

ABSTRACT
Immunostimulatory receptors belonging to the tumor necrosis factor receptor (TNFR) superfamily are emerging as promising targets for cancer immunotherapies. To optimize the agonism of therapeutic antibodies to these receptors, Fc engineering of antibodies was applied to facilitate the clustering of cell surface TNFRs to activate downstream signaling pathways. One engineering strategy is to identify Fc mutations that facilitate antibody multimerization on the cell surface directly. From the analyses of the crystal packing of IgG1 structures, we identified a novel set of Fc mutations, T437R and K248E, that facilitated antibody multimerization upon binding to antigens on cell surface. In a NF-κB reporter assay, the engineered T437R/K248E mutations could facilitate enhanced agonism of an anti-OX40 antibody without the dependence on FcγRIIB crosslinking. Nonetheless, the presence of cells expressing FcγRIIB could facilitate a boost of the agonism of the engineered antibody with mutations on IgG1 Fc, but not on the silent IgG2σ Fc. The Fc engineered antibody also showed enhanced effector functions, including antibody-dependent cell-meditated cytotoxicity, antibody-dependent cellular phagocytosis, and complement-dependent cytotoxicity, depending on the IgG subtypes. Also, the engineered antibodies showed normal FcRn binding and pharmacokinetic profiles in mice. In summary, this study elucidated a novel Fc engineering approach to promote antibody multimerization on a cell surface, which could enhance agonism and improve effector function for anti-TNFR antibodies as well as other therapeutic antibodies.
KEYWORDS: Antibody engineering, anti-cancer drug, Fc receptor, immunotherapy, tumor necrosis factor, OX40, agonism, effector function
Abbreviations
- ADCC
antibody-dependent cell-meditated cytotoxicity
- ADCP
antibody-dependent cellular phagocytosis
- BRET
bioluminescence resonance energy transfer
- CDC
complement-dependent cytotoxicity
- CTLA-4
cytotoxic T lymphocyte antigen 4
- GITR
glucocorticoid-induced TNFR-related protein
- LDH
lactate dehydrogenase
- MSD
Meso Scale Discovery
- PD-1
programmed cell death 1
- PPI
protein-protein interaction
- R-PE
R-phycoerythrin
- SEC
size-exclusion chromatography
- TBST
Tris buffered saline with Tween20
- TNF
tumor necrosis factor
- TNFR
tumor necrosis factor receptor
Introduction
Therapeutic antibodies targeting the immune checkpoint receptors can stimulate antitumor immunity and have emerged as promising cancer therapeutics in recent years.1,2 Blocking antibodies for inhibitory receptors, such as CTLA-4 and PD-1, have been approved as single agent therapies for several types of tumors, including advanced melanoma and lung cancer. In addition, agonist antibodies directed against the immunostimulatory receptors are under extensive investigation as alternative approaches for cancer immunotherapies.3
The anti-tumor activities of antibodies to immunostimulatory receptors depend on their agonistic activities to promote the activation of critical players of immune modulation, such as effector T cells and antigen-presenting cells.4-6 Many immunostimulatory receptors, including OX40, CD27, 4–1BB and GITR on T cells and CD40 on antigen-presenting cells, belong to the tumor necrosis factor receptor (TNFR) superfamily. Recent studies demonstrated that the concomitant engagement to the inhibitory FcγRIIB receptor can be critical for the activity of agonistic antibodies to these TNFR targets.7-12 The crosslinking of IgG Fc to FcγRIIB receptors can multimerize more than one antibody molecule that in turn can facilitate the clustering of enough TNFR molecules to activate downstream signaling pathways. On the other hand, several TNFRs, such as OX40 and GITR, have elevated expression on regulatory T cells (Treg), which negatively modulate tumor immunity. 13,14 In this case, the effector functions of the anti-OX40 and anti-GITR antibodies may facilitate the selective elimination of regulatory T cells in the tumor microenvironment.13,14 Such antibody-mediated cell killing depends on the interactions of the antibodies with various activating Fcγ receptors. Clearly, immunomodulatory antibodies engage different types of Fc receptors for their agonistic activities and effector functions. However, except for FcγRI, human IgG antibodies have poor binding affinities to most human Fc receptors. Thus, Fc engineering approaches are needed to optimize the antitumor activity of agonist antibodies to immunostimulatory TNFRs.
In this regard, we recently evaluated several Fc engineering approaches to optimize the agonism of an anti-OX40 antibody, SF2.12,15 The agonism of the SF2 antibody was quantitated by an OX40-driven HEK-Blue NF-κB reporter assay, which is based on the major mechanism of action of OX40 receptor activation.16 Similar surrogate assays were adopted to assess the agonism of therapeutic antibodies to other TNFRs with good correlation to peripheral blood mononuclear cell-based functional assays.17,18 The soluble SF2 antibody with native IgG1 Fc barely had agonistic activity in this assay while the immobilized antibody did,12 confirming that crosslinking is needed for the agonism. To facilitate FcγRIIB crosslinking, SF2 antibodies were engineered with Fc mutations that were reported to enhance antibody binding to FcγRIIB, such as the S267E/L328F mutations and a set of 6 mutations collectively known as V12 mutations.19,20 Both engineered SF2 antibodies showed enhanced agonism dependent on FcγRIIB engagement. However, such engineering approaches depend heavily on the Fcγ receptor expression in the local microenvironment, and the efficacy of such an antibody may be limited to the anatomic site of action. Besides, both engineering approaches affect the IgG1 antibody binding to FcγRIIIA, and the engineered SF2 antibodies abrogate antibody-dependent cell-mediated cytotoxicity (ADCC) activities.12 On the other hand, SF2 antibodies were also engineered with mutations that can facilitate the formation of an IgG antibody hexamer upon binding targets on the cell surface.21 Such an engineered SF2 antibody showed increased agonism without the dependence on FcγRIIB crosslinking since the binding of multimerized antibodies could cluster enough OX40 receptors for activation.12 Nonetheless, the engineered SF2 antibody with IgG1 Fc showed an FcγRIIB-dependent boost of the agonism and enhanced effector functions such as ADCC and complement-dependent cytotoxicity (CDC), likely due to the increased interactions of multimerized antibodies with distinct Fc receptors.12
In this study, we expand the Fc engineering tool box by the identification and structural characterization of a novel set of Fc mutations that can facilitate antibody multimerization upon binding target antigens on cell surfaces. SF2 antibody with such engineered mutations showed enhanced agonism and effector functions that depended on antibody multimerization driven by the mutations. This study offers another Fc engineering approach for antibody optimization to improve therapeutic activities.
Results
Design of mutations that promote IgG multimerization
Diebolder et al.21 reported that a hexameric arrangement of IgG molecules similar to that observed in the crystal packing of anti-gp120 IgG1 molecules (Protein Data Bank (PDB) id 1HZH22) well described cryo-electron tomography density maps generated from IgG-C1 complexes. Among the set of mutations identified that facilitate a similar hexameric association of IgG molecules in solution,21 we found that the E345R mutation may enhance the agonism of SF2 antibody by facilitating the clustering of OX40 receptors.12 In an effort to identify additional mutations of human IgG that might enhance the agonistic activity by facilitating antibody multimerization, we further interrogated the crystal packing of IgG molecules in the crystal structure 1HZH. The majority of contacts stabilizing the closed-ring arrangement of molecules were found to exist between CH3 domains of neighboring Fc molecules. It also was noted that such an arrangement of Fc molecules would be restrictive of the conformations accessible to the CH2 domain. Indeed, when a prototypical crystal structure of an IgG1 Fc (PDBid 3AVE23) was aligned to the CH3 domains present in the closed-ring arrangement, a clash of CH2 domains associated with neighboring Fcs resulted. We hypothesized that a perturbation of contacts thought to stabilize the CH2:CH3 interface would facilitate the adoption of a CH2 conformation allowed within the context of the closed ring association of Fc molecules. Thus, by using the programs Coot24 and PyMol,25 the crystal packing was inspected to identify mutations that might facilitate multimerization either by enhancing the association of CH3 dimers or by destabilizing the intramolecular CH2:CH3 interface. To assess whether mutations predicted to promote antibody multimerization can facilitate enhanced agonism, these mutations were engineered into SF2 antibody. The agonism of the engineered antibodies were screened by the OX40-driven HEK-Blue NF-κB reporter assay that demonstrated that the E345R mutation facilitated higher agonism of SF2 by promoting antibody multimerization.12
T437R and K248E double mutations facilitate SF2 antibody with enhanced agonism
One mutation predicted to facilitate the association of CH3 dimers was the mutation of T437 to arginine. In silico modeling demonstrated that T437R would be sterically allowed at the inter-CH3 dimer interface and could potentially engage E382 on a neighboring Fc in a salt bridge interaction (Supplemental Fig. 1). K248E was identified as a mutation that might facilitate multimerization by destabilizing the intramolecular CH2:CH3 interface. This mutation would prevent formation of a commonly observed salt bridge interaction thought to stabilize the IgG CH2:CH3 interface.26 In the reporter assay, the T437R mutation facilitated SF2 with higher reporter gene expression in a dose-dependent manner, in contrast to SF2 with a native IgG1 Fc (SF2IgG1), which had little agonistic activity (Fig. 1A). Furthermore, when this mutation was combined with K248E mutation, which itself barely led to agonism enhancement, a synergistic effect was observed and the double-mutated SF2 (SF2IgG1_RE) showed much higher agonism across testing concentrations, with over 30% efficacy compared with OX40 ligand at 1000 ng/mL (Fig. 1A). In the same assay, the SF2IgG1 antibody engineered with the E345R mutation showed 66% activity relative to OX40 ligand at 1000 ng/mL. To exclude the possibility that the enhanced agonism was due to the increased affinity of the engineered antibody to OX40 receptors, the binding of both SF2IgG1 and SF2IgG1_RE to OX40 receptors expressed on HEK-Blue:OX40 cells were assessed by flow cytometry. Both antibodies showed comparable binding affinities to OX40 receptors and the insignificant difference in their binding could not account for the difference in their agonism (Fig. 1B).
Figure 1.
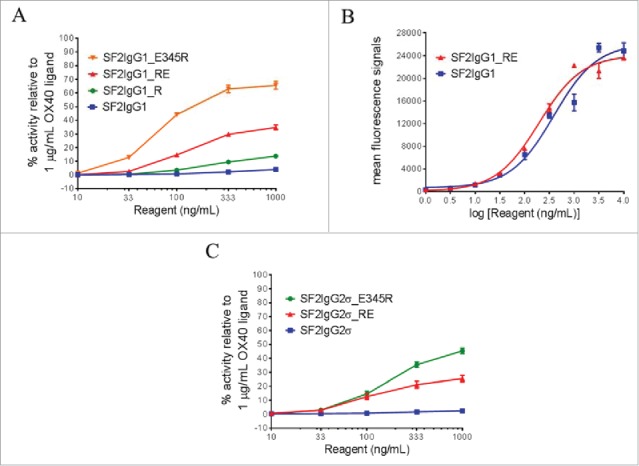
Agonistic activities of SF2 antibodies by the HEK-Blue NF-κB reporter assay. (A) Increasing concentrations (10 ng/mL to 1000 ng/mL) of SF2IgG1, SF2IgG1_R, SF2IgG1_RE and SF2IgG1_E345R antibodies were applied to HEK-Blue: OX40 cells, and agonistic activities of the antibodies were assessed by the HEK-Blue NF-κB reporter assay. The agonistic activities of SF2 antibodies, normalized as percent activity relative to that driven by 1 μg/mL OX40 ligand, were plotted against the concentrations of test antibodies (Data expressed as mean ± SEM, n). (B) Increasing concentrations (1 ng/mL to 10000 ng/mL) of SF2IgG1 and SF2IgG1_RE antibodies were assessed for binding to OX40 receptors expressed on HEK-Blue: OX40 cells by flow cytometry assays. Mean fluorescence signals were plotted against the concentrations of test antibodies (Data expressed as mean ± SEM, n 2). (C) Similar HEK-Blue NF-κB reporter assays were set up to study the agonistic activities of SF2IgG2σ, SF2IgG2σ_RE and SF2IgG2σ_E345R antibodies (Data expressed as mean ± SEM, n 2).
Both T437 and K248 are conserved residues among the IgG subtypes. T437R and K248E mutations were also engineered into an SF2 variant with an IgG2σ silent Fc (SF2IgG2σ_RE).27 HEK-Blue NF-κB reporter assay revealed that while SF2IgG2σ had little agonistic activity, the SF2IgG2σ_RE variant showed agonism in a dose-dependent manner, with about 25% efficacy compared with OX40 ligand at 1000 ng/mL (Fig. 1C). The SF2IgG2σ_E345R antibody showed 45% activity relative to OX40 ligand at 1000 ng/mL.
To assess whether T437R and K248E mutations can facilitate agonism enhancement to other agonistic antibodies, these mutations were engineered on another anti-OX40 IgG1 antibody (O2IgG1) recognizing an OX40 epitope distinct from that of SF2. These mutations were also engineered on agonistic IgG1 antibodies against 2 other TNFR targets, target A (AIgG1) and target B (BIgG1). Similar HEK-Blue NF-κB reporter assays were established to assess the agonistic activities of engineered antibodies to TNFR target A and B. In all cases, while the antibodies with a native IgG1 Fc (O2IgG1, AIgG1, BIgG1) had little agonistic activity, the antibodies with the T437R and K248E mutations (O2IgG1_RE, AIgG1_RE, BIgG1_RE) had much higher levels of dose-dependent agonism (Supplemental Fig. 2), similar to those antibodies with the E345R mutation (O2IgG1_E345R, AIgG1_E345R, BIgG1_E345R).
Multimerization of antibodies with T437R and K248E mutations upon antigen binding at the cell surface
Size-exclusion chromatography (SEC) analysis revealed that, like SF2IgG1, the SF2IgG1_RE was a monomer in solution (Fig. 2A). The multimeric model predicted that the T437R and K248E mutations facilitated the engineered antibody multimerization upon binding to cell surface antigens. To demonstrate this, we conducted a NanoBRET protein-protein interaction (PPI) assay, which was a proximity-based assay that can detect protein-protein interactions by measuring energy transfer from a bioluminescent protein donor to a fluorescent protein acceptor. For this assay, both SF2IgG1 and SF2IgG1_RE were engineered to have either the Nanoluc or the Halotag attached at the C-termini of the light chains to serve as the donor and acceptor antibodies, respectively. The tagged antibodies were monomers in solution and showed comparable functional activities to the corresponding un-tagged antibodies in a HEK-Blue NF-κB reporter assay (data not shown). NanoBRET PPI assays were performed by applying the donor and acceptor antibodies to HEK-Blue:OX40 cells, and the Corrected NanoBRET ratios were calculated to reflect the degree of association of multimerized antibody. While the SF2IgG1 antibody just had the background Corrected NanoBRET ratio, SF2IgG1_RE showed much higher Corrected NanoBRET ratios across the concentrations ranging from 10 to 1000 ng/mL (Fig. 2B), indicating a higher degree of antibody association driven by the mutations upon binding the antigens on the cell surface.
Figure 2.
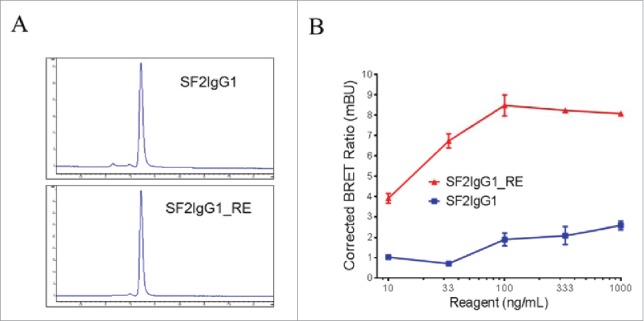
Assessment of monodispersity and multimerization of SF2IgG1 and SF2IgG1_RE. (A) SEC profiles of SF2IgG1 and SF2IgG1_RE antibodies in solution. The y axes are absorbance values at 280 nm. The x axes are retention times in minutes. (B) NanoBRET PPI assay for SF2IgG1 and SF2IgG1_RE. Increasing concentrations (from 10 ng/mL to 1000 ng/mL) of both Nanoluc donor antibody and Halotag receptor antibody were applied to HEK-Blue: OX40 cells and NanoBRET PPI assays were conducted. Mean Corrected BRET ratio were plotted against the concentrations of test antibodies (Data expressed as mean ± SEM, n 2).
Similar NanoBRET PPI assays were performed to assess whether T437R and K248E mutations could facilitate multimerization of antibodies against TNFR target A upon binding target A on the cell surface. While the AIgG1 antibody just had the background Corrected NanoBRET ratio, AIgG1_RE showed much higher levels of dose-dependent Corrected NanoBRET ratios (Supplemental Fig. 3), similar to AIgG1_E345R.
FcγRIIB crosslinking further boost the agonism of SF2 antibody with T437R/K248E mutations depending on IgG subtype
Although T437R/K248E mutations can facilitate the agonism of SF2 with either IgG1 or IgG2σ Fc independent of FcγRIIB crosslinking, the effect of FcγRIIB crosslinking was nonetheless studied by HEK-Blue NF-κB reporter assay in which the engineered antibodies were applied to HEK-Blue:OX40 cells co-cultured with Raji cells. Such an assay system was used previously demonstrating the boost of agonism of SF2 antibody with E345R hexamerization mutation in a FcγRIIB crosslinking-dependent fashion.12 Similarly, it was observed that the presence of Raji cells could further boost the agonism of SF2IgG1_RE, with over 3-fold increases in activity across test concentrations compared with the case without Raji cell co-culturing (Fig. 3A). When FcγRIIB-specific 2B6 antibody28 was added to pre-block FcγRIIB receptors on Raji cells, the Raji cell-mediated boost of agonism for SF2IgG1_RE was abrogated nearly completely, with agonistic activity reduced to a level similar to the case without Raji cell co-culturing, implying that the boost of agonism was driven by FcγRIIB crosslinking.
Figure 3.
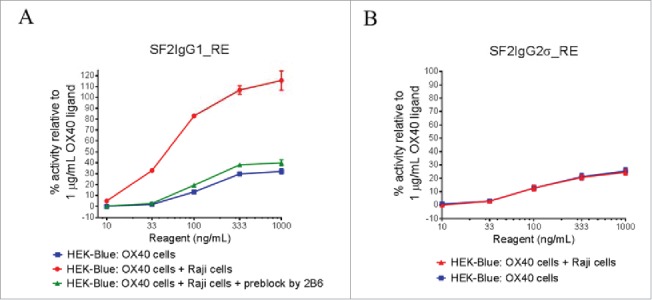
The effects of FcγRIIB crosslinking on the agonistic activities of engineered SF2 antibodies by HEK-Blue NF-κB reporter assay. (A) Increasing concentrations (10 ng/mL to 1000 ng/mL) of SF2IgG1_RE were incubated with HEK-Blue: OX40 cells with or without co-culturing with Raji cells. Another set of assays were set up in which Raji cells were pre-incubated with 5 μg/mL 2B6 antibody before co-culturing with HEK-Blue: OX40 cells to test the effect of blocking FcγRIIB-mediated crosslinking. Agonistic activities of the antibodies were assessed by the HEK-Blue NF-κB reporter assay. Agonistic activities of SF2 antibodies, normalized as percent activity relative to that driven by 1 μg/mL OX40 ligand, were plotted vs. the concentrations of test antibodies (Data expressed as mean ± SEM, n). (B) Similar assays were set up to study the effects of co-culturing with Raji cells on the agonistic activities of SF2IgG2σ_RE (Data expressed as mean ± SEM, n).
Similar assays were set up to evaluate FcγRIIB crosslinking on the agonism of SF2IgG2σ_RE. The presence of Raji cells failed to boost the agonistic activity of SF2IgG2σ_RE antibody (Fig. 3B). This indicated that, similar to the case of the E345R hexamerization mutation,12 Raji cell-mediated boost of agonism for SF2 with T437R/K248E mutations depended on the IgG subtype, with an effect on IgG1 Fc but no effect on the silent IgG2σ Fc.
Fc effector functions for SF2 antibodies with T437R/K248E mutations
The ADCC activities of the SF2IgG1_RE and SF2IgG2σ_RE were studied by the FcγRIIIA-mediated ADCC reporter bioassay. SF2IgG1 showed ADCC activity, driving the reporter gene expression dose dependently with an EC50 value of 155 ng/mL (Fig. 4A). The T437R/K248E mutations could enhance SF2IgG1 with even more potent ADCC activity with an EC50 value of 81 ng/mL. In contrast, SF2IgG2σ did not have ADCC activity in this assay due to the silent nature of this Fc, while SF2IgG2σ_RE maintained Fc silencing in the ADCC activity.
Figure 4.
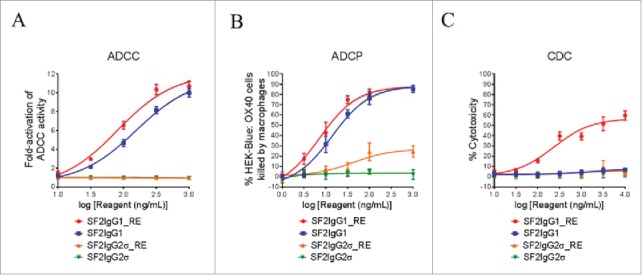
Effector functions of engineered SF2 antibodies. (A) ADCC activities of SF2 antibodies. Increasing concentrations (10 ng/mL to 1000 ng/mL) of SF2IgG1, SF2IgG1_RE, SF2IgG2σ and SF2IgG2σ_RE were incubated with HEK-Blue: OX40 cells co-cultured with effectors cells and the ADCC reporter bioassays were performed. Fold- activation of ADCC activities were plotted against the concentrations of test antibodies (Data expressed as mean ± SEM, n). (B) ADCP activities of SF2 antibodies. Increasing concentrations (1 ng/mL to1000 ng/mL) of SF2IgG1, SF2IgG1_RE, SF2IgG2σ and SF2IgG2σ_RE were incubated with GFP positive HEK-Blue: OX40 cells co-cultured with differentiated macrophages and the phagocytosis of GFP positive HEK-Blue: OX40 target cells were evaluated by flow cytometry assay. The percentages of GFP positive cells eliminated, which reflected the ADCP activities, were plotted vs. concentrations of test antibodies (Data expressed as mean ± SEM, n ). (C) CDC activities of SF2 antibodies. Increasing concentrations (10 ng/mL to10000 ng/mL) of SF2IgG1, SF2IgG1_RE, SF2IgG2σ and SF2IgG2σ_RE were incubated with HEK-Blue: OX40 cells in the presence of rabbit complement. CDC activities were quantitated by measuring lactate dehydrogenase (LDH) activity released from the cytosol of lysed HEK-Blue: OX40 cells, and expressed as percent cytotoxicity relative to that of cells lysed by Triton X-100 (Data expressed as mean ± SEM, n).
The antibody-dependent cellular phagocytosis (ADCP) activities of the SF2IgG1_RE and SF2IgG2σ_RE were studied by the phagocytosis of green fluorescent protein (GFP)-expressing HEK-Blue:OX40 cells using differentiated macrophages. SF2IgG1 could dose-dependently mediate efficient killing of HEK-Blue: OX40 target cells by the macrophages, while the T437R/K248E mutations marginally enhanced the potency of ADCP activity of SF2IgG1 with an EC50 value of 7.2 ng/mL (Fig. 4B). Although the antibodies with the silent IgG2σ Fc barely showed ADCP activity, the ADCP activity for SF2IgG2σ_RE was elevated at higher concentrations relative to that observed for SF2IgG2σ.
The CDC activities of the engineered SF2 antibodies were studied by a complement-mediated cell killing assay. While SF2IgG1 did not mediate significant CDC activity toward HEK-Blue:OX40 target cells, SF2IgG1_RE showed a dose-dependent elevation of CDC activity with an EC50 value of 229 ng/mL (Fig. 4C). In contrast, SF2IgG2σ did not have CDC activity in this assay, and mutations in SF2IgG2σ_RE did not change its silencing in CDC activity.
In vitro binding to FcRn for the engineered SF2 antibodies
Since the T437R and K248E mutations are both located close to the known binding sites to FcRn,29,30 possible interference from these mutations with the proper interaction of our variant antibodies with FcRn were evaluated using an in vitro competitive binding assay. Figure 5A depicts the relative binding affinities of engineered SF2 antibodies to a recombinant human FcRn extracellular domain at pH 6.0. Both SF2IgG1_RE and SF2IgG1_R exhibited apparent similar binding affinities relative to SF2IgG1. In a binding and release assay, both SF2IgG1_RE and SF2IgG1_R released from FcRn binding at similar levels relative to SF2IgG1 when the pHs of the buffers were raised from 6.0 to 6.6, 7.0 or 7.4 (Supplemental Fig. 4). These data suggested little to no apparent effect on FcRn binding of the engineered arginine mutation at position 437, in contrast to an arginine at residue 435 of IgG3 antibody that impaired its FcRn binding and half-life relative to other human IgGs.31 This result, in the absence of other potential pharmacokinetics (PK)-impacting influences, predicted that the T437R and K248E mutations would not have a significant effect on serum half-life.
Figure 5.
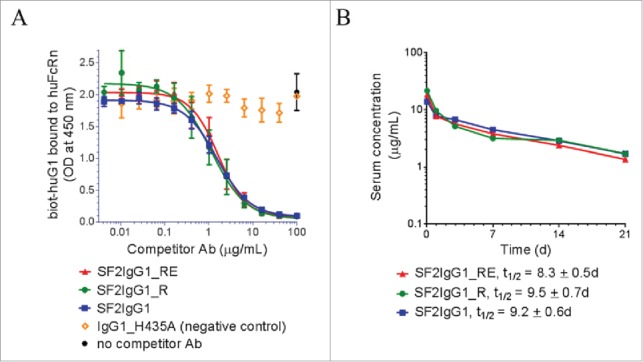
FcRn binding and pharmacokinetic analysis of engineered SF2 antibodies. (A) Competitive binding of SF2 antibodies to human FcRn at pH 6.0. Serially-diluted test antibodies were co-incubated at pH 6.0 with a constant concentration of biotinylated human IgG1 (biot-huIgG1) on a plate coated with human FcRn extracellular domain. Remaining bound biot-huIgG1 was detected with streptavidin-HRP developed with TMB substrate and read at 450 nm. (Data expressed as mean OD450 ± SEM, n = 2). (B) Pharmacokinetic analysis of SF2 antibodies in Tg32 SCID mice. Test antibodies were administered at an intravenous dose of 2 mg/kg, and serial retro-orbital bleeds were taken from each animal at indicated time points. PK profiles of each Ab were displayed as a semi-log plot serum concentration vs time. (Data expressed as mean ± SEM, n ).
A pharmacokinetic (PK) study of the engineered SF2 antibodies in FcRn transgenic mice
A PK study was performed to evaluate the SF2 antibodies containing novel Fc engineered mutations in FcRn transgenic severe combined immunodeficient (SCID) mice, which are deficient in functional B and T lymphocytes, and hence have minimal immune responses to test antibodies. FcRn transgenic mouse models are used routinely for early assessment and prediction of human PK of therapeutic antibodies.32-35 Tg32 homozygous SCID mice (5 mice/group) were injected intravenously with a 2 mg/kg dose of antibodies. Serial retro-orbital bleeds from each animal were obtained at 1 h, and 1, 3, 7, 14 and 21 d after injection. Sera were prepared and the amounts of human IgG were determined by an electrochemiluminescent immunoassay. Mean serum concentrations for each antibody were shown in Fig. 5B. For all samples, there was a linear decline of serum concentration over the course of 21 d and no significant differences (p <0.05) were observed among the test groups. Half-life values, t1/2, were determined to be: SF2IgG1_R, t1/2 = 9.5 ± 0.7 d; SF2IgG1_RE, t1/2 = 8.3 ± 0.5 d; SF2IgG1, t1/2 = 9.2 ± 0.6 d. These data revealed comparable PK profiles of the engineered antibodies to that with native IgG1 Fc.
Structure of IgG1 Fc with T437R/K248E mutations
The crystal structure of the hIgG1 Fc with T437R/K248E mutations was determined to a resolution of 3.1 Å. The Fc crystallized in space group P61 (Table 1) with one Fc dimer present in the asymmetric unit. For the 2 Fc chains: chain A was modeled from G237 to L443 and chain B was modeled from V240 to V323 and from I332 to L443. However, poor or absent electron density precluded the building of residues 324 to 331 of the FG loop of chain B. An N297-linked G0F glycan was modeled for the A chain; however, weak electron density did not allow modeling of an N-acetylglucosamine residue in the α(1–3) arm of the chain B glycan. The average B factors over protein atoms of the CH2 domains of chains A and B were 94 Å2 and 113 Å2, respectively. Generally, a poorer quality of the electron density, particularly for the BC, DE, and FG loops, and elevated temperature factors indicated an enhanced disorder within the crystal lattice for the CH2 domain of chain B relative to that of chain A.
Table 1.
Crystal and X-ray data and refinement statistics.
| Crystal data |
|
| Space group | P61 |
| Unit cell parameters | |
| a, b, c (Å) | 115.0, 115.0, 73.6 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| Asymmetric unit content | 1 dimer |
| Vm (A3/Da) | 2.80 |
| Solvent content (%) | 56 |
| X-ray data | |
| Resolution (Å) | 50–3.10 (3.18–3.10)* |
| Number of reflections | |
| Measured | 102,289 (7,718) |
| Unique | 10,183 (724) |
| Completeness (%) | 99.9 (100.0) |
| Redundancy | 10.0 (10.7) |
| Rmerge | 0.068 (0.782) |
| <I/σ> | 24.46 (3.21) |
| CC1/2 | 0.999 (0.907) |
| Refinement statistics | |
| Resolution (Å) | 34.51–3.10 |
| Number of reflections | 10,163 |
| Rwork / Rfree (%) | 22.9 / 28.5 |
| Number atoms | 3,206 |
| Protein | 3,022 |
| Carbohydrate | 184 |
| Mean B-factor (Å2) | 92.7 |
| Protein | 90.0 |
| Carbohydrate | 137.3 |
| RMSD | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.404 |
| Ramachandran | |
| Favored (%) | 98.5 |
| Allowed (%) | 1.5 |
| Outliers (%) | 0.0 |
Values for the high resolution shell are shown in parentheses
Within the lattice, Fc molecules are arranged in a corkscrew fashion with the CH3 domains of neighboring molecules juxtaposed and the dimer pseudo-2-fold directed toward the crystallographic 6-fold screw axis (Fig. 6A). The helical pitch defined by the length of the unit cell c-axis was 73.6 Å, a distance allowing for adjacent spirals to interdigitate. The relationship of juxtaposed CH3 dimers in the crystal structure differed from that in the hexameric model derived from crystal structure 1HZH by only a slight rotation of 7° about a dihedral angle defined by the Cα atoms of residues 424 and 426 within strand F on opposing chains and a translation of 3 Å (Supplemental Fig. 5). Omitting contributions from the CH2 domain, an average of 487 Å2 of accessible surface area per chain was buried at the interface of adjacent Fc dimers within the spiral, approximately half the surface area buried per chain at the intra-Fc CH3 dimer interface (1,047 Å2). When the CH2 contribution was considered, the average per chain surface area buried between adjacent Fcs within the spiral was 767 Å2. The CH2 domains within the Fc were found to be closed in about the pseudo-2-fold axis through the CH3 dimer (Fig. 6B), resulting in an extensive interaction and hydrogen bonding between glycans on opposing chains including contacts between the bifurcated mannose residues, as well as close association of the fucose residue in the chain B glycan with the mannose and N-acetylglucosamine residues in the α(1–3) arm of the glycan on chain A (Fig. 6C).
Figure 6.
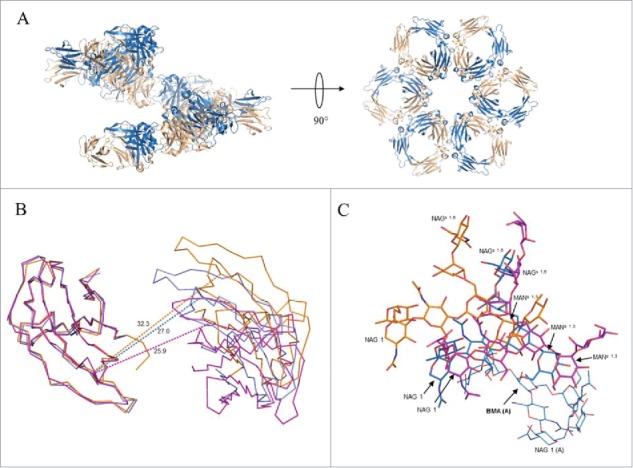
Association of Fc dimers and relative CH2 domain orientation within crystals of SF2IgG1_RE Fc. (A) Left panel shows a spiral of SF2IgG1_RE Fcs (cartoon) generated by application of the crystallographic symmetry operators to the Fc dimer (colored wheat and blue). Right panel shows a rotated view with projection down the 6-fold screw axis. (B) A prototypical wild-type IgG1 Fc structure (PDBid 3AVE, orange, ribbon) and wild-type IgG1 Fc crystallized under high salt conditions (PDBid 1H3Y, magenta, ribbon) were aligned to the crystal structure of SF2IgG1_RE Fc (blue, ribbon) based on the chain A CH2 domain. CH2 domains from both chains A and B are shown oriented looking down the pseudo 2-fold axis though the SF2IgG1_RE Fc CH3 dimer. CH3 domains are omitted for clarity. Dashed lines corresponding in color to Fc connect Cα atoms of residue R301 in opposing chains. Distances given in Ångstroms. (C) Fcs from PDBs 3AVE (orange) and 1H3Y (magenta) aligned to the bifurcated mannose residue (BMA, labeled with chain indicated in parentheses, bold) in the chain A glycan of the SF2IgG1_RE Fc structure (blue). The chain A glycan is shown in line for the latter structure only. Chain B glycans are shown in stick for all 3 Fc structures with select residues labeled.
The K248E side chain was found to fold back toward its own backbone nitrogen in both Fc chains (Fig. 7A). The E380 side chain, which typically forms a salt bridge interaction with K248 in structures of the wild-type molecule, was directed toward the FG loop of a neighboring Fc dimer making salt bridge and hydrogen bond interactions with H433 and N434, respectively. As the interface is not perfectly symmetric, the latter hydrogen bond was not observed for E380 in chain B (Fig. 7A). A hydrogen bond additionally observed between the side chains of N434 in chain A and S254 in chain B represents the only other specific interaction between neighboring Fcs within the spiral. With respect to the T437R mutation, slightly different conformations were observed in the 2 chains present in the asymmetric unit (Fig. 7B). For the more poorly ordered instance in chain A, T437R packs against G385 in a neighboring Fc molecule and additionally engages in an intrachain salt bridge interaction with E345. In chain B, the T437R guanidinium group packs against the peptide bond between N384 and G385.
Figure 7.
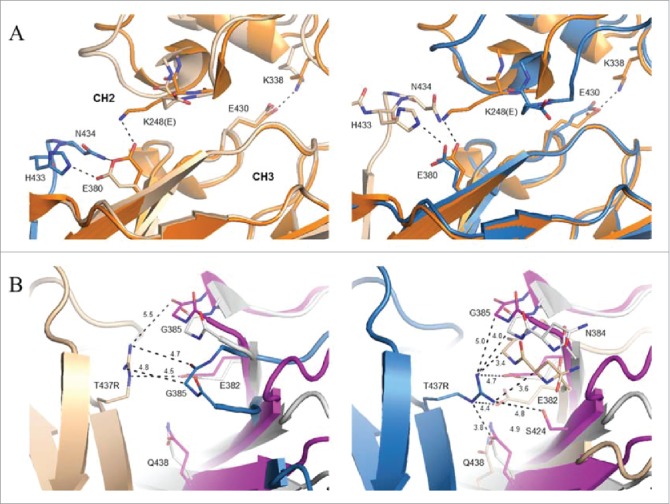
Local environment about K248E and T437R mutations in crystal structure of SF2IgG1_RE Fc. (A) Chain A of a prototypical wild-type IgG1 Fc (PDB 3AVE, orange, cartoon) shown for comparison aligned to the CH3 domain of chains A (left, wheat, cartoon) and B (right, blue, cartoon) from SF2IgG1_RE Fc. CH3 domain FG loop and G strand of a symmetry related SF2IgG1_RE Fc molecule shown in stick and cartoon and colored blue (left) or wheat (right). Select residues at the CH2:CH3 interface additionally shown in stick (labeled). Dashed lines indicate salt bridge or hydrogen bond interactions. CH2 and CH3 domains are labeled. (B) Inter-Fc interface about T437R (stick, labeled) in chains A (left, wheat cartoon) and B (right, blue cartoon) in crystal structure of SF2IgG1_RE Fc (wheat and blue). The equivalent interface for chains H and K from PDBid 1HZH were aligned to the CH3 domain of SF2IgG1_RE Fc chain A (left) or chain B (right), and the non-aligned interfacial CH3 domains are shown in gray and magenta cartoon. Residues within 5 Å of labeled T437R are shown in stick (labeled). Select distances given in Ångstroms.
Discussion
Receptor multimerization is a prerequisite for the activation of immunostimulatory TNFRs, which play pivotal roles in anti-tumor immunity.36 This unique property obligates agonistic therapeutic antibodies against TNFRs to multimerize at the cell surface to promote the clustering of bound receptors. Through structure analysis, we identified T437R/K248E as a novel pair of Fc mutations that facilitated antibody multimerization upon binding antigens at the cell surface. Such mutations could facilitate increased agonism of an anti-OX40 antibody without the dependence on crosslinking to FcγRIIB. In addition, the multimerized antibody may have increased binding affinities to different types of Fc receptors that lead to the further boost of agonism and elevated effector functions.
The design of the T437R/K248E mutations was based on interrogation of crystal structures of hexamerically associated Fcs and intact monoclonal antibodies. Davies et al. noted that a CH3-based overlay of many Fc structures deposited in the PDB onto the closed, hexameric arrangement of deglycosylated IgG4 Fc observed in crystal structure 4D2N resulted in a clash of CH2 domains.37 This is true for both chains A and B of 3AVE (hIgG1),23 4HAF (hIgG2),26 4C54 (hIgG4),38 and 4W4O (hIgG1) in complex with FcγR1.39 Similarly, the clash is seen for this set of Fcs aligned to the hexameric association of Fcs present in structures 1HZH (Supplemental Fig. 6) and 4NHH.40 Davies et al. further noted a conformational adjustment within the CH2 domain AB loop for Fcs arranged into hexamers in structures 1HZH and 4D2N37 relative to the conformation existing in non-hexamerically associated Fcs.37 In those cases, the conformational change was necessary to prevent clash between juxtaposed Fcs. However, the observed alteration in AB loop conformation alone appears not to be the sole requirement for hexamer formation. Alignment of the CH2 domain from 4D2N having the altered AB loop conformation onto Fc chains from the structures 3AVE, 4HAF, 4C54, or 4W4O hexamerically arranged as in PDBid 1HZH also resulted in a clash between the CH2 domain AB loop and in many cases the EF loop of neighboring molecules (Supplemental Fig. 7). Thus, the arrangement of Fcs into a closed, hexameric arrangement places certain restrictions on the relative CH2:CH3 domain orientations.
Two intra-chain salt bridge interactions, commonly observed at the CH2:CH3 interface of Fc structures, have been suggested to be important for the stabilization of the interface.26 K248 located in the short helical segment intervening CH2 domain strands A and B forms a salt bridge with E380 in strand C of the CH3 domain, and K338 C-terminal to CH2 strand G interacts with E430 in the CH3 domain FG loop (Fig. 6B). Molecular dynamics simulation of an Fc domain having the mutations E380A and E430A resulted in a slightly broadened distribution of the CH2-CH3 domain angle relative to wild-type, and additional deletion of the hinge resulted in a further broadening of the distribution and a shift to larger CH2-CH3 angles relative to the point mutation or hinge deletion constructs alone.41 In this study, the K248E mutation was designed to test the hypothesis that disruption of one of these salt bridge interactions might allow the CH2 domain to more facilely adopt a conformation necessary for optimal self-association of Fc dimers. Consistent with this idea, E430G, one of three mutations in a human IgG1 construct shown to exist largely as a hexamer in solution, would abolish the second of the two salt bridge interactions present at the CH2:CH3 interface.21
Interestingly, the crystal structure of IgG1 Fc with T437R/K248E mutations revealed the CH2 domains of the Fc dimer to be in a highly “closed” form (Figs. 6B and 6C). Teplyakov et al. suggested a method for comparing the relative disposition of CH2 domains among Fc structures by referencing the distance between Cα atoms at positions 238, 241, 301, and 329 on opposing chains.26 For the mutated Fc structure, only 2 of these positions, 241 and 301, were resolved in both Fc chains, and the associated inter-chain distances corresponding to these positions were 20.0 Å and 27.0 Å, respectively. These values were similar to other closed-form Fc structures, which included Fcs with altered glycosylation (e.g., PDBid 3S7G) and the structure of a wild-type Fc obtained under high salt conditions (PDBid 1H3Y42) (Fig. 7B). In the case of the latter, the high salt condition could serve to screen the electrostatic interaction between side chains to destabilize the CH2:CH3 interface similar to what was accomplished in our work through mutation. 1H3Y was a low-resolution structure (4.1 Å), and inspection of electron density maps generated from the deposited structure factors revealed no density for the side chain of E380 in either chain nor for K248 in one of the two chains present in the asymmetric unit. These considerations are consistent with the idea that K248E functions to destabilize the CH2:CH3 interface, allowing the CH2 domains to more easily access limited orientations compatible with Fc self-association.
We also sought to strengthen the interface between CH3 dimers in a possible hexameric arrangement of antibodies with the T437R mutation. The crystal structure of the mutated Fc revealed that the T437R side chain adopted different conformations in chains A and B, likely resulting from the similar but non-identical environments about the respective residues (Fig. 7B). Furthermore, although structurally the association of Fcs into a helical spiral observed in the present crystal structure was broadly similar to the closed, hexameric forms observed in structures 1HZH and 4D2N, the inter-Fc CH3:CH3 interfaces were non-identical (Fig. 7B). Thus, although in the present structure T437R was not observed to engage E382 on a neighboring Fc in a salt bridge interaction as was the rationale for the design (Supplemental Fig. 1), such interaction might occur in a closed ring form. Nonetheless, T437R was in proximity to E382, likely providing a favorable electrostatic interaction at the inter-CH3 dimer interface, and did make contacts with the neighboring Fc that would not be possible with a native threonine.
Although we cannot specify precisely how the antibodies were organizing, multimerization of the SF2 antibody and antibody against TNFR target A driven by T437R/K248E mutations upon binding to corresponding receptors on the cell surface were demonstrated by the NanoBRET protein-protein interaction assays (Fig. 2B, Supplemental Fig. 3). Besides, the functional effects of T437R/K248E mutations on the agonism and effector functions of anti-TNFR antibodies were analogous to the effects of E345R mutation, which facilitated antibody hexamerization.12
The antibody multimerization driven by the T437R/K248E mutations had several unique utilities. First, the multimerized antibodies may facilitate increased clustering of bound receptors, a process necessary for the activation of TNFRs such as the OX40 receptor. Consequently, SF2IgG1_RE showed enhanced agonism without the need to crosslink to FcγRIIB (Fig. 1A). The agonism enhancement was not epitope-specific, since similar effects were observed on another anti-OX40 antibodies (O2IgG1_RE) and on engineered antibodies against 2 other TNFR targets (Supplemental Fig. 2). The agonism enhancement also applied to engineered SF2 antibody with the silent IgG2σ Fc (Fig. 1C), suggesting that the agonism enhancement was not due to increased engagement to a serum factor. The mechanism for agonism enhancement driven by the T437R/K248E mutations was similar to that driven by the E345R mutation, which facilitated antibody hexamerization.21 By the NF-κB reporter assay, the degree of agonism enhancement driven by the E345R mutation seemed to be larger than that driven by the T437R/K248E mutations, likely due to tighter antibody multimerization driven by the E345R mutation inferred from the NanoBRET protein-protein interaction assays and structural analysis.12 Compared to those Fc engineering approaches to improve the engagement with FcγRIIB, the independence on FcγRIIB crosslinking characterized by the T437R/K248E mutations may equip therapeutic antibodies with agonism enhancement regardless of FcγR expression levels in the local microenvironment. This may be particularly advantageous for tumor microenvironments with low levels of infiltration of FcγR expressing cells. However, the non-dependence of FcγRIIB crosslinking may stimulate agonism non-specifically, which may lead to undesired off-target effects. Thus, the design of highly specific targeting would be critical for the use of such Fc engineering.
Although T437R/K248E mutations can enhance agonism independent of FcγRIIB crosslinking, the degree of enhancement appeared to be moderate. The presence of Raji cells, however, could significantly boost the agonism enhancement to a level comparable to that driven by OX40 ligand (Fig. 3A). Two observations indicated that the further boost of agonism depended on the engineered antibody interaction with FcγRIIB expressed on Raji cells. First, the boost of agonism was only observed on an antibody with engineered IgG1 Fc, which had weak binding affinity to FcγRIIB, but not on an antibody with the silent IgG2σ Fc (Fig. 3). Second, the boost of agonism could be completely reversed by pre-blocking FcγRIIB expressed on Raji cells (Fig. 3A). By flow cytometry assay, no increased binding on Raji cells were observed for monomer SF2IgG1_RE antibody compared with that with native IgG1 Fc (data not shown), ruling out the possibility that the boost in agonism was due to the increased FcγRIIB affinity for the engineered antibody. However, although SF2IgG1_RE antibodies existed as monomers in solution, they multimerized upon binding OX40 receptors on cell surface. It was reported that the multimerized antibody has much higher affinity to Fcγ receptors compared with antibody in monomer.43 Therefore, the oligomerized SF2IgG1_RE antibody bound to OX40 receptors on the cell surface likely have increased binding to FcγRIIB on Raji cells, which in turn further stabilizes antibody multimerization and facilitates receptor clustering that leads to the boost of agonism.
The multimerized antibodies on the cell surface driven by the T437R/K248E mutations not only have increased affinity to FcγRIIB, they may also have increased bindings to other Fcγ receptors, although the monomer SF2IgG1_RE antibodies in solution did not show increased affinities to major cell surface Fcγ receptors by flow cytometry assays (data not shown). As a consequence, it was observed that the T437R/K248E mutations further boost the potency of the FcγRIIIA-dependent ADCC activity of the SF2 antibody (Fig. 4A). The effect was specific for engineered antibody with IgG1 Fc, which was capable of binding to FcγRIIIA, but not for the antibody with the IgG2σ Fc, which was silent in FcγRIIIA binding.27 Similarly, the T437R/K248E mutations resulted in marginally higher ADCP activity, on SF2IgG1_RE likely due to the increased binding of multimerized antibodies to Fcγ receptors including FcγRIIA, which mediates ADCP (Fig. 4B). Surprisingly, the T437R/K248E mutations conferred elevated ADCP activity to the otherwise silent SF2IgG2σ. A similar effect was also observed for SF2IgG2σ with the E345R mutation that promotes antibody hexamerization.12 Perhaps IgG2σ Fc still maintained weak binding capacity to certain Fcγ receptor mediating ADCP, which was amplified upon binding to the multimerized antibodies with the engineered mutations. The T437R/K248E mutations also facilitated significantly elevated CDC activity to SF2 antibody with IgG1 Fc but not with the silent IgG2σ Fc (Fig. 4C), likely due to the increased binding of multimerized antibody to C1q, a component of the C1 complex that could trigger the initiation of complement cascade. The effects of T437R/K248E mutations on ADCC, ADCP and CDC were similar to those of E345R mutations, which facilitated antibody hexamerization.12,21 Both cases indicated that the effector functions of therapeutic antibodies could be readily enhanced with Fc engineering approaches that facilitated antibody multimerization.
In summary, a novel pair of Fc mutations that facilitated antibody multimerization was identified in this study and their enhancement of agonism and effector functions of an anti-OX40 antibody were evaluated. In general, the effects of engineered mutations depended on the choice of IgG subtypes. T437R/K248E mutations with a silent IgG Fc could instill anti-TNFR antibodies with modest but defined agonistic activity regardless of FcγR expression levels in the local microenvironment. In addition, the engineered antibodies remained silent in ADCC and CDC effector functions. In contrast, T437R/K248E mutations engineered onto IgG1 Fc could significantly enhance both the agonism and effector functions depending on the type and quantity of FcγR-expressing cells infiltrated. In addition to modulating the agonism and effector functions of therapeutic antibodies against immunostimulatory TNFRs, this Fc engineering approach could be more broadly applied to modulate monoclonal antibodies to other receptors whose activities rely on protein oligomerization. To choose an Fc engineering approach appropriately, many other factors need to be considered besides therapeutic activities, including the PK, developability, manufacturability and the immunogenicity of the engineered biologics. In this context, it was observed that T437R/K248E mutations did not affect the binding affinity to FcRn (Fig. 5A) or the PK profile of SF2 antibody in mice (Fig. 5B), suggesting that incorporating these mutations into human IgG would render a relatively long half-life in humans similar to the antibody with native IgG. In addition, the expression and purification levels of antibodies with the T437R/K248E mutations were not different from standard human IgG1 molecules, and the engineered antibodies were stable upon concentration greater than 50 mg/mL and were monodisperse in solution for more than 1 month (data not shown). The engineering approach evaluated in this study should be a valuable addition to the antibody engineering tool box, and more rigorous studies are needed for further evaluation.
Materials and methods
Fc engineering of SF2 antibody
Plasmids encoding the heavy chain (HC) and light chain (LC) of a humanized anti-OX40 antibody SF215 were constructed for the expression of SF2 antibody with either human IgG1 Fc (SF2IgG1) or IgG2σ Fc (SF2IgG2σ).27 Gene syntheses were performed by Genewiz to introduce further mutations (EU numbering) on the Fc of the HC constructs to express the following engineered SF2 antibodies described in this study: 1) SF2IgG1_R: SF2IgG1 antibody with T437R mutation; 2) SF2IgG1_RE: SF2IgG1 antibody with mutations T437R and K248E; and 3) SF2IgG2σ_RE: SF2IgG2σ antibody with mutations T437R and K248E
Antibody expression and purification
Plasmids encoding antibody HC and LC were co-transfected at a 1:3 (HC: LC) molar ratio into Expi293F cells following the transfection kit instructions (Thermo Scientific). Cells were centrifuged 5 d post transfection and the supernatant were passed through a 0.2 µm filter. The titer of antibody expression was quantified using Octet (ForteBio). Antibody purification was performed by affinity chromatography over MabSelect SuRe column (GE Healthcare Life Sciences). The purified antibody was buffer-exchanged into Dulbecco's phosphate-buffered saline (DPBS), pH7.2 by dialysis and protein concentration was determined by UV absorbance at 280 nm. Quality was assessed by SEC over a TSKgel G3SW column (Tosoh Bioscience) in 1X DPBS pH 7.2 buffer and SDS-PAGE of reduced and non-reduced samples.
HEK-Blue NF-κB reporter assay
The HEK-Blue reporter cell line stably expressing human OX40 (HEK-Blue:OX40) and the establishment of a HEK-Blue NF-κB reporter assay to study the agonistic activity of anti-OX40 antibody were described previously.12 Briefly, the OX40 ligand or antibodies were applied to 1 × 105 HEK-Blue:OX40 cells re-suspended in 200 μL culture media in each well of the 96-well assay plate. To test the crosslinking effect, 1 × 105 Raji cell was co-cultured in the same assay well. After incubation at 37°C overnight, the agonistic activities of the antibodies were evaluated by the quantification of the induced secreted alkaline phosphatase (SEAP) reporter gene expression using Quanti-Blue detection kit (Invivogen). The agonistic activity of anti-OX40 antibody was normalized as percent activity relative to that induced by 1 μg/mL OX40 ligand. Similar stable cell lines and HEK-Blue NF-κB reporter assays were established to assess the agonism of antibodies to TNFR targets A and B.
NanoBRET protein-protein interaction assay
The coding sequence for the light chain of SF2 was cloned into pNLF-C and pHTC halotag vectors (Promega) in frame with C-terminal Nanoluc and Halotag sequences, respectively. These LCs were paired with the HCs for SF2IgG1 and SF2IgG1_RE antibodies to express antibodies with either Nanoluc or Halotag attached at the C-termini of the light chains. Standard affinity chromatography over MabSelect SuRe column was used to purify these modified antibodies. The NanoBRET protein-protein interaction assay (Promega) to study antibody multimerization on the cell surface was described previously.12 Briefly, equal concentrations of Nanoluc-tagged antibody (donor) and Halotag-tagged antibody (acceptor) were applied to HEK-Blue:OX40 cells seeded in each well of the 96-well assay plate. Halotag 618 ligands were added in experimental wells and no ligand control wells were also set up. After incubation at 37°C for 30 min, the cells were washed twice and mixed with Nano-Glo substrate. The donor emission (460 nm) and acceptor emission (618 nm) were measured by Envision (PerkinElmer). Raw NanoBRET ratio values with milliBRET units (mBU) were calculated as RawBRET = 618nmEm/460nmEm * 1000. To factor in donor-contributed background or bleed through, Corrected NanoBRET ratio values were calculated as CorrectedBRET = RawBRET of experimental sample – RawBRET of no-ligand control sample, which reflects energy transfer from a bioluminescent protein donor to a fluorescent protein acceptor due to protein-protein interactions. Tagged antibody to TNFR target A were also made to quantitate the multimerization of engineered AIgG1 antibodies using similar NanoBRET protein-protein interaction assay.
Flow cytometry binding assay
Two × 105 cells per well were seeded in 96-well plate and blocked in bovine serum albumin (BSA) Stain Buffer (BD Biosciences) for 30 min at 4°C. Cells were incubated with test antibody on ice for 1.5 hour at 4°C. After being washed twice with BSA stain buffer, the cells were incubated with R-PE labeled anti-human IgG secondary antibody (Jackson ImmunoResearch Laboratories, catalog number 109–116–097) for 45 min at 4°C. The cells were washed twice in stain buffer and then re-suspended in 150 µL of stain buffer containing 1:200 diluted DRAQ7 live/dead stain (Cell Signaling Technology). PE and DRAQ7 signals of the stained cells were detected by Miltenyi MACSQuant flow cytometer (Miltenyi Biotec). Live cells were gated on DRAQ7 exclusion and the geometric mean fluorescence signals were determined for at least 10,000 live events collected. FlowJo software (Tree Star) was used for analysis. Data was plotted as the logarithm of antibody concentration versus mean fluorescence signals. Nonlinear regression analysis was performed by GraphPad Prism 6 (GraphPad Software) and EC50 values were calculated.
ADCC assay
The ADCC activities of SF2 antibodies were evaluated by an ADCC reporter bioassay as instructed by the manufacturer (Promega). Briefly, 25,000 HEK-Blue:OX40 cells per well plated in 96-well plate overnight were mixed with the engineered effector cells in which the activation of FcγRIIIA receptor leads to the expression of a luciferase reporter. SF2 antibodies were added to the cells and incubated at 37°C for 6 hours. Then, Bio-Glo luciferase reagent was added and the luciferase signals were quantitated by Envision. The ADCC activities of SF2 antibodies were expressed as fold of activation of luciferase signals over that without testing antibody added.
ADCP assay
The ADCP assay for SF2 antibody was described previously.12 Briefly, macrophages were differentiated from CD14+CD16+ monocytes isolated from peripheral blood mononuclear cells (Biological Specialty) using a negative human monocyte enrichment kit (StemCell Technologies). For the ADCP assay, differentiated macrophages were mixed with GFP-expressing HEK-Blue:OX40 cells (4: 1 ratio) in 96-well U-bottom plates. The test antibodies were added and the phagocytosis of target cells was performed for 24 hours. The macrophages were stained with anti-CD11b and anti-CD14 antibodies (BD Biosciences, catalog number 555385 and 555395) coupled to Alexa Fluor 647 (Thermo Scientific). GFP-positive HEK-Blue:OX40 target cells and Alexa647-positive macrophages were identified by flow cytometry using Miltenyi MACSQuant flow cytometer. The data were analyzed using FlowJo software (Tree Star) and ADCP-mediated cell killing was determined by measuring the reduction in GFP fluorescence using the following equation: Percentage of target cells killed = ((Percentage of GFP+, CD11b−, CD14− cells with the lowest concentration of antibody) − (Percentage of GFP+, CD11b−, CD14− cells with the test concentration of antibody))/(Percentage of GFP+, CD11b−, CD14− cells with the lowest concentration of antibody) × 100.
CDC assay
The CDC activities of the SF2 antibodies were evaluated by a complement-mediated cell killing assay. Briefly, 1 × 105 HEK-Blue:OX40 cells were incubated with diluted rabbit complement (Cedar Lane Labs) and testing antibodies for one hour. The lactate dehydrogenase (LDH) activity released from the cytosol of lysed HEK-Blue:OX40 cells into the supernatant were measured by Cytotoxicity Detection Kit per manufacturer's instruction (Roche Diagnostics). The complement-mediated cytotoxicity was expressed as percent cytotoxicity relative to that lysed by 2% (v/v) Triton X-100.
In vitro assays for FcRn binding
A competitive binding assay was used to assess relative affinities of different antibody samples to an in-house recombinant human FcRn extracellular domain with a poly-histidine affinity tag (FcRn-His6). Ninety-six-well copper-coated plates (Thermo Scientific) were used to capture FcRn-His6 at 5 µg/mL in PBS, after which plates were washed with 0.15 M NaCl, 0.02% (w/v) Tween 20, and then incubated with blocking reagent (0.05M MES [2-(N-morpholino) ethanesulfonic acid], 0.025% (w/v) BSA, 0.001% (v/v) Tween-20, pH 6.0, 10% (v/v) chemiBLOCKER (Millipore)). Plates were washed as above, and then serial dilutions of competitor test antibodies in blocking reagent were added to the plate in the presence of a fixed 1 µg/mL concentration of an indicator antibody (a biotinylated human IgG1 monoclonal antibody prepared in-house). Plates were incubated at room temperature for 1 hour, washed 3 times as above, and then incubated with a 1: 10,000 dilution of streptavidin-horseradish peroxidase (HRP) (Jackson ImmunoResearch Laboratories) at room temperature for 30 minutes to bind biotinylated antibody. Plates were washed 5 times as above, and bound streptavidin-HRP detected by adding TMB (3,3´,5,5´-tetramethylbenzidine) peroxidase substrate with stable stop (Fitzgerald Industries International) and incubating for 4 minutes. Color development was stopped by addition of 0.5 M HCl. Optical densities were determined with a SpectraMax Plus384 plate reader (Molecular Devices) at 450 nm wavelength.
A ‘bind and release’ assay was performed to compare how the different variants dissociated from human FcRn at increased pH. Ninety-six-well plates were coated, blocked, and washed in the same manner as for the competitive binding assay except the concentration of chemiBLOCKER in the blocking reagent was increased to 20%. Several dilutions of test antibody in blocking reagent were added to the plate, incubated at room temperature for 1–2 hours, washed 3 times as above, and then quickly washed 3 times again with 0.1M phosphate, 0.05% (w/v) Tween 20 at one of 4 different pHs: 6.0, 6.6, 7.0, and 7.4. Plates were immediately washed 3 times using the normal wash reagent (0.15 M NaCl, 0.02% (w/v) Tween 20) to quickly remove any residual phosphate buffer, and then incubated with a 1:5,000 dilution of HRP-labeled goat F(ab′)2 fragment anti-human IgG F(ab′)2 fragment-specific antibody (Jackson ImmunoResearch Laboratories, cat.no. 109–036–097) at room temperature for 1 hour to bind to FcRn-bound test antibody. Plates were washed 5 times with normal wash buffer and bound HRP-labeled antibodies were detected as above.
PK study
For the antibody PK studies, female Tg32 homozygous SCID mice, 4–8 weeks old (Stock 018441, Jackson Laboratory) were injected with test antibodies (SF2IgG1_R, SF2IgG1_RE or SF2IgG1) intravenously via tail vein (2 mg/kg doses, 5 animals per group). Serial retro-orbital bleeds were obtained from CO2-anesthesized mice at the indicated time points and terminal bleeds were taken by cardiac puncture. After 30 min at room temperature, blood samples were centrifuged 3,000 × g for 15 min and serum collected for analyses. The PK study was approved by the Institutional Animal Care and Use Committee at Janssen Research & Development, LLC.
Human IgG in mouse sera was determined using an electrochemiluminescent immunoassay. Streptavidin Gold 96-well plates (Meso Scale Diagnostics) were coated with 50 µL/well of 2.5 µg/mL biotinylated goat anti human IgG F(ab’)2 antibody (Jackson ImmunoResearch Laboratories, cat.no. 109–065–097) in Starting Block T20 (Thermo Scientific) overnight at 4°C. Plates were washed with Tris-buffered saline with 0.5% (v/v) Tween 20 (TBST); samples and standards diluted in 2% (w/v) BSA-TBST were added to plates and incubated for 2 h on a shaker at room temperature. Bound antibody was detected using Sulfo-TAG-labeled R10Z8E9, an anti-human Fcγ-pan antibody.44 Plates were washed and 200 µL MSD Read Buffer was added and plates read on the MSD Sector Imager 6000 (Meso Scale Diagnostics). Serum concentrations of the antibodies were determined from a standard curve using a 5-parameter non-linear regression program in GraphPad Prism 6 (GraphPad Software).
Terminal half-life (t1/2) calculations of the elimination phase (β phase) for PK studies were determined using the 1-phase exponential decay model fitted by a non-linear regression of natural log concentration vs. time using GraphPad Prism 6 (GraphPad Software). Half-life of the elimination phase (β phase) was calculated using the formula t1/2 = ln2/β, where β is the negative slope of the line fitted by the least square regression analysis starting after day 1. The terminal antibody half-life value was the average of the t1/2 values within the test group. Values for each antibody vs IgG1 were compared by T-tests, and a p value < 0.05 indicated a significant difference.
Structure determination
The plasmid encoding the Fc segment of human IgG1 with the T437R and K248E mutations were constructed by Genewiz. The plasmid was transfected into Expi293F cells and the mutated Fc fragment expressed was purified by standard affinity chromatography over MabSelect SuRe column (GE Healthcare Life Sciences).
Crystallization experiments using the sitting drop vapor diffusion method were performed in Corning 3550 plates. 300 nL drops were set up at room temperature using a Mosquito liquid handling robot (TTP Labtech) by mixing the mutated hIgG1 Fc at 15 mg/mL in 50 mM NaCl, 20 mM Tris, pH 7.4, with reservoir solution at a 1:1 (v:v) ratio. Diffraction quality crystals were grown at 20°C using a reservoir solution of 35.5% (w/v) PEG 300, 0.1 M HEPES, pH 7.0. Crystals were flash frozen in liquid nitrogen ahead of data collection. Diffraction data were collected at the Advanced Photon Source beamline 17-ID (IMCA-CAT) equipped with a DECTRIS Pilatus 6M pixel array detector, and processed using XDS.45 Initial phases were determined by the method of molecular replacement with the program Phaser46 implemented in the CCP4 software suite47 using CH2 and CH3 domains from PDBid 1L6X48 as search models. The model underwent subsequent rounds of rebuilding and refinement with the programs Coot24 and Phenix,49 respectively. Toward the end of structure determination, the model was processed with PDB_REDO,50 which uses Refmac551 for refinement. Final refinement was performed using Phenix. Data collection and refinement statistics are given in Table 1. The final coordinates and structure factors have been deposited in the PDB52 under accession number 5VME.
Supplementary Material
Disclosure statement
Janssen Research & Development, LLC provided funding for the research. All authors are employees of Janssen Research & Development, LLC.
Acknowledgments
We thank Michelle Kinder, Shixue Shen, Adam Zwolak, Matthew Bunce, Mehabaw Derebe, Stephen Jarantow and Natalie Fursov for their excellent contributions to this work. Use of the IMCA-CT beamline 17-ID at the Advanced Photon Source was supported by the companies of the Industrial Macromolecular Crystallography Association through a contract with Hauptman-Woodward Medical Research Institute. This research used resources of the Advanced Photon Source, a US. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
References
- 1.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480-9. doi: 10.1038/nature10673. PMID:22193102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013;13:227-42. doi: 10.1038/nri3405. PMID:23470321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaer DA, Hirschhorn-Cymerman D, Wolchok JD. Targeting tumor-necrosis factor receptor pathways for tumor immunotherapy. J Immunother Cancer. 2014;2:7. doi: 10.1186/2051-1426-2-7. PMID:24855562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kanamaru F, Youngnak P, Hashiguchi M, Nishioka T, Takahashi T, Sakaguchi S, Ishikawa I, Azuma M. Costimulation via glucocorticoid-induced TNF receptor in both conventional and CD25+ regulatory CD4+ T cells. J Immunol. 2004;172:7306-14. doi: 10.4049/jimmunol.172.12.7306 [DOI] [PubMed] [Google Scholar]
- 5.He LZ, Prostak N, Thomas LJ, Vitale L, Weidlick J, Crocker A, Pilsmaker CD, Round SM, Tutt A, Glennie MJ, et al.. Agonist anti-human CD27 monoclonal antibody induces T cell activation and tumor immunity in human CD27-transgenic mice. J Immunol. 2013;191:4174-83. doi: 10.4049/jimmunol.1300409 [DOI] [PubMed] [Google Scholar]
- 6.Khalil M, Vonderheide RH. Anti-CD40 agonist antibodies: preclinical and clinical experience. Update Cancer Ther. 2007;2:61-5. doi: 10.1016/j.uct.2007.06.001. PMID:19587842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li F, Ravetch JV. Inhibitory Fcgamma receptor engagement drives adjuvant and anti-tumor activities of agonistic CD40 antibodies. Science. 2011;333:1030-4. doi: 10.1126/science.1206954. PMID:21852502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White AL, Chan HT, Roghanian A, French RR, Mockridge CI, Tutt AL, Dixon SV, Ajona D, Verbeek JS, Al-Shamkhani A, et al.. Interaction with FcgammaRIIB is critical for the agonistic activity of anti-CD40 monoclonal antibody. J Immunol. 2011;187:1754-63. doi: 10.4049/jimmunol.1101135 [DOI] [PubMed] [Google Scholar]
- 9.Li F, Ravetch JV. Apoptotic and antitumor activity of death receptor antibodies require inhibitory Fcgamma receptor engagement. Proc Natl Acad Sci U S A. 2012;109:10966-71. doi: 10.1073/pnas.1208698109. PMID:22723355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson NS, Yang B, Yang A, Loeser S, Marsters S, Lawrence D, Li Y, Pitti R, Totpal K, Yee S, et al.. An Fcgamma receptor-dependent mechanism drives antibody-mediated target-receptor signaling in cancer cells. Cancer Cell. 2011;19:101-13. doi: 10.1016/j.ccr.2010.11.012. PMID:21251615 [DOI] [PubMed] [Google Scholar]
- 11.Xu Y, Szalai AJ, Zhou T, Zinn KR, Chaudhuri TR, Li X, Koopman WJ, Kimberly RP. Fc gamma Rs modulate cytotoxicity of anti-Fas antibodies: implications for agonistic antibody-based therapeutics. J Immunol. 2003;171:562-8. doi: 10.4049/jimmunol.171.2.562 [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Goldberg MV, Chiu ML. Fc Engineering Approaches to Enhance the Agonism and Effector Functions of an Anti-OX40 Antibody. J Biol Chem. 2016;291:27134-46. doi: 10.1074/jbc.M116.757773. PMID:27856634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL. Activating Fc gamma receptors contribute to the antitumor activities of immunoregulatory receptor-targeting antibodies. J Exp Med. 2013;210:1685-93. doi: 10.1084/jem.20130573. PMID: 23897982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulliard Y, Jolicoeur R, Zhang J, Dranoff G, Wilson NS, Brogdon JL. OX40 engagement depletes intratumoral Tregs via activating FcgammaRs, leading to antitumor efficacy. Immunol Cell Biol. 2014;92:475-80. doi: 10.1038/icb.2014.26. PMID:24732076 [DOI] [PubMed] [Google Scholar]
- 15.Simons PJ, Boon L, Luo J, Brezski RJ, Goldberg M. Humanized anti-CD134 (OX40) antibodies and uses thereof. 2014. Patent US 2014/0377284 A1, WO 2014/ 148895 A1 [Google Scholar]
- 16.Kawamata S, Hori T, Imura A, Takaori-Kondo A, Uchiyama T. Activation of OX40 signal transduction pathways leads to tumor necrosis factor receptor-associated factor (TRAF) 2- and TRAF5-mediated NF-kappaB activation. J Biol Chem. 1998;273:5808-14. doi: 10.1074/jbc.273.10.5808. PMID:9488716 [DOI] [PubMed] [Google Scholar]
- 17.Fisher TS, Kamperschroer C, Oliphant T, Love VA, Lira PD, Doyonnas R, Bergqvist S, Baxi SM, Rohner A, Shen AC, et al.. Targeting of 4-1BB by monoclonal antibody PF-05082566 enhances T-cell function and promotes anti-tumor activity. Cancer Immunol Immunother. 2012;61:1721-33. doi: 10.1007/s00262-012-1237-1. PMID:22406983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Williams GS, Mistry B, Guillard S, Ulrichsen JC, Sandercock AM, Wang J, González-Muñoz A, Parmentier J, Black C, Soden J, et al.. Phenotypic screening reveals TNFR2 as a promising target for cancer immunotherapy. Oncotarget. 2016;7:68278-91. doi: 10.18632/oncotarget.11943. PMID: 27626702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, Joyce PF, Szymkowski DE, Desjarlais JR. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol. 2008;45:3926-33. doi: 10.1016/j.molimm.2008.06.027. PMID:18691763 [DOI] [PubMed] [Google Scholar]
- 20.Mimoto F, Katada H, Kadono S, Igawa T, Kuramochi T, Muraoka M, Wada Y, Haraya K, Miyazaki T, Hattori K. Engineered antibody Fc variant with selectively enhanced FcgammaRIIb binding over both FcgammaRIIa(R131) and FcgammaRIIa(H131). Protein Eng Des Sel. 2013;26:589-98. doi: 10.1093/protein/gzt022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, et al.. Complement is activated by IgG hexamers assembled at the cell surface. Science. 2014;343:1260-3. doi: 10.1126/science.1248943. PMID:24626930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saphire EO, Parren PW, Pantophlet R, Zwick MB, Morris GM, Rudd PM, Dwek RA, Stanfield RL, Burton DR, Wilson IA. Crystal structure of a neutralizing human IGG against HIV-1: a template for vaccine design. Science. 2001;293:1155-9. doi: 10.1126/science.1061692. PMID:11498595 [DOI] [PubMed] [Google Scholar]
- 23.Matsumiya S, Yamaguchi Y, Saito J, Nagano M, Sasakawa H, Otaki S, Satoh M, Shitara K, Kato K. Structural comparison of fucosylated and nonfucosylated Fc fragments of human immunoglobulin G1. J Mol Biol. 2007;368:767-79. doi: 10.1016/j.jmb.2007.02.034. PMID:17368483 [DOI] [PubMed] [Google Scholar]
- 24.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126-32. doi: 10.1107/S0907444904019158. PMID:15572765 [DOI] [PubMed] [Google Scholar]
- 25.The PyMOL Molecular Graphics System https://www.pymol.org/citing Version 1.8 Schrödinger, LLC. [Google Scholar]
- 26.Teplyakov A, Zhao Y, Malia TJ, Obmolova G, Gilliland GL. IgG2 Fc structure and the dynamic features of the IgG CH2-CH3 interface. Mol Immunol. 2013;56:131-9. doi: 10.1016/j.molimm.2013.03.018. PMID:23628091 [DOI] [PubMed] [Google Scholar]
- 27.Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, Scallon B, Teplyakov A, Malia TJ, Strohl WR. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014;65:114-26. doi: 10.1016/j.ymeth.2013.06.035. PMID:23872058 [DOI] [PubMed] [Google Scholar]
- 28.Veri MC, Gorlatov S, Li H, Burke S, Johnson S, Stavenhagen J, Stein KE, Bonvini E, Koenig S. Monoclonal antibodies capable of discriminating the human inhibitory Fcgamma-receptor IIB (CD32B) from the activating Fcgamma-receptor IIA (CD32A): biochemical, biological and functional characterization. Immunology. 2007;121:392-404. doi: 10.1111/j.1365-2567.2007.02588.x. PMID:17386079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burmeister WP, Huber AH, Bjorkman PJ. Crystal structure of the complex of rat neonatal Fc receptor with Fc. Nature. 1994;372:379-83. doi: 10.1038/372379a0. PMID:7969498 [DOI] [PubMed] [Google Scholar]
- 30.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'Acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem. 2014;289:7812-24. doi: 10.1074/jbc.M113.537563. PMID:24469444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, Zhao Y, Kleijer M, Sandlie I, de Haas M, et al.. Competition for FcRn-mediated transport gives rise to short half-life of human IgG3 and offers therapeutic potential. Nat Commun. 2011;2:599. doi: 10.1038/ncomms1608. PMID:22186895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tam SH, McCarthy SG, Brosnan K, Goldberg KM, Scallon BJ. Correlations between pharmacokinetics of IgG antibodies in primates vs. FcRn-transgenic mice reveal a rodent model with predictive capabilities. MAbs. 2013;5:397-405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petkova SB, Akilesh S, Sproule TJ, Christianson GJ, Al Khabbaz H, Brown AC, Presta LG, Meng YG, Roopenian DC. Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int Immunol. 2006;18:1759-69. doi: 10.1093/intimm/dxl110. PMID:17077181 [DOI] [PubMed] [Google Scholar]
- 34.Haraya K, Tachibana T, Nanami M, Ishigai M. Application of human FcRn transgenic mice as a pharmacokinetic screening tool of monoclonal antibody. Xenobiotica; the fate of foreign compounds in biological systems. 2014;44:1127-34. doi: 10.3109/00498254.2014.941963. PMID:25030041 [DOI] [PubMed] [Google Scholar]
- 35.Avery LB, Wang M, Kavosi MS, Joyce A, Kurz JC, Fan YY, Dowty ME, Zhang M, Zhang Y, Cheng A, et al.. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8:1064-78. doi: 10.1080/19420862.2016.1193660. PMID:27232760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wajant H. Principles of antibody-mediated TNF receptor activation. Cell Death Differ. 2015;22:1727-41. doi: 10.1038/cdd.2015.109. PMID:26292758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies AM, Jefferis R, Sutton BJ. Crystal structure of deglycosylated human IgG4-Fc. Mol Immunol. 2014;62:46-53. doi: 10.1016/j.molimm.2014.05.015. PMID:24956411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies AM, Rispens T, Ooijevaar-de Heer P, Gould HJ, Jefferis R, Aalberse RC, Sutton BJ. Structural determinants of unique properties of human IgG4-Fc. J Mol Biol. 2014;426:630-44. doi: 10.1016/j.jmb.2013.10.039. PMID:24211234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiyoshi M, Caaveiro JM, Kawai T, Tashiro S, Ide T, Asaoka Y, Hatayama K, Tsumoto K. Structural basis for binding of human IgG1 to its high-affinity human receptor FcgammaRI. Nat Commun 2015;6:6866. doi: 10.1038/ncomms7866. PMID:25925696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu Y, West AP Jr., Kim HJ, Thornton ME, Ward AB, Bjorkman PJ. Structural basis for enhanced HIV-1 neutralization by a dimeric immunoglobulin G form of the glycan-recognizing antibody 2G12. Cell Rep. 2013;5:1443-55. doi: 10.1016/j.celrep.2013.11.015. PMID:24316082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frank M, Walker RC, Lanzilotta WN, Prestegard JH, Barb AW. Immunoglobulin G1 Fc domain motions: implications for Fc engineering. J Mol Biol. 2014;426:1799-811. doi: 10.1016/j.jmb.2014.01.011. PMID:24522230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Krapp S, Mimura Y, Jefferis R, Huber R, Sondermann P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J Mol Biol 2003;325:979-89. doi: 10.1016/S0022-2836(02)01250-0. PMID:12527303 [DOI] [PubMed] [Google Scholar]
- 43.Luo Y, Lu Z, Raso SW, Entrican C, Tangarone B. Dimers and multimers of monoclonal IgG1 exhibit higher in vitro binding affinities to Fcgamma receptors. MAbs. 2009;1:491-504. doi: 10.4161/mabs.1.5.9631. PMID:20065648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stubenrauch K, Wessels U, Lenz H. Evaluation of an immunoassay for human-specific quantitation of therapeutic antibodies in serum samples from non-human primates. J Pharm Biomed Anal. 2009;49:1003-8. doi: 10.1016/j.jpba.2009.01.030. PMID:19250787 [DOI] [PubMed] [Google Scholar]
- 45.Kabsch W Xds. Acta crystallographica Section D, Biological crystallography. 2010;66:125-132. doi: 10.1107/S0907444909047337. PMID:20124692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658-74. doi: 10.1107/S0021889807021206. PMID:19461840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 2011;67:235-42. doi: 10.1107/S0907444910045749. PMID:21460441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Idusogie EE, Presta LG, Gazzano-Santoro H, Totpal K, Wong PY, Ultsch M, Meng YG, Mulkerrin MG. Mapping of the C1q binding site on rituxan, a chimeric antibody with a human IgG1 Fc. J Immunol 2000;164:4178-84. doi: 10.4049/jimmunol.164.8.4178 [DOI] [PubMed] [Google Scholar]
- 49.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213-21. doi: 10.1107/S0907444909052925. PMID:20124702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Joosten RP, Long F, Murshudov GN, Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213-20. doi: 10.1107/S2052252514009324. PMID:25075342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355-67. doi: 10.1107/S0907444911001314. PMID:21460454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235-42. doi: 10.1093/nar/28.1.235. PMID:10592235 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.