

ABSTRACT
During manufacturing and storage process, therapeutic proteins are subject to various post-translational modifications (PTMs), such as isomerization, deamidation, oxidation, disulfide bond modifications and glycosylation. Certain PTMs may affect bioactivity, stability or pharmacokinetics and pharmacodynamics profile and are therefore classified as potential critical quality attributes (pCQAs). Identifying, monitoring and controlling these PTMs are usually key elements of the Quality by Design (QbD) approach. Traditionally, multiple analytical methods are utilized for these purposes, which is time consuming and costly. In recent years, multi-attribute monitoring methods have been developed in the biopharmaceutical industry. However, these methods combine high-end mass spectrometry with complicated data analysis software, which could pose difficulty when implementing in a quality control (QC) environment. Here we report a multi-attribute method (MAM) using a Quadrupole Dalton (QDa) mass detector to selectively monitor and quantitate PTMs in a therapeutic monoclonal antibody. The result output from the QDa-based MAM is straightforward and automatic. Evaluation results indicate this method provides comparable results to the traditional assays. To ensure future application in the QC environment, this method was qualified according to the International Conference on Harmonization (ICH) guideline and applied in the characterization of drug substance and stability samples. The QDa-based MAM is shown to be an extremely useful tool for product and process characterization studies that facilitates facile understanding of process impact on multiple quality attributes, while being QC friendly and cost-effective.
KEYWORDS: monoclonal antibody, multi-attribute method, process development, product quality attributes, quality control
Abbreviations
- BLA
biologics license application
- CDR
complementarity-determining region
- CE-SDS
capillary electrophoresis sodium dodecyl sulfate
- CEX
cation exchange chromatography
- CM
clarified medium
- DS
drug substance
- DTT
dithiothreitol
- EDTA
EDTA
- Fab
fragment antigen-binding
- Fc
fragment crystallizable
- FcRn
neonatal Fc receptor
- GuHCl
guanidine hydrochloride
- HILIC
hydrophilic interaction chromatography
- HL
heavy chain-light chain
- IAM
iodoacetamide
- ICH
international conference on harmonization
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- LOD
limit of detection
- LOQ
limit of quantitation
- Lys-C
lysyl endopeptidase
- m/z
mass-to-charge ratio
- mAbs
monoclonal antibodies
- MAM
multi-attribute method
- Man
high mannose
- MS
mass spectrometry
- NaCl
sodium chloride
- NEM
N-ethylmaleimide
- PBS
phosphate-buffered saline
- pCQAs
potential critical quality attributes
- PTMs
post-translational modifications
- QbD
quality by design
- QC
quality control
- QDa
quadrupole dalton
- SIR
selected ion recording
- TFA
trifluoroacetic acid
Introduction
Therapeutic monoclonal antibodies (mAbs) are subject to various post-translational modifications (PTMs) during cell culture process, purification, formulation, and long-term storage. These modifications may affect the biologic activity, pharmacokinetics and pharmacodynamics, immunogenicity and safety of the drug product, and therefore may be classified as potential critical quality attributes (pCQAs).1 The process parameters that affect the pCQAs must be identified, characterized and controlled.2 In-depth and thorough knowledge of these product variants and their link to manufacturing process are essential for the Quality by Design (QbD) approach.1,3
Fc glycosylation is a common PTM known to play a pivotal role in the safety and activity of mAbs by affecting immunogenicity,4 serum half-life,5 and effector functions.6 Several multi-attribute methods have been developed to monitor the glycosylation profiles of mAbs. The most popular one is a bottom-up approach to analyze protease-digested glycopeptides using liquid chromatography and high resolution tandem mass spectrometry (LC-MS/MS). This approach provided site-specific quantitation of N-linked glycan distribution on the constant region (Fc) and variable region (Fab), and showed good correlation with the traditional hydrophilic interaction chromatography (HILIC)-based oligosaccharide analysis.7-9 Another method estimates the relative abundance of glycoforms from the intact molecule or its subunits using high resolution MS.10
In addition to glycosylation, other PTMs identified as pCQAs also need to be monitored and evaluated to understand their effect on process and product quality. Asn deamidation into Asp and iso-Asp is a common degradation pathway in mAbs mainly affected by buffer pH and temperature.11 The deamidation level is traditionally measured through charge heterogeneity analysis. However, although deamidation is a main contributor of the acidic species, other modifications such as sialic acid, trisulfide bond, cysteinylation, and glycation can also result in acidic species.12 Isomerization of Asp residue to iso-Asp is another widely reported modification. Although no charge difference is caused by isomerization, the antibody containing Asp and iso-Asp can be separated by cation exchange chromatography (CEX), possibly due to structural differences.13 The Asp isomerization species have been found in the basic eluting peaks of CEX.12 The effect of oxidation on product quality largely depends on the location in protein sequence. It has been demonstrated that Met-252 oxidation in the IgG1 Fc region is the main contributor to impaired neonatal Fc receptor (FcRn) binding, which leads to shorter serum half-life.14,15 A Met-252 oxidized variant has also been found enriched in the basic peaks of CEX.16,17
Several comprehensive reviews have discussed the analytical approaches commonly used for characterizing protein therapeutics. A combination of liquid chromatography, electrophoresis, and MS methods have been used at all stages of mAbs discovery and development.18-20 Although chromatography and electrophoresis methods are widely used as release assays for therapeutic proteins, they typically monitor product variants at the intact protein level. However, the QbD approach requires direct monitoring of pCQAs at the amino acid level.21 As a result, peptide mapping-MS-based analytical methods are gaining popularity in recent years due to the site-specific identification and quantitation of various PTMs. A recent evaluation of the biologics license application (BLAs) found that use of MS for the analysis of individual attributes, especially deamidation and oxidation, has risen steadily from 20% to 80% in recent years. In addition, the average number of attributes analyzed by MS also increased from 2 to 11 per BLA.22 We can anticipate that use of MS for therapeutic proteins will continue to progress.
To gain more comprehensive understanding of the product and process with better efficiencies, several pharmaceutical companies have developed and implemented MS-based methods that are capable of quantifying several attributes at once. Such an approach is typically referred to as multi-attribute method (MAM).7-10,23,24 Most of these methods combine high accuracy / high resolution MS with sophisticated data analysis software to identify and quantify various PTMs. These methods have shown comparable results with the traditional methods such as HILIC oligosaccharide analysis, CEX, and capillary electrophoresis sodium dodecyl sulfate (CE-SDS).7,9,24 However, there is continued debate about whether MS-based protein analysis, such as MAM, should replace the conventional electrophoresis and chromatography methods.25 MS is not widely used in quality control (QC) laboratories in current pharmaceutical industry due to high-maintenance equipment and complicated data analysis. In addition, the high-end mass spectrometers are expensive and pose difficulty in global QC application.
We present here a QC-friendly multi-attribute monitoring method that combines non-reduced peptide mapping with Quadrupole Dalton (QDa) detection. ACQUITY QDa is a single-quadrupole mass detector with an electrospray ionization source. Compared to traditional high-end mass spectrometers, QDa is cost effective and very easy to operate and maintain. QDa has been applied in quantitative analysis of amino acids26 and additives in drug candidates.27 It has also been used in the QC environment for analyzing peptide drugs,28 and testing complementarity-determining region (CDR) peptides of a mAb.29 In this study, we show the development of a QDa-based MAM for targeted quantitation of PTMs in a therapeutic mAb (MAB1). These PTMs include deamidation of Asn and isomerization of Asp in the CDR region; deamidation of PENNY peptide, oxidation of Met-252, and glycosylation in the Fc region; and disulfide bond modifications (trisulfide bond and thioether bond between light and heavy chains, and free thiol and cysteinylation on the light chain terminal cysteine). Most of these PTMs are considered pCQAs for MAB1.
This method has been implemented to support cell culture process development, downstream process development, and protein characterization in serum incubation studies. This single assay can replace several traditional assays for monitoring these product quality attributes, substantially reducing time and cost. In addition, to ensure future application in QC, this method has been qualified according to the ICH guidelines and applied to characterizing drug substance and stability samples.
Results
Development of QDa-based MAM
The sample preparation and chromatography conditions for QDa-based MAM were optimized from the traditional non-reduced Lys-C disulfide bond mapping procedure for MAB1. Lys-C digestion at pH 7.0 can reduce the artificial disulfide-bond scrambling and deamidation that was normally introduced due to the slight alkaline pH in the traditional tryptic peptide mapping.30 Chromatographic separation was performed on an ACQUITY I-class UPLC system coupled with a QDa mass detector. The detailed method is described in the Materials and Methods section. To increase sensitivity and generate straightforward result output, we used the Selected Ion Recording (SIR) function of QDa to monitor and quantify specific peaks. Fig. 1 shows the comparison of UV absorbance spectrum at 220 nm and the SIR plots of each peptide. The mass-to-charge ratio (m/z) of each peptide was used for the SIR. As shown in Fig. 1B, one m/z was used to monitor both the native peptide and its isomerized variant because there was no mass change. Deamidation and oxidation caused mass shift, and therefore were monitored in separate channels. In Fig. 1A, light chain Cys-214 cysteinylation, heavy chain-light chain (HL) thioether bond and HL disulfide bond peaks overlap in retention time. The SIR specifically monitored these variants and provided much cleaner peak profiles. SIR is also much higher in sensitivity as demonstrated by the identification of Cys-214 free thiol, which was too low to be detected in the UV spectrum. The higher specificity and sensitivity of SIR can reduce the quantitation variability caused by peak integration. Commonly, in a complicated protein digest matrix, 2 or more peptides can have very similar masses. This is exemplified in the detection of more than one peak in the SIR channel where HL trisulfide bond was monitored. In addition, QDa has a 0.7 Da mass resolution, which may also contribute to the unspecific detection of species with small mass difference. To ensure correct peak assignment, all peaks monitored by QDa were confirmed by LC-MS/MS for identity and relative retention times.
Figure 1.
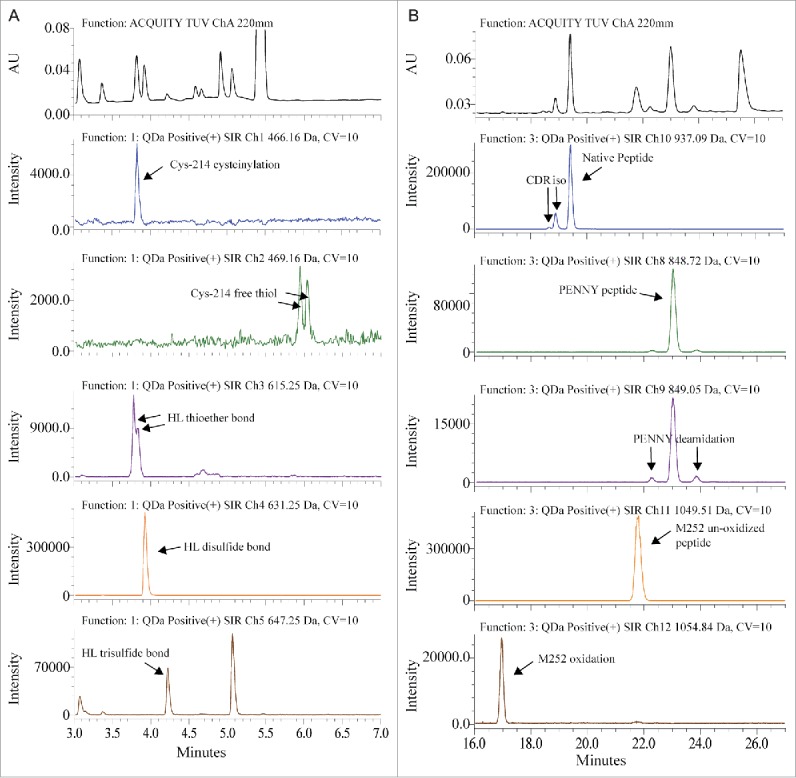
Overlay of UV spectrum with SIR channels. (A) Disulfide bond modifications were monitored between 3 to 7 min. (B) CDR isomerization, PENNY peptide deamidation and Met-252 oxidation were monitored between 16 to 27 min.
Detection and quantitation of PTMs for the QDa-based MAM were fully automated in the associated software Empower 3. Peaks were detected based on the specific SIR channel and relative retention time. The level of each PTM, except for Fc glycosylation, was calculated by using the variant peak area divided by the sum of native and variant peak area.
The mechanism of action for MAB1 includes antibody-dependent cell-mediated cytotoxicity; therefore, afucosylation is an important pCQA to monitor for this molecule.31-33 In the high-end MS-based MAM approach and HILIC oligosaccharide profiling, the total afucosylation level was calculated using the area of all afucosylated glycans divided by the summed area of all detected glycans. The most abundant glycan in MAB1 is the fucosylated complex-type oligosaccharide G0f, while other fucosylated glycans (e.g., G0f-GN, G1f-GN, G1f, G2f) are present at lower levels. The major afucosylated glycans in MAB1 are G0, G0-GN, and M5, while other afucosylated glycans account for less than 2% of total glycans. However, due to the QDa mass range (30 – 1250 Da), not all MAB1 glycopeptides can be monitored by QDa using the same charged state. Therefore, we were not able to calculate the afucosylation level in the same way as HILIC or other MAM methods.
Interestingly, an analysis of HILIC oligosaccharide profiling data collected from over 70 process development samples of MAB1 showed a linear correlation between the “Total afucosylation by HILIC” (sum of all afucosylated glycans) and the “Relative afucosylation to G0f” calculated as follows: (G0 + G0-GN + M5) ÷ (G0 + G0-GN + M5 + G0f) (Fig. 2). R2 of the linear regression is 0.9930, indicating a highly linear correlation between the 2 sets of data.
Figure 2.
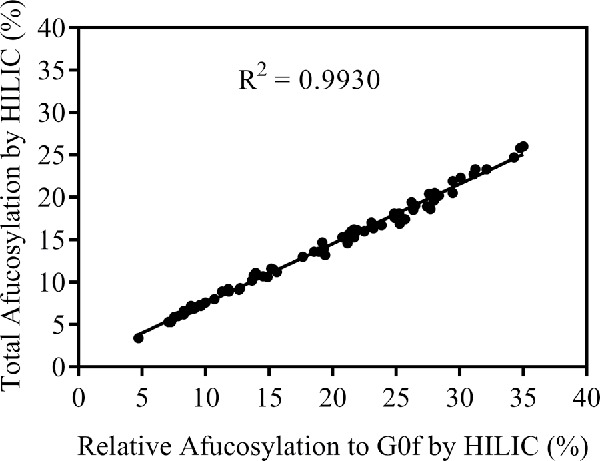
Correlation between total afucosylation (sum of all afucosylated glycans) and relative afucosylation to G0f [(G0 + G0-GN + M5) ÷ (G0 + G0-GN + M5 + G0f)] using HILIC oligosaccharide profiling data.
Based on this information, the QDa was configured to monitor and quantify glycopeptides with G0, G0-GN, M5 and G0f glycans (Fig. 3A). The “Relative afucosylation to G0f by MAM” is calculated by summed areas of G0, G0-GN and M5 divided by the summed area of all 4 glycopeptides. Nine MAB1 samples with different afucosylation levels were selected and evaluated using this method. The resulting “Relative afucosylation to G0f by MAM” was plotted with the “Total afucosylation by HILIC." Similar to the linear correlation observed from HILIC oligo profiling data, a linear correlation (R2 = 0.9911) was also observed between the MAM data and the total afucosylation (Fig. 3B). The linear regression equation can be used to predict the total afucosylation level from MAM results (Predicted total afucosylation = 0.8118 × Measured relative afucosylation by MAM – 0.1586).
Figure 3.
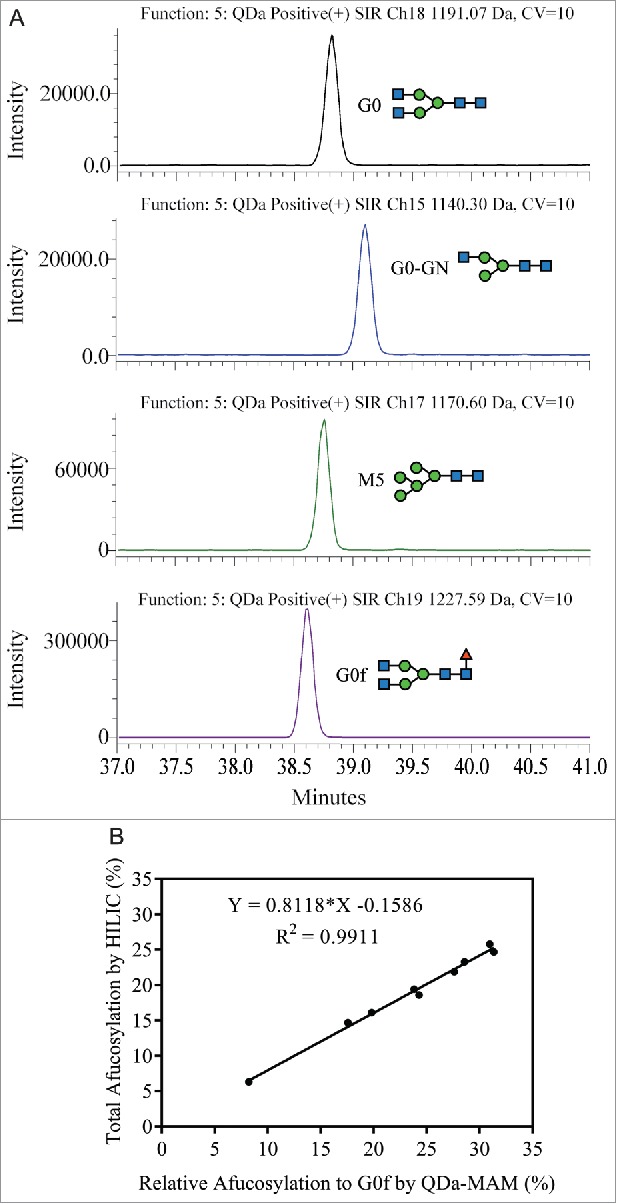
(A) Glycopeptides with G0, G0-GN, M5, and G0f were monitored by SIR between 37 to 41 min. (B) Linear correlation was confirmed between the total afucosylation measured by HILIC and the relative afucosylation to G0f measured by QDa-based MAM.
Characterization of stability and photo-stressed samples by QDa-based MAM
In traditional protein characterization, multiple analytical methods are often used to measure pCQAs. For MAB1, site-specific deamidation and oxidation were quantified from extracted ion chromatogram in reduced tryptic peptide mapping. CDR isomerization was quantified from UV spectrum using a focused peptide mapping method with optimized mobile phase and gradient. Disulfide bond modifications were quantified using non-reduced Lys-C disulfide bond mapping. Thioether bonds can also be quantified from reducing gel electrophoresis.34 Fc region N-linked glycans were quantified using a HILIC separation of 2-AB labeled released glycan method. To evaluate comparability of QDa-based MAM with traditional methods, we characterized MAB1 pCQAs in stability and photo-stressed samples and compared with historical data.
Stability
Stability samples were generated by incubating MAB1 in the original formulation buffer at 4°C, 25°C and 40°C for 3 months. The modification levels quantified using QDa-based MAM are summarized in Fig. 4A. CDR isomerization increased 27.5% after 3 months incubation at 25°C. The rate was even faster at 40°C, where 61% increase was observed after 3 months. The isomerization levels measured by QDa-MAM were comparable with that measured by the traditional UV-based focused peptide mapping method (Fig. 4B). Deamidation in both the CDR region and the PENNY peptide also increased at 40°C, but at much slower rates (< 8% increase for each site after 3 months). The deamidation levels measured by QDa-MAM were lower than the traditional tryptic peptide mapping method (Fig. 4C, 4D). This is likely because this method used a pH 7.0 buffer during sample preparation, which reduced artificial deamidation caused by the slight alkaline pH in traditional tryptic digestion.30 A small increase (∼1%) in HL thioether at 40°C was also observed. This was consistent with the level observed in reducing gel electrophoresis (Fig. 4E). These results indicate that CDR isomerization is a major degradation pathway for MAB1 at elevated temperatures.
Figure 4.
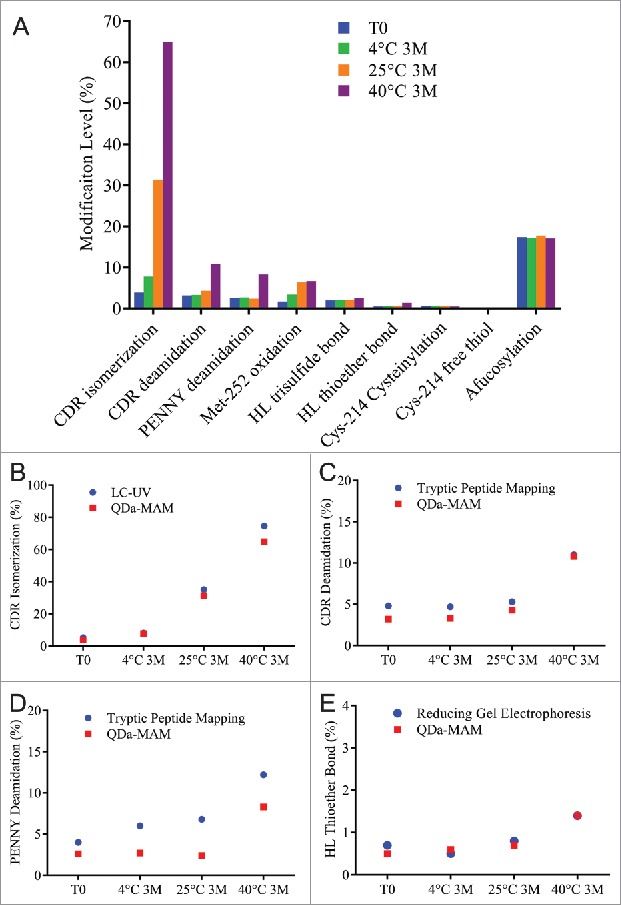
(A) Modification levels measured by QDa-based MAM in MAB1 stability samples at initial time point (blue), 3 months at 4°C (green), 3 months at 25°C (orange), and 3 months at 40°C (purple). (B) CDR isomerization quantified by LC-UV-based method (blue) and by QDa-MAM (red). (C) CDR deamidation quantified by tryptic peptide mapping (blue) and QDa-MAM (red). (D) PENNY peptide deamidation quantified by tryptic peptide mapping (blue) and QDa-MAM (red). (E) HL thioether bond quantified by reducing gel electrophoresis (blue) and QDa-MAM (red).
Photo-stress
Photo-stressed samples were generated by exposing MAB1 to cool-white light and UV light at 1X or 3X dosage of the ICH guideline Q1B. The detailed experiment set up is described in the Materials and Methods section. The photo-stressed samples were evaluated by QDa-based MAM and summarized in Fig. 5A. As expected, the most noteworthy impact by photo-stress was Met-252 oxidation. About 45% of Met-252 was oxidized at 1X ICH dosage of light. Close to 100% oxidation was reached under 3X ICH dosage of light. The oxidation level measured by QDa-MAM was comparable to that obtained using traditional peptide mapping method (Fig. 5B). The results also showed substantial reduction of HL trisulfide bond from 12.5% to 0.3% after photo-stress, and Cys-214 free thiol level increased 4.6% under 3X ICH dosage of light. These results indicated UV-induced disulfide bond cleavage and reformation that has been reported previously.35 No changes in isomerization, deamidation, afucosylation and other disulfide bond modifications were observed. Interestingly, 5% increase of the non-reducible peak at ∼92 Da (data not shown) was observed in 1X ICH photo-stressed sample by reducing gel electrophoresis, but not by MAM, indicating this photo-induced crosslinking is not thioether related.
Figure 5.
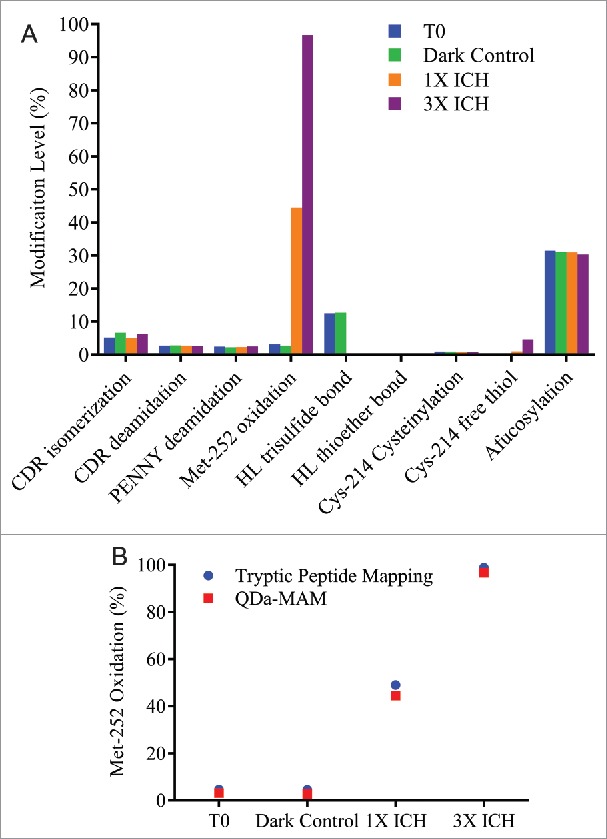
(A) Modification levels measured by QDa-based MAM in MAB1 photo-stressed samples at initial time point (blue), dark control (green), light irradiated at the dosage per ICH guideline (1X ICH, orange), and light irradiated at 3 times the strength per ICH guideline (3X ICH, purple). (B) Met-252 oxidation quantified by tryptic peptide mapping (blue) and QDa-MAM (red).
Qualification of QDa-based MAM
To support further application of the QDa-based MAM as a stability-indicating assay in the QC environment, this method was qualified based on the validation design described in the ICH guideline Q2R1. The ability of this method to quantify MAB1 quality attributes, including isomerization, deamidation, oxidation, disulfide modifications and afucosylation, was evaluated based on specificity, precision, linearity, accuracy, limit of quantitation (LOQ), and limit of detection (LOD). The qualification results are summarized in Table 1 and 2.
Table 1.
Summary of qualification results for repeatability and intermediate precision.
| Intermediate precision |
||||||
|---|---|---|---|---|---|---|
| Post-Translational Modifications (PTMs) | Precision-Repeatability (6 preps × 1 injection) | Day-to-Day (3 preps × 1 injection / day, 2 days) | Analyst-to-Analyst (3 preps × 1 injection / analyst, 2 analysts) | Column-to-Column (3 preps × 1 injection / column, 2 columns) | Instrument-to-Instrument (3 preps × 1 injection / instrument, 2 instruments) | |
| CDR isomerization (%) | Mean | 3.8 | 3.9 | 3.6 | 3.9 | 3.9 |
| % CV | 1.9 | 1.2 | 7.4 | 1.7 | 1.4 | |
| CDR deamidation (%) | Mean | 3.1 | 3.2 | 3.2 | 3.2 | 3.2 |
| % CV | 1.4 | 2.3 | 2.4 | 2.4 | 5.0 | |
| PENNY deamidation (%) | Mean | 2.7 | 2.7 | 2.7 | 2.6 | 2.7 |
| % CV | 3.1 | 2.8 | 1.9 | 7.9 | 2.4 | |
| Met-252 oxidation (%) | Mean | 1.8 | 1.8 | 1.8 | 1.9 | 2.0 |
| % CV | 0.9 | 3.3 | 7.0 | 5.0 | 8.0 | |
| HL trisulfide bond (%) | Mean | 2.0 | 2.0 | 2.0 | 2.0 | 1.9 |
| % CV | 3.3 | 1.4 | 1.6 | 2.6 | 2.0 | |
| HL thioether bond (%) | Mean | 0.5 | 0.5 | 0.5 | 0.5 | 0.5 |
| % CV | 9.3 | 7.1 | 7.6 | 7.5 | 6.4 | |
| Afucosylation (%) | Mean | 17.0 | 17.4 | 17.4 | 17.2 | 16.9 |
| % CV | 0.3 | 2.2 | 2.8 | 1.6 | 0.9 | |
Table 2.
Summary of qualification results for linearity, accuracy, Limit of Quantitation (LOQ), and Limit of Detection (LOD).
| Linearity and Range |
Accuracy |
||||||
|---|---|---|---|---|---|---|---|
| R2 | Range | LOQ, from calibration curve, 10 (SD/S) | LOD, from calibration curve, 3.3 (SD/S) | Level | % Recovery | % CV | |
| CDR isomerization (%) | 0.9995 | 4.1 – 65.7 | 3.3 | 1.1 | Low | 107.0 | 1.7 |
| Middle | 99.0 | 0.4 | |||||
| High | 100.0 | 0.8 | |||||
| CDR deamidation (%) | 0.9981 | 3.2 – 10.7 | 1.2 | 0.4 | Low | 106.0 | 0.3 |
| Middle | 102.0 | 0.9 | |||||
| High | 103.0 | 0.5 | |||||
| PENNY deamidation (%) | 0.9996 | 2.5 – 50.1 | 2.6 | 0.8 | Low | 103.0 | 1.7 |
| Middle | 101.0 | 2.9 | |||||
| High | 99.0 | 0.9 | |||||
| Met-252 oxidation (%) | 0.9977 | 3.1 – 46.2 | 4.8 | 1.6 | Low | 104.0 | 3.9 |
| Middle | 96.0 | 2.4 | |||||
| High | 100.0 | 0.0 | |||||
| HL trisulfide bond (%) | 0.9970 | 2.0 – 11.1 | 1.6 | 0.5 | Low | 96.0 | 0.6 |
| Middle | 105.0 | 4.5 | |||||
| High | 103.0 | 8.8 | |||||
| HL thioether bond (%) | 0.9979 | 0.5 – 13.6 | 1.4 | 0.5 | Low | 96.0 | 1.1 |
| Middle | 106.0 | 0.1 | |||||
| High | 102.0 | 2.6 | |||||
| Cys-214 free thiol (%) | 0.9936 | 0 – 3.1 | 0.6 | 0.2 | Low | 160.0 | 3.6 |
| Middle | 99.0 | 1.3 | |||||
| High | 104.0 | 6.0 | |||||
| Afucosylation (%) | 0.9917 | 8.3 – 31.4 | 6.7 | 2.2 | Low | 99.0 | 0.9 |
| Middle | 98.0 | 0.3 | |||||
| High | 101.0 | 0.4 | |||||
Specificity
Specificity of the assay was assessed by analyzing formulation buffer, digestion buffer, 3 months at 40°C stressed sample, and samples collected from each downstream purification step. No responses were observed from formulation buffer or digestion buffer in all monitored channels. Expected increases in isomerization and deamidation were observed in 3 months at 40°C stressed sample. The in-process samples and MAB1 drug substance (DS) had comparable peak profiles, indicating little impact from the sample matrix.
Precision
Precision includes repeatability and intermediate precision. Repeatability was evaluated by analyzing 6 independently prepared MAB1 DS digests. Intermediate precision was evaluated by analyzing 2 sets of 3 independently prepared digests from 2 different days (day-to-day), 2 different analysts (analyst-to-analyst), 2 different column lots (column-to-column), and 2 different instruments (instrument-to-instrument). The percent coefficient of variation (%CV) for isomerization, deamidation, oxidation, trisulfide bond, thioether bond, and afucosylation were less than 10% (Table 1).
Linearity, accuracy, limit of quantitation, and limit of detection
Linearity was analyzed by spiking 0%, 25%, 50%, 75%, and 100% of stressed materials into MAB1 DS. The assessment was performed in duplicate. Heat-stressed samples were used to evaluate CDR isomerization and deamidation linearity range. Photo-stressed samples were used to evaluate Met-252 oxidation and Cys-214 free thiol. High pH-stressed samples were used to evaluate PENNY deamidation and HL thioether bond. Two samples with different levels of trisulfide bond and afucosylation were used to evaluate trisulfide and afucosylation linearity and range. The results are summarized in Table 2. Calibration curves were generated based on the measured modification level to the spike percentage. The R2 of the calibration curves for all attributes evaluated were above 0.99, demonstrating a good fit of the linear regression. The LOQ and LOD were calculated based on the standard deviation and slope of the calibration curve as described in ICH guideline Q2B. Accuracy was assessed by spiking 10%, 45% and 90% of stressed material into MAB1 DS, representing low, middle, and high values in the linear range. Recovery was calculated by comparing the experimentally determined modification level with the expected level calculated from a calibration curve. For all attributes assessed, the % CV at all 3 levels were below 10%, and the recoveries were between 96–107%, except for Cys-214 free thiol at low level. This was likely because the results were below LOQ. Overall, the results demonstrated good linear range and quantitation limit, as well as high accuracy of the QDa-based MAM.
Application of QDa-based MAM for characterization and process development
Serum incubation study
A serum incubation study was designed to assess MAB1 PTM changes under physiologic conditions that mimic the in vivo environment. MAB1 incubated in 1X phosphate-buffered saline (PBS) pH 7.4 at 37°C was used as a control. As shown in Fig. 6A, incubating in human serum and PBS resulted in isomerization increase, which was likely due to the high temperature (37°C). We also observed increases in deamidation, and the levels were higher in serum than in PBS at 2 weeks. The exact cause for faster deamidation in serum was unclear. Because Asn deamidation can be affected by buffer type, pH and temperature,36 it is likely that the sample matrix and the pH difference between human serum and PBS played a role in the different deamidation rate. Other changes observed during serum incubation were the disulfide bond modifications. Decreases in trisulfide and Cys-214 cysteinylation levels were observed in human serum, but not in PBS. The conversion from trisulfide to disulfide was likely due to the redox reactions, which also cleaved the disulfide bond of Cys-214 cysteinylation. A previously published study showed complete conversion of trisulfide to disulfide in vivo within 24 hours.37 The slight increase of thioether bond in serum was consistent with previously observed formation of thioether in vitro and in vivo.38 Another interesting observation was the slight increase in afucosylation upon serum incubation. We found that the increase was mainly contributed by the increase in Man-5 level (Fig. 6B), which was likely due to the presence of soluble α-mannosidase in human serum that trimmed the other high mannose species (Man-6, Man-7, Man-8) to Man-5.39
Figure 6.
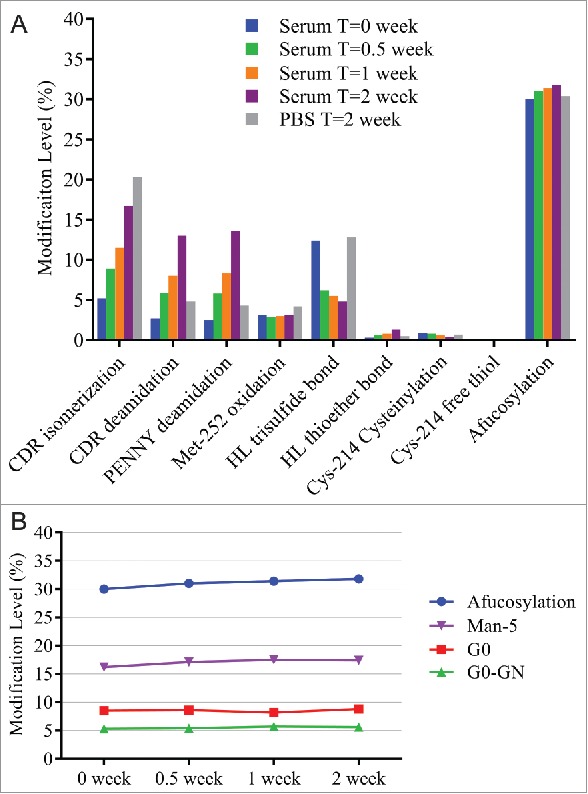
(A) Modification levels measured by QDa-based MAM in serum incubated samples at initial time point (blue), after 0.5 week at 37°C (green), after 1 week at 37°C (orange), and after 2 weeks at 37°C (purple). MAB1 incubated in PBS after 2 weeks at 37°C was tested side by side as control (gray). (B) Slight afucosylation increase in serum incubated samples was mainly contributed by the increase in Man-5.
Cell culture process development
The QDa-based MAM can also be implemented as a tool for upstream process development. As shown in Fig. 7, we used this method to monitor the changes in MAB1 product attributes over multiple days of cell culture. Harvest materials from day 4 to last day of culture (day 14) were first centrifuged to remove cells, and then filtered to yield clarified medium (CM) samples. The CM samples were then purified using an automated protein A purification platform on a Tecan liquid handling system. The purified samples were directly digested and analyzed using the QDa-MAM procedure.
Figure 7.
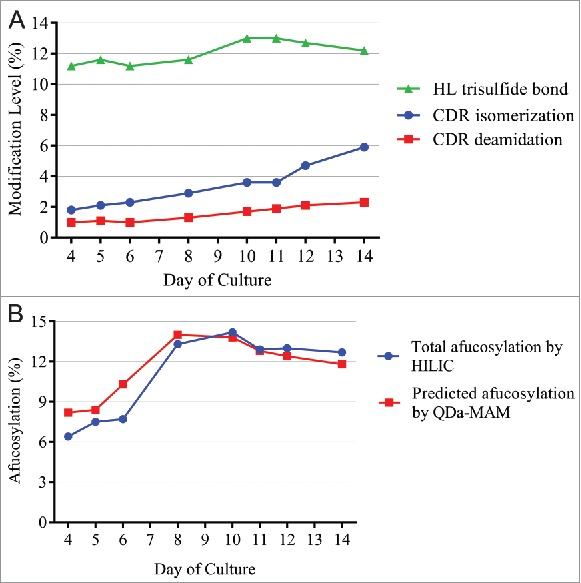
(A) Modification levels measured by QDa-based MAM in cell culture daily samples. (B) Total afucosylation by HILIC (blue) and predicated afucosylation by QDa-MAM (red, Predicted afucosylation = 0.8118 × Measured relative afucosylation by MAM – 0.1586) in cell culture daily samples.
As shown in Fig. 7A, from day 4 to day 14, CDR isomerization and deamidation both increased steadily. Trisulfide levels increased initially and peaked at day 10–11, and then slowly decreased to 12% at day 14. Levels of other attributes were below the LOQ. Fig. 7B shows the changes of afucosylation predicted from QDa-MAM, using the linear correlation equation shown in Fig. 3B. Total afucosylation was also measured by the HILIC method. The afucosylation started at 6∼8% in early days of culture, quickly increased and peaked at ∼14% on day 10, and then slowly decreased to ∼12% at day 14. These results demonstrated that QDa-MAM can monitor the changes of MAB1 afucosylation during cell culture. The predicted afucosylation level using the linear correlation equation was highly comparable with the HILIC method.
Purification process development
We also applied the QDa-based MAM to monitor product attribute changes during downstream processing. As shown in Fig. 8, a decrease was observed in CDR isomerization after CEX. Antibody containing iso-Asp is known to enrich in the CEX basic peaks,12 and was therefore removed during this process. Although a small increase in Met-252 oxidation after low pH treatment was observed, the values were below LOQ of this method (4.8% for Met-252 oxidation). Met-252 oxidation at low levels is not a major concern because only high levels of Met-252 oxidation (> 40%) will affect serum half-life.15 No changes in other modifications were observed.
Figure 8.
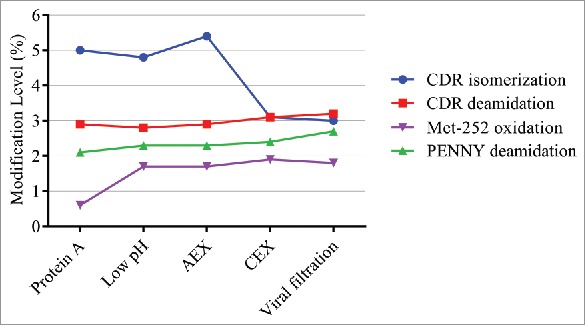
Modification levels measured by QDa-based MAM in in downstream purification process.
Discussion
With the increased expectation from regulatory agencies that a QbD approach will be followed in pharmaceutical development, MS-based MAMs are gaining momentum because they can provide more comprehensive information on PTMs in complex protein drugs in a single assay compared with conventional methods.21 The information acquired by MAMs provides deeper and better understanding of the product and process, which is the basis of the QbD approach. However, due to the complexity of MS instrument and data analysis, there remains debate on whether MS-based techniques should be applied for releasing drug product in QC laboratories. As described here, we designed a QC-friendly, multi-attribute monitoring method that uses a QDa mass detector with SIR for product attribute detection and quantitation. Unlike the other MAMs that detect PTMs on all possible amino acids,7,8 the QDa-based MAM is designed to monitor selected PTMs based on the product-specific pCQA list, which was determined through forced degradation and attribute enrichment studies in the early stage of product development. Although the results are not as comprehensive as the other methods, the targeted pCQA monitoring provides more straightforward information on the important PTMs. The SIR mode is also more sensitive than full scanning because the mass spectrometer can spend all available time on the ion of interest, which is especially useful when the modification level is low. In addition, the QDa-based method is simple to use, robust, and cost-effective.
The QDa-based MAM can be applied throughout the later stage of the drug development life cycle. As demonstrated in this study, we have used this method to characterize a therapeutic IgG, MAB1. The results obtained for DS, stability samples, and photo-stressed samples were comparable with the traditional methods, including tryptic peptide mapping, focused peptide mapping, reducing gel electrophoresis, and HILIC oligosaccharide profiling. By using a non-reducing Lys-C digestion method, the QDa-based MAM can also provide information on disulfide bond-related modifications, such as trisulfide, thioether, free thiols and cysteinylation. Although we did not perform disulfide bond mapping for the stressed materials, historical data of trisulfide levels in MAB1 samples was highly comparable to the results obtained from the QDa-based MAM. We have also observed light-induced disulfide bond cleavage that provided new information about the photo-stability of the molecule.
In addition to protein characterization, we implemented the QDa-based MAM to support upstream and downstream process development. Afucosylation is an important pCQA for MAB1, which needs to be monitored and controlled. However, due to the limited mass range, only glycopeptides with certain glycans can be monitored using the QDa-based method. To solve this problem, we used a prediction model derived from the observed linear correlation between total afucosylation measured by HILIC oligosaccharide analysis and the relative afucosylation measured by QDa-MAM. The relative afucosylation was calculated using the summed area of major afucosylated species (G0, G0-GN, M5) divided by the summed area of G0, G0-GN, M5 and G0f. Although we have not been able to explain this correlation from a biologic perspective, the linear relationship was statistically significant as demonstrated by the high R2 value (> 0.99). Using this prediction model (Predicted total afucosylation = 0.8118 × Measured relative afucosylation by MAM – 0.1586), we analyzed the cell culture daily samples from an upstream process. The predicted afucosylation levels were highly comparable with that measured by HILIC method, demonstrating good prediction of this model and application of QDa-based MAM for upstream process development. It is important to note that this prediction model may not be directly applied to other molecules. New correlations need to be established for new molecules to be studied. In addition, because the QDa-based MAM monitors targeted pCQAs, new modifications to the sample due to forced degradation or unexpected change in process development still require other methods, including chromatography, electrophoresis, high-resolution MS and bioassay for comprehensive characterization.18-20
In this study, we applied an automated Protein-A purification for the CM samples. Our next step is to also automate the sample digestion for QDa-based MAM. The fully automated workflow will enable process analytical technology application to establish correlation between critical quality attributes and critical process parameters more efficiently, and to meet higher regulatory expectations. Since the QDa-based approach is automated and the software platforms is suitable for the regulatory environment, we expect increased application of QDa-based MAM in biologics development, including quality control and comparability studies.
Materials and methods
Materials
MAB1 (IgG1) was expressed in Chinese hamster ovary cells and purified through a multi-column purification process at MedImmune. MAB1 was stored at −80°C in its formulation buffer (pH 6.0) until use.
Automated protein A purification
The mAb products from conditioned cell culture media samples were isolated using a high-throughput, automated protein A-based purification platform. Briefly, the Tecan Evo Freedom 200 liquid handling system (PN 30020020, Tecan) was used to cyclically aspirate and dispense 150 μL of cell culture sample through protein A ProPlus Resin PhyTips (PTM 92–20–07, PhyNexus) to bind antibody to the resin. Following 8 cycles, the tips were washed in 1X PBS for 4 cycles and then normal saline for 4 cycles. After washing, the antibody was eluted off the tip packed with protein A resin using a single cycle of 100 μL 50 mM sodium acetate pH 3.7. Finally, the resulting eluate was neutralized to pH about 6.0 by adding 38.5 μL 2 M Tris pH 8.0.
Sample preparation for QDa-based MAM
The antibody samples were first diluted in 100 mM sodium phosphate pH 7.0 buffer to 1mg/mL, then 100 µg of samples were alkylated and denatured at 37°C for 30 minutes in a 100 µL mixture of 5.5 mM sodium phosphate pH 7.0, 0.2 mM N-ethylmaleimide (NEM, E1271–5G, Sigma-Aldrich), 50 mM sodium chloride (NaCl, 71376, Sigma) and 3.2 M guanidine HCl (GuHCl, 24115, ThermoFisher Scientific). The alkylated and denatured antibodies were then diluted using 100 mM sodium phosphate pH 7.0 and 0.1 mM ethylenediaminetetraacetic acid (EDTA, E5391, Sigma) to reduce guanidine concentration to 1.7 M. Lysyl endopeptidase (Lys-C, 125–05061, Wako) was added at 1:5 protease: protein ratio and incubated at 37°C for 4 hours. After digestion, the samples were either stored at −80°C or transferred to HPLC vials and placed in a temperature controlled (6–8°C) auto sampler for analysis.
Chromatography method for QDa-based MAM
Digested peptide mixture was analyzed using a Waters I-class UPLC system coupled with a QDa mass detector. Mobile phase A contained 0.1% trifluoroacetic acid (TFA) in water and mobile phase B contained 0.1% TFA in acetonitrile. Waters ACQUITY UPLC BEH300 C18 column (1.7 um, 2.1 × 150 mm) was temperature controlled at 47°C. Flow rate was 0.2 mL / minute. A focused separation gradient was developed to optimize peak separation and shorten total run time. Phase B first increased from 5% to 15% in 3 minutes, then to 21% at 12 minutes. This was followed by a shallow increase to 22% B at 25 minutes. Next, phase B ramped up to 24% at 26 minutes and then to 26% at 32 minutes, followed by another quick increase to 28% at 34 minutes and a gradual increase to 30% B at 40 minutes. The column was then washed with 95% B followed by equilibration at 5% B. The total run time per sample was 47 minutes.
Traditional reduced tryptic peptide mapping
Reduced tryptic peptide mapping for MAB1 was performed based on the manufacturer's recommendations with some modifications.40 Briefly, 50 µg of antibody sample at 10 mg/mL was denatured and reduced at 37°C for 30 minutes in 20 µL of 6.3 M urea, 1 M GuHCl, 100 mM Tris pH 8.0, and 5 mM dithiothreitol (DTT, 20291, Thermo Scientific) mixture. Then, the sample was alkylated with 15 mM iodoacetamide (IAM, 786–078, G-Biosciences) for 30 minutes in the dark at room temperature. Subsequently, the reaction was diluted with 3 volumes of 100 mM Tris pH 7.5 to allow for trypsin digestion. Trypsin (V5280, Promega) was added at 1:12.5 protease: protein ratio and incubated at 37°C for 3–4 hours. The reaction was quenched by adding 1 µLTFA (T6508, Sigma-Aldrich). The digests were analyzed by LC-MS using a Waters ACQUITY UPLC system equipped with a Waters ACQUITY BEH C18 column (1.7 µm, 2.1x150 mm), mobile phase A (0.02% TFA in HPLC water), mobile phase B (0.02% TFA in acetonitrile), and an Orbitrap Fusion mass spectrometer (Thermo).
UV-based focused peptide mapping (LC-UV) for CDR isomerization
Sample digest preparation was the same as described in tryptic peptide mapping. The digests were analyzed using an optimized gradient by an Agilent 1290 system equipped with a Waters ACQUITY BEH C18 column (1.7 µm, 2.1x150 mm), mobile phase A (0.1% TFA in HPLC water), and mobile phase B (0.1% TFA in acetonitrile).
Reducing gel electrophoresis
The reducing gel electrophoresis for MAB1 was performed by first diluting the antibody samples to 0.5 mg/mL in a 50 mM sodium phosphate pH 6.5 buffer containing 2% SDS (BP2436200, Fisher Scientific) and 5% 2-mercaptoethanol (M6250, Sigma). The mixture was heated at 70°C for 5 minutes. Then, the denatured and reduced samples were analyzed on a Sciex PA800 plus CE system.
HILIC 2-AB oligosaccharide profiling
HILIC oligosaccharide profiling of the MAB1 samples was conducted through UPLC analysis. Briefly, 100 µg of antibody was digested overnight with PNGaseF (V4831, Promega) at 37°C, and subsequently labeled with 2-aminobenzamide (PN 654213, Sigma-Aldrich) for 30 minutes at 37°C. The labeled oligosaccharides were extracted using GlykoClean SPE Cartridges (GC210, Prozyme), which were then injected onto a Waters ACQUITY UPLC outfitted with a Glycoprotein Amide Column (186007963, Waters).
Photo-stress
The cool white fluorescent lamp specified in ISO 10977(1933) and a near UV fluorescent lamp with spectral distribution from 320 nm to 400 nm were used as light sources in the photo-stress study. The light chamber temperature was maintained at 25 ℃. 1 mL of MAB1 was put in a 3 mL glass vial. The vials were sealed with rubber stoppers and aluminum crimp caps, laid down on the side on the stainless steel tray in the test chamber. The samples were exposed to about 1.2 million Lux hours of cool white light and about 200 Watt hours/m2 of UV light according to ICH guidelines to get a 1-fold dose (1X ICH). Proportionally, about 3.6 million Lux hours of cool white light and a total of about 600 Watt hours/m2 of UV light exposure was used to get a 3-fold dose (3X ICH). To distinguish between the effect of light and temperature, a dark control sample was wrapped in aluminum foil and left with other samples to be exposed to 3X ICH dose of light exposure.
Serum incubation
Normal human serum (BioreclamationIVT) was passed through a Protein A (MabSelect SuRe) column twice to fully deplete the endogenous IgGs that bind to protein A. MAB1 was spiked into the IgG-depleted serum or 1X PBS pH 7.4 (10010023, Thermo Fisher Scientific) to a final concentration at 1 mg/mL. The mixture was incubated at 37°C for up to 2 weeks. Samples were kept at −80°C at the end of each time point. Once samples from all time points were collected, the serum mixtures were thawed and the antibody was recovered by protein A chromatography purification. Briefly, a protein A column was sanitized using 100 mM NaOH, then equilibrated in 1X PBS. Serum mixture was then loaded to the column, and washed in 1X PBS. The antibody was eluted using 100 mM glycine pH 3.0. After elution, the antibody samples were buffer exchanged to formulation buffer without PS80 using Vivaspin 10 KD spin columns (28–9323–60, GE).
Acknowledgments
We would like to thank George Chacko, Anthony Shannon, Pin Ren – MedImmune and Kenneth Blakeslee – Waters Corporation for their support in the development and application of this method.
References
- 1.Alt N, Zhang TY, Motchnik P, Taticek R, Quarmby V, Schlothauer T, Beck H, Emrich T, Harris RJ. Determination of critical quality attributes for monoclonal antibodies using quality by design principles. Biologicals. 2016;44:291-305. doi: 10.1016/j.biologicals.2016.06.005. PMID:27461239. [DOI] [PubMed] [Google Scholar]
- 2.Finkler C, Krummen L. Introduction to the application of QbD principles for the development of monoclonal antibodies. Biologicals. 2016;44:282-90. doi: 10.1016/j.biologicals.2016.07.004. PMID:27546184 [DOI] [PubMed] [Google Scholar]
- 3.Luciani F, Galluzzo S, Gaggioli A, Kruse NA, Venneugues P, Schneider CK, Pini C, Melchiorri D. Implementing quality by design for biotech products: Are regulators on track? mAbs. 2015;7:451-5. doi: 10.1080/19420862.2015.1023058. PMID:25853461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung CH, Mirakhur B, Chan E, Le Q-T Berlin J, Morse M, Murphy BA, Satinover SM, Hosen J, Mauro D, et al.. Cetuximab-Induced Anaphylaxis and IgE Specific for Galactose-α-1,3-galactose. New England journal of medicine. 2008;358:1109-7. doi: 10.1056/NEJMoa074943. PMID:18337601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goetze AM, Liu YD, Zhang Z, Shah B, Lee E, Bondarenko PV, Flynn GC. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology. 2011;21:949-59. doi: 10.1093/glycob/cwr027. PMID:21421994 [DOI] [PubMed] [Google Scholar]
- 6.Kolbeck R, Kozhich A, Koike M, Peng L, Andersson CK, Damschroder MM, Reed JL, Woods R, Dall'acqua WW, Stephens GL, et al.. MEDI-563, a humanized anti–IL-5 receptor α mAb with enhanced antibody-dependent cell-mediated cytotoxicity function. J Allergy Clin Immunol. 2010;125:1344-53.e2. doi: 10.1016/j.jaci.2010.04.004. PMID:20513525 [DOI] [PubMed] [Google Scholar]
- 7.Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. mAbs. 2015;7:881-90. doi: 10.1080/19420862.2015.1069454. PMID:26186204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Li X, Liu YH, Richardson D, Li H, Shameem M, Yang X. Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion. MAbs. 2016;8:1477-86. doi: 10.1080/19420862.2016.1226715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, Chu L, Li W, Lawson K, Apostol I, Eris T. Application of a Quantitative LC–MS Multiattribute Method for Monitoring Site-Specific Glycan Heterogeneity on a Monoclonal Antibody Containing Two N-Linked Glycosylation Sites. Analytical Chemistry. 2017;89:3562-7. doi: 10.1021/acs.analchem.6b04856. PMID:28199092 [DOI] [PubMed] [Google Scholar]
- 10.Dong J, Migliore N, Mehrman SJ, Cunningham J, Lewis MJ, Hu P. High-Throughput, Automated Protein A Purification Platform with Multiattribute LC-MS Analysis for Advanced Cell Culture Process Monitoring. Anal Chem. 2016;88:8673-9. doi: 10.1021/acs.analchem.6b01956. PMID:27487007 [DOI] [PubMed] [Google Scholar]
- 11.Pace AL, Wong RL, Zhang YT, Kao Y-H, Wang YJ. Asparagine Deamidation Dependence on Buffer Type, pH, and Temperature. J Pharm Sci. 2013;102:1712-23. doi: 10.1002/jps.23529. PMID:23568760 [DOI] [PubMed] [Google Scholar]
- 12.Du Y, Walsh A, Ehrick R, Xu W, May K, Liu H. Chromatographic analysis of the acidic and basic species of recombinant monoclonal antibodies. mAbs. 2012;4:578-85. doi: 10.4161/mabs.21328. PMID:22820257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vlasak J, Bussat MC, Wang S, Wagner-Rousset E, Schaefer M, Klinguer-Hamour C, Kirchmeier M, Corvaïa N, Ionescu R, Beck A. Identification and characterization of asparagine deamidation in the light chain CDR1 of a humanized IgG1 antibody. Analytical Biochemistry. 2009;392:145-54. doi: 10.1016/j.ab.2009.05.043. PMID:19497295 [DOI] [PubMed] [Google Scholar]
- 14.Gao X, Ji JA, Veeravalli K, John Wang Y, Zhang T, McGreevy W, Zheng K, Kelley RF, Laird MW, Liu J, et al.. Effect of Individual Fc Methionine Oxidation on FcRn Binding: Met252 Oxidation Impairs FcRn Binding More Profoundly than Met428 Oxidation. J Pharm Sci. 2015;104:368-77. doi: 10.1002/jps.24136. PMID:25175600 [DOI] [PubMed] [Google Scholar]
- 15.Wang W, Vlasak J, Li Y, Pristatsky P, Fang Y, Pittman T, Roman J, Wang Y, Prueksaritanont T, Ionescu R, et al.. Impact of methionine oxidation in human IgG1 Fc on serum half-life of monoclonal antibodies. Molecular Immunology. 2011; 48:860-6. doi: 10.1016/j.molimm.2010.12.009. PMID:21256596 [DOI] [PubMed] [Google Scholar]
- 16.Zhang T, Bourret J, Cano T. Isolation and characterization of therapeutic antibody charge variants using cation exchange displacement chromatography. J Chromatogr A. 2011;1218:5079-86. doi: 10.1016/j.chroma.2011.05.061. PMID:21700290 [DOI] [PubMed] [Google Scholar]
- 17.Chumsae C, Gaza-Bulseco G, Sun J, Liu H. Comparison of methionine oxidation in thermal stability and chemically stressed samples of a fully human monoclonal antibody. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;850:285-94. doi: 10.1016/j.jchromb.2006.11.050. PMID:17182291 [DOI] [PubMed] [Google Scholar]
- 18.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A Sanglier-Cianférani S. Characterization of Therapeutic Antibodies and Related Products. Analytical Chemistry. 2013;85:715-36. doi: 10.1021/ac3032355. PMID:23134362 [DOI] [PubMed] [Google Scholar]
- 19.Fekete S, Guillarme D, Sandra P, Sandra K. Chromatographic, Electrophoretic, and Mass Spectrometric Methods for the Analytical Characterization of Protein Biopharmaceuticals. Analytical Chemistry. 2016;88:480-507. doi: 10.1021/acs.analchem.5b04561. PMID:26629607 [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz SA, Engen JR, Mazzeo JR, Jones GB. Analytical tools for characterizing biopharmaceuticals and the implications for biosimilars. Nat Rev Drug Discov. 2012;11:527-40. doi: 10.1038/nrd3746. PMID:22743980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiaobin X, Haibo Q, Li N. LC-MS multi-attribute method for characterization of biologics. Journal of Applied Bioanalysis. 2017;3:5. [Google Scholar]
- 22.Rogstad S, Faustino A, Ruth A, Keire D, Boyne M, Park J. A Retrospective Evaluation of the Use of Mass Spectrometry in FDA Biologics License Applications. J Am Soc Mass Spectrom. 2017;28:786-94. doi: 10.1007/s13361-016-1531-9. PMID:27873217 [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Huang Y, Ferrant J, Lyubarskaya Y, Zhang Y, Li S, Wu SL. Assessing in vivo dynamics of multiple quality attributes from a therapeutic IgG4 monoclonal antibody circulating in cynomolgus monkey. mAbs. 2016;8:961-8. doi: 10.1080/19420862.2016.1167298. PMID:27030286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bomans K, Haberger M, Bonnington L, Wagner K, Kailich T, Wegele H, et al.. Multi-Attribute Monitoring of Antibody Modifications by Semi-Automated Liquid Chromatography Mass Spectrometry Peptide Mapping. American Pharmaceutical Review; 2016. [Google Scholar]
- 25.Haberger M, Bonnington L, Bomans K, Maier M, Eltner S, Dembowski M, et al.. Application of Electrospray Ionization Mass Spectrometry in a Quality Control Laboratory. American Pharmaceutical Review; 2016. [Google Scholar]
- 26.Galba J, Michalicova A, Parrak V, Novak M, Kovac A. Quantitative analysis of phenylalanine, tyrosine, tryptophan and kynurenine in rat model for tauopathies by ultra-high performance liquid chromatography with fluorescence and mass spectrometry detection. J Pharm Biomed Anal. 2016;117:85-90. doi: 10.1016/j.jpba.2015.08.026. PMID:26344382 [DOI] [PubMed] [Google Scholar]
- 27.Lemasson E, Bertin S, Hennig P, Boiteux H, Lesellier E, West C. Development of an achiral supercritical fluid chromatography method with ultraviolet absorbance and mass spectrometric detection for impurity profiling of drug candidates. Part I: Optimization of mobile phase composition. Journal of Chromatography A. 2015;1408:217-26. [DOI] [PubMed] [Google Scholar]
- 28.Stalmans S, Gevaert B, Verbeke F, D'Hondt M, Bracke N, Wynendaele E, De Spiegeleer B. Quality control of cationic cell-penetrating peptides. J Pharm Biomed Anal. 2016;117:289-97. doi: 10.1016/j.jpba.2015.09.011. PMID:26397208 [DOI] [PubMed] [Google Scholar]
- 29.Zhang J, Qin T, Xu L, Liu B, Li Y, Guo H, Lankun S, Guo Qingcheng, Xu Jin. Development and Validation of a Peptide Mapping Method for the Characterization of Adalimumab with QDa Detector. Chromatographia. 2016;79:395-403. doi: 10.1007/s10337-016-3046-8 [DOI] [Google Scholar]
- 30.Ren D, Pipes GD, Liu D, Shih L-Y, Nichols AC, Treuheit MJ, Brems DN, Bondarenko PV. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Analytical Biochemistry. 2009;392:12-21. doi: 10.1016/j.ab.2009.05.018. PMID:19457431 [DOI] [PubMed] [Google Scholar]
- 31.Chung S, Quarmby V, Gao X, Ying Y, Lin L, Reed C, Fong C, Lau W, Qiu ZJ, Shen A, et al.. Quantitative evaluation of fucose reducing effects in a humanized antibody on Fcγ receptor binding and antibody-dependent cell-mediated cytotoxicity activities. mAbs. 2012;4:326-40. doi: 10.4161/mabs.19941. PMID:22531441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abès R, Teillaud J-L. Impact of Glycosylation on Effector Functions of Therapeutic IgG. Pharmaceuticals. 2010;3:146-57.doi: 10.3390/ph3010146. PMID:27713246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reusch D, Tejada ML. Fc glycans of therapeutic antibodies as critical quality attributes. Glycobiology. 2015;25:1325-34. doi: 10.1093/glycob/cwv065. PMID:26263923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tous GI, Wei ZP, Feng JH, Bilbulian S, Bowen S, Smith J, Strouse R, McGeehan P, Casas-Finet J, Schenerman MA. Characterization of a novel modification to monoclonal antibodies: Thioether cross-link of heavy and light chains. Analytical Chemistry. 2005;77:2675-82. doi: 10.1021/ac0500582. PMID:15859580 [DOI] [PubMed] [Google Scholar]
- 35.Wongkongkathep P, Li H, Zhang X, Ogorzalek Loo RR, Julian RR, Loo JA. Enhancing protein disulfide bond cleavage by UV excitation and electron capture dissociation for top-down mass spectrometry. Int J Mass Spectrom. 2015;390:137-45. doi: 10.1016/j.ijms.2015.07.008. PMID:26644781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pace AL, Wong RL, Zhang YT, Kao Y-H, Wang YJ. Asparagine Deamidation Dependence on Buffer Type, pH, and Temperature. J Pharm Sci. 2013;102:1712-23. doi: 10.1002/jps.23529. PMID:23568760 [DOI] [PubMed] [Google Scholar]
- 37.Gu S, Wen D, Weinreb PH, Sun Y, Zhang L, Foley SF, Kshirsagar R, Evans D, Mi S, Meier W, et al.. Characterization of trisulfide modification in antibodies. Analytical Biochemistry. 2010;400:89-98. doi: 10.1016/j.ab.2010.01.019. PMID:20085742 [DOI] [PubMed] [Google Scholar]
- 38.Zhang Q, Schenauer MR, McCarter JD, Flynn GC. IgG1 Thioether Bond Formation in Vivo. J Biol Chem. 2013;288:16371-82. doi: 10.1074/jbc.M113.468397. PMID:23625924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porwoll S, Fuchs H, Tauber R. Characterization of a soluble class I α-mannosidase in human serum. FEBS Letters. 1999;449:175-8. doi: 10.1016/S0014-5793(99)00422-6. PMID:10338126 [DOI] [PubMed] [Google Scholar]
- 40.Technical Bulletin for Trypsin Gold https://www.promega.com/-/media/files/resources/protocols/technical-bulletins/101/trypsin-gold-mass-spectrometry-grade-protocol.pdf Promega.