RN function does not confer susceptibility to psoriasis vulgaris. J Invest Dermatol 2014; 134:271-3; PMID:23792462; https://doi.org/ 10.1038/jid.2013.285 [DOI] [PubMed] [Google Scholar]
- 31.Setta-Kaffetzi N, Navarini AA, Patel VM, Pullabhatla V, Pink AE, Choon SE, Allen MA, Burden AD, Griffiths CE, Seyger MM, et al.. Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol 2013; 133:1366-9; PMID:23303454; https://doi.org/ 10.1038/jid.2012.490 [DOI] [PubMed] [Google Scholar]
- 32.Navarini AA, Valeyrie-Allanore L, Setta-Kaffetzi N, Barker JN, Capon F, Creamer D, Roujeau JC, Sekula P, Simpson MA, Trembath RC, et al.. Rare variations in IL36RN in severe adverse drug reactions manifesting as acute generalized exanthematous pustulosis. J Invest Dermatol 2013; 133:1904-7; PMID:23358093; https://doi.org/ 10.1038/jid.2013.44 [DOI] [PubMed] [Google Scholar]
- 33.Li M, Lu Z, Cheng R, Li H, Guo Y, Yao Z. IL36RN gene mutations are not associated with sporadic generalized pustular psoriasis in Chinese patients. Br J Dermatol 2013; 168:452-5; PMID:22862555; https://doi.org/ 10.1111/j.1365-2133.2012.11195.x [DOI] [PubMed] [Google Scholar]
- 34.Korber A, Mossner R, Renner R, Sticht H, Wilsmann-Theis D, Schulz P, Sticherling M, Traupe H, Hüffmeier U. Mutations in IL36RN in patients with generalized pustular psoriasis. J Invest Dermatol 2013; 133:2634-7; PMID:23648549; https://doi.org/ 10.1038/jid.2013.214 [DOI] [PubMed] [Google Scholar]
- 35.Kanazawa N, Nakamura T, Mikita N, Furukawa F. Novel IL36RN mutation in a Japanese case of early onset generalized pustular psoriasis. J Dermatol 2013; 40:749-51; PMID:23834760; https://doi.org/ 10.1111/1346-8138.12227 [DOI] [PubMed] [Google Scholar]
- 36.Farooq M, Nakai H, Fujimoto A, Fujikawa H, Matsuyama A, Kariya N, Aizawa A, Fujiwara H, Ito M, Shimomura Y. Mutation analysis of the IL36RN gene in 14 Japanese patients with generalized pustular psoriasis. Human mutat 2013; 34:176-83; PMID:22903787; https://doi.org/ 10.1002/humu.22203 [DOI] [PubMed] [Google Scholar]
- 37.Capon F. IL36RN mutations in generalized pustular psoriasis: just the tip of the iceberg? J Invest Dermatol 2013; 133:2503-4; PMID:24129779; https://doi.org/ 10.1038/jid.2013.361 [DOI] [PubMed] [Google Scholar]
- 38.Singh S, Kroe-Barrett RR, Canada KA, Zhu X, Sepulveda E, Wu H, He Y, Raymond EL, Ahlberg J, Frego LE, et al.. Selective targeting of the IL23 pathway: Generation and characterization of a novel high-affinity humanized anti-IL23A antibody. mAbs 2015; 7:778-91; PMID:25905918; https://doi.org/ 10.1080/19420862.2015.1032491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu H, An LL. Tailoring kinetics of antibodies using focused combinatorial libraries. Methods Mol Biol 2003; 207:213-33; PMID:12412477; https://doi.org/ 10.1385/1-59259-334-8:213 [DOI] [PubMed] [Google Scholar]
- 40.De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol 2007; 28:482-90; PMID:17964218; https://doi.org/ 10.1016/j.it.2007.07.011 [DOI] [PubMed] [Google Scholar]
- 41.Urlaub G, Kas E, Carothers AM, Chasin LA. Deletion of the diploid dihydrofolate reductase locus from cultured mammalian cells. Cell 1983; 33:405-12; PMID:6305508; https://doi.org/ 10.1016/0092-8674(83)90422-1 [DOI] [PubMed] [Google Scholar]
- 42.Arduin E, Arora S, Bamert PR, Kuiper T, Popp S, Geisse S, Grau R, Calzascia T, Zenke G, Kovarik J. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol Immunol 2015; 63:456-63; PMID:25451975; https://doi.org/ 10.1016/j.molimm.2014.09.017 [DOI] [PubMed] [Google Scholar]
- 43.Baudino L, Shinohara Y, Nimmerjahn F, Furukawa J, Nakata M, Martinez-Soria E, Petry F, Ravetch JV, Nishimura S, Izui S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J Immunol 2008; 181:6664-9; PMID:18941257; https://doi.org/23064362 10.4049/jimmunol.181.9.6664 [DOI] [PubMed] [Google Scholar]
- 44.Tortola L, Rosenwald E, Abel B, Blumberg H, Schafer M, Coyle AJ, Renauld JC, Werner S, Kisielow J, Kopf M. Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J Clin Invest 2012; 122:3965-76; PMID:23064362; https://doi.org/ 10.1172/JCI63451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gkalpakiotis S, Arenberger P, Gkalpakioti P, Hugo J, Sticova E, Tesinsky P, Arenbergerová M. A case of acute generalized pustular psoriasis of von Zumbusch treated with adalimumab. J Eur Acad Dermatol Venereol 2015; 29:2063-4; PMID:24981161; https://doi.org/ 10.1111/jdv.12597 [DOI] [PubMed] [Google Scholar]
- 46.Bohner A, Roenneberg S, Eyerich K, Eberlein B, Biedermann T. Acute Generalized Pustular Psoriasis Treated With the IL-17A Antibody Secukinumab. JAMA Dermatol 2015:1-2; PMID:26650014; https://doi.org/23909475 10.1001/jamadermatol.2015.4686 [DOI] [PubMed] [Google Scholar]
- 47.Huffmeier U, Watzold M, Mohr J, Schon MP, Mossner R. Successful therapy with anakinra in a patient with generalized pustular psoriasis carrying IL36RN mutations. Br J Dermatol 2014; 170:202-4; PMID:23909475; https://doi.org/ 10.1111/bjd.12548 [DOI] [PubMed] [Google Scholar]
- 48.Rondeau JM, Ramage P, Zurini M, Gram H. The molecular mode of action and species specificity of canakinumab, a human monoclonal antibody neutralizing IL-1beta. mAbs 2015; 7:1151-60; PMID:26284424; https://doi.org/ 10.1080/19420862.2015.1081323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Groot AS, Martin W. Reducing risk, improving outcomes: bioengineering less immunogenic protein therapeutics. Clini Immunol 2009; 131:189-201; PMID:19269256; https://doi.org/ 10.1016/j.clim.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 50.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J 2000; 78:1606-19; PMID:10692345; https://doi.org/ 10.1016/S0006-3495(00)76713-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen SB, Proudman S, Kivitz AJ, Burch FX, Donohue JP, Burstein D, Sun YN, Banfield C, Vincent MS, Ni L, et al.. A randomized, double-blind study of AMG 108 (a fully human monoclonal antibody to IL-1R1) in patients with osteoarthritis of the knee. Arthritis Res Ther 2011; 13:R125; PMID:21801403; https://doi.org/ 10.1186/ar3430 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.