

Abstract
Purpose
Glioblastoma (GBM) is the most malignant primary brain tumor, with a median survival of less than two years. More effective therapeutic approaches are needed to improve clinical outcomes.
Experimental Design
Glioblastoma patient-derived cells (GPDCs) were isolated from patient GBMs and implanted in mice to form xenografts. Immunohistochemistry was performed for hERG expression and tumor proliferation. Sphere-forming assays with hERG blocker E-4031 were performed on a highand low hERG expressing lines. A GBM TMA (115 patients) was used to correlate hERG expression with patient survival. Clinical data was analyzed to determine if patient survival was affected by incidental administration of hERG inhibitory drugs, and the correlative effect of patient GBM hERG expression levels.
Results
hERG expression was upregulated in GBM xenografts with higher proliferative indices. High hERG-expressing GPDCs showed a reduction in sphere formation when treated with hERG inhibitors compared to low hERG-expressing GPDCs. GBM TMA analysis showed worse survival for GBM patients with high hERG expression versus low expression, 43.5 vs. 60.9 weeks respectively (p= 0.022). Furthermore, patients who received at least one hERG blocker had a better survival rate compared to patients who did not (p=0.0015). Subgroup analysis showed that GBM patients with high hERG expression who received hERG blockers had improved survival (p=0.0458). There was no difference in survival for low hERG-expressing GBM patients who received hERG blockers (p=0.4136).
Conclusions
Our findings suggest that hERG is a potential GBM survival marker, and that already approved drugs with non-torsadogenic hERG inhibitory activity may potentially be re-purposed as adjuvant GBM therapy in high hERG-expressing GBM patients.
Keywords: Glioblastoma, HERG, Cancer stem-like cells, Biomarkers, K+ channels
Introduction
In the United States each year, more than 14,000 new cases of glioblastoma (GBM), the World Health Organization grade IV brain cancer, will be diagnosed (1). Despite the current standard of maximal safe surgical resection followed by radiotherapy and chemotherapy with the alkylating agent temozolomide, patients have an overall median survival of less than two years (2). Thus, there is an urgent need to find new prognostic biomarkers to inform clinical management and therapies to improve outcome.
Ion channels, transmembrane proteins regulating ion flux across the plasma membrane, have been reported to influence progression of GBM and other cancers (3–8). Kv.11.1, encoded by the human Ether-à-go-go Related Gene (hERG, or KCNH2), is well characterized for its role in cardiac repolarization (9, 10). It is the primary target for acquired or drug-induced long QT syndrome, leading in some cases to sudden cardiac death. However, many hERG blockers are in clinical use and known to be generally safe (11). hERG was shown to be overexpressed in leukemia, gastric cancer, colon cancer and glioblastoma cell lines (8, 12–15). In other tumor cells, altered hERG expression was associated with cell cycle regulation and proliferation (16, 17). Recent work was reported showing a possible role for hERG inhibition in pediatric brain cancer, medulloblastoma (18). The few studies that implicate hERG in GBM have used traditional cell lines or serum-cultured cells that lack the diverse genotypic and phenotypic characteristics observed in patient-derived tumors (19). Furthermore, many prior studies were conducted in vitro and direct consequences of hERG expression on GBM tumors in situ or patient survival have not been reported.
We set out to determine whether in vivo hERG expression of tumor xenografts initiated from glioblastoma patient-derived cells (GPDCs) (20). We also sought to determine whether GPDC proliferation rates and GBM patient survival were affected by hERG channel inhibition. We showed hERG expression levels are correlated with higher proliferation rates in GPDC-derived xenografts and that patients with high hERG expressing GBM had worse survival rates. We also demonstrate that hERG blockers reduced GPDC proliferation, and improved survival in patients who received one or more hERG blocking drugs but only if their tumors exhibited high hERG expression levels.
Materials and Methods
Isolation of GBM stem-like cancer cells (GPDCs)
GPDCs were isolated following protocols previously reported (19, 21–23) with some modifications. Tumor tissue was collected directly from the operating room, weighed, coarsely minced with a scalpel blade, and subsequently chopped twice to 200 µm using a tissue chopper (Sorvall TC-2 Smith-Farquahar). Chopped tissue was directly plated in suspension at 10 mg/ml in growth medium (passage medium [PM]: 70% Dulbecco modified Eagle medium-high glucose, 30% Ham's F12, 1× B27 supplement, 5 µg/ml heparin, penicillin-streptomycin-amphotericin (PSA), and 20 ng/ml each of EGF and bFGF). Cultures were passaged approximately every 7 to 10 days by tissue chopping twice at 200 µm. For these studies, we used four different GPDC lines: 12.1 22, 112, and 114. 12.1 and 22 displayed markers consistent with a mesenchymal phenotype, while 112 and 114 displayed markers consistent with a proneural phenotype (data unpublished). Each cell line was validated for self-renewal by neurosphere formation, multipotency, and tumor initiation before experiments were performed. Establishing of cell cultures came from cryopreservation of cell cultures ranging from passages 10–22. Cells used for experiments ranged from passage 20–25.
Sphere formation assay
GPDCs were enzymatically dissociated to single cells and seeded at 100 cells/well into 96-well plates. After cell recovery overnight, media was exchanged with drug-containing media at increasing concentrations o E-4031. After two weeks GPDC spheres were counted, with drug-treated conditions normalized to vehicle controls.
GPDC orthotopic xenograft model
GBM orthotopic xenografts were initiated as previously described (24). GPDCs were enzymatically dissociated to single cells, and 2*105 cells were suspended in 5 µl of media. Using a Hamilton syringe, the cells were stereotactically implanted into the right striatum of anesthetized non-obese diabetic severe combined immunodeficient (Nod-SCID) mice between the ages of 8–10 weeks at 0.33 µl/min at the following coordinates referenced from bregma: 0 mm anteroposterior, +2.5 mm mediolateral, and −3.5 mm dorsoventral. At either 3 months or the onset of neurological symptoms, tumor formation was verified using magnetic resonance imaging (MRI). Mice were anesthetized, contrast enhanced using 10 mmol/kg of intraperitoneal gadodiamide (Omniscan; GE Healthcare, Piscataway, NJ), and placed onto a small animal MRI scanner (4.7-T horizontal bore imaging/spectroscopy system; Varian, Palo Alto, CA), and T1- and T2-weighted images were obtained. As per our approved animal protocol, once neurological symptoms were observed and the mice were moribund, which occurred between 80–120 days, implanted Nod-SCID mice were immediately euthanized by perfusion fixation with 4% paraformaldehyde.. Brains were then excised, embedded in paraffin, and processed for general histology.
Immunohistochemistry (IHC)
IHC was performed on GBM mouse xenografts and a human TMA as previously described (22). Immunohistochemistry was performed on Nod-SCID mouse brains implanted with 200,000 GPDCs from lines 12.1, 22, 112 and 114. IHC was also performed on the human GBM tissue microarray. Formalin-fixed, paraffin embedded tissue sections were mounted on positively charged microscope slides. Tissue sections were then deparaffinized and rehydrated to water, microwaved in antigen unmasking solution (Vector Laboratories, Burlingham, CA) to retrieve epitopes, and blocked for endogenous peroxidase and biotin before the application of the primary antibody. Incubation of antibodies was performed overnight at 4°C. The anti-hERG primary antibody was used at the concentration of 1:1600 (Enzo Life Sciences, ALX-215-049-R100). Subsequent immunodetection was performed using the Elite Vector Stain ABC System (Vector Laboratories, Burlingham, CA). Color visualization was performed using 3–3’-diaminobenzidine (DAB) as the chromagen substrate (Sigma Chemical Company, St. Louis, MO). Tissues were counterstained with hematoxylin to visualize cellular morphology. Images were acquired with a Nikon TE-2000 (Tokyo, Japan) and EVOS XL Core (Advanced Microscopy Group, Bothell, WA) brightfield microscopes. To determine hERG expression levels, positive cells (brown) were counted at 100× and compared to the total number of cells present in three random fields in the tumor. Black boxes in whole-brain images indicate the corresponding location of acquisition for high magnification photomicrographs.
Ki-67 indexing
After Ki-67 immunolabeling, positive cells were defined as those with nuclei of any brown color, regardless of the intensity or pattern of staining. Human versus mouse nuclei were distinguished using 4',6-diamidino-2-phenylindole (DAPI) labeling. Mouse nuclei were small, regular and round displaying concentrated aggregate DAPI labeling, indicative of characteristic mouse chromocenters. Human GPDC nuclei were comparatively large and atypically oblong with homogenous DAPI labeling. Five random high power fields were chosen using a 100× objective and the number of Ki-67 positive cells was counted as well as the total number of human cells. Ki-67 percentage was determined from the average of the five random fields chosen for each GSC xenograft. High hERG expression xenografts and low hERG expression xenografts were then grouped together to determine the Ki-67 positive percentage in each group. Counting of Ki-67 positive nuclei began at the site of implantation until 500 random human nuclei were identified with a 60× objective.
Tissue microarray (TMA)
With IRB approval, we created a clinically annotated GBM TMA from 205 GBMs resected between 1999 and 2009, archived in the UW Department of Pathology and Laboratory Medicine and clinical data accessed from UW patient records. One to three representative tissue punches/cores were obtained for each tumor sample depending on morphologic heterogeneity and tissue availability. Neuropathology designated patient diagnosis and tissue punch locations was determined based on the most representative section of the whole GBM specimen. Each additional tissue punch contained classic GBM features (nuclear atypia, high mitotic indices, vascular endothelial proliferation, and/or necrosis). Grades II and III astrocytoma, grade II oligodendroglioma, meningioma, hippocampus and neocortex tissue punches were used as controls. Of the 205 patients on the TMA, 149 of them have corresponding clinical records including survival information. Based on histological processing 115 samples of 149 were usable. IHC was performed using the aforementioned anti-hERG antibody. Punches were blindly scored as having high hERG expression or low hERG expression. In order to take into account the heterogeneity of GBM samples even from the same tumor, for patients that had multiple punches, if one punch had high hERG expression, the patient was denoted as having high expression levels. Survival analysis was done using a log-rank test and presented as a Kaplan-Meier survival plot with the addition of hazard ratio analysis. P-values <0.05 were considered statistically significant. Plots were generated using GraphPad Prism 6.
Study approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the University of Wisconsin-Madison (UW) Institutional Review Board and in compliance with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study. All procedures performed in studies involving animals were in accordance with the ethical standards of UW and approved by the Animal Care and use Committee.
Results
HERG expression levels in GBMs correlate with tumor proliferation rates
We evaluated whether hERG expression levels in vivo correlated with proliferation in a tumor xenograft assay, where four distinct GPDC lines were orthotopically implanted into the right striatum of non-obese diabetic severe combined immunodeficient (NOD-SCID) mice. H&E staining was used to visualize tumor xenografts in mouse brain (Fig. 1A1–D1), and tumor proliferation rate was determined via immunohistochemistry for the proliferation protein marker, Ki-67, on sections of GBM xenografts. Five random fields at 100× were chosen and Ki-67-positive cells were counted and compared to the total number of tumor cells. Based on the Ki-67 index, two groups emerged from the mouse xenografts – a higher proliferation group (mean of 70.29 ± 4.837% Ki-67-positive cells) and a lower proliferation group (mean of 46.60 ± 2.744% Ki-67-positive cells) (p=0.0005) (Fig. 1E). Then, immunohistochemistry using a pan-hERG antibody on serial xenograft sections (Fig. 1A2–D2) was performed on both groups. Higher Ki-67 index xenografts [12.1 GSC (66%) and 22 GSC (75%)] were also positive for high hERG levels [12.1 GSC (91% of cells) and 22 GSC (85% of cells)], whereas lower Ki-67 index xenografts [112 GSC (47%) and 114 GSC (46%)] had low hERG levels [112 GSC (21% of cells) and 114 GSC (33% of cells)], suggesting that hERG expression is associated with GBM proliferation (Fig.1F). Interestingly, this data also shows that although hERG is overexpressed in GBMs with higher proliferation rates, not all GBM exhibit high hERG expression levels.
Fig.1. HERG expression levels are correlated with proliferation.

(1A–1D) Mouse brains from GPDC xenografts were stained with H&E. (2A–2D). An anti-hERG antibody was used on GPDC mouse xenograft sections. The scale bar represents 250 um. (E) Ki-67 cell percentages from 5 random fields for each GPDC xenograft with Ki-67 cell percentages are grouped based on high hERG levels and low hERG levels. (F) Fraction of hERG positive cells for each xenograft. ***=<0.001, ****=<0.0001
Lower patient survival rates associated with hERG expression
To determine whether hERG expression correlated with patient survival, we examined a human tissue microarray (TMA) consisting of GBMs resected from the years 1999 to 2009 with 149 patients that had clinically accessible records for correlation. Of note, many of these patients were treated before the current standard temozolomide adjuvant chemoradiotherapy was established, and many current molecular markers (e.g. MGMT methylation, IDH mutation) were tested. Each patient had one to three 1-mm GBM specimen punches that were chosen by a neuropathologist to be the most representative areas of the entire patient GBM specimen. Immunohistochemistry was performed on the TMA using the same pan-hERG antibody mentioned above, followed by blind scoring for low or high hERG expression levels. The blind scoring revealed that high hERG expression correlated with a worse survival rate compared with low hERG expression, based on the log-rank test (p=0.022) with a median survival of 43.5 weeks (N=45) compared to 60.9 weeks (N=70), respectively (Fig. 2). Furthermore, patients with high hERG levels had a 61% greater chance of dying before patients with low hERG levels (log-rank hazard ratio of 1.536). These findings indicate that hERG may be an informative survival biomarker for GBM patients.
Fig. 2. HERG expression is correlated with GBM patient survival.

An anti-hERG antibody was used on a GBM tissue microarray with 1mm specimen punches. A Kaplan-Meier survival curve, based on hERG expression, was generated with a survival difference that was statistically significant (p= 0.022). Scale bar represents 250um.
Univariate and Multivariate Analysis of hERG expression
Univariate and multivariate analysis were used to determine whether the correlation of hERG expression with survival was influenced by any confounding variables. Patient gender, age, KPS score, temozolomide treatment, radiation treatment, and tobacco use were assessed as potential confounding variables. Univariate analysis was conducted to determine which factors influence patient survival rates. There was not a statistically significant difference in survival based on patient gender, age, or tobacco use; however, there was a statistically significant difference in survival based on KPS score (0.024), temozolomide treatment (<0.001), and radiation treatment (<0.001). Multivariate survival analysis using Cox’s regression model was used to determine whether hERG expression is an independent biomarker for survival taking into account KPS score, temozolomide treatment, and radiation treatment. This analysis indicated that hERG expression level is an independent marker for GBM patient survival, with reduced survival rates for patients exhibiting high hERG expression levels compared to those with low hERG expression levels (p=0.003) (Table 1).
Table 1.
Univariate and multivariate cox regression analysis of GBM hERG expression and confounding variables
| Univariate Analysis | Multivariate Analysis | |||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| N | Median Survival (Weeks) |
95% CI | P-Value (Log Rank) |
Hazard Ratio (HR) |
HR 95% CI | P-Value | ||
| hERG Expression | 0.022 | 2.122 | 1.247–3.610 | 0.003 | ||||
|
| ||||||||
| High | 45 | 43.5 | 33.8–53.1 | |||||
|
|
||||||||
| Low | 71 | 60.9 | 48.2–73.6 | |||||
|
| ||||||||
| Gender | 0.463 | NS | ||||||
|
| ||||||||
| Male | 81 | 52.2 | 44.5–59.9 | |||||
| Female | 35 | 56.5 | 34.5–78.5 | |||||
|
| ||||||||
| Age | 0.957 | NS | ||||||
|
| ||||||||
| ≤ 55 | 51 | 52.2 | 39.5–64.8 | |||||
| > 55 | 58 | 52.2 | 44.1–60.3 | |||||
|
| ||||||||
| KPS | 0.024 | 1.143 | 0.769–1.700 | 0.508 | ||||
|
| ||||||||
| ≤ 70 | 55 | 60.9 | 48.3–73.5 | |||||
| > 70 | 60 | 43.5 | 29.7–57.3 | |||||
|
| ||||||||
| Temozolomide | <0.001 | 2.845 | 1.849–4.378 | <0.001 | ||||
|
| ||||||||
| Yes | 66 | 69.6 | 61.6–77.6 | |||||
| No | 50 | 34.8 | 27.3–42.3 | |||||
|
| ||||||||
| Radiation | <0.001 | 2.122 | 1.247–3.610 | 0.006 | ||||
|
| ||||||||
| Yes | 95 | 60.9 | 9.7–25.1 | |||||
| No | 21 | 17.4 | 49.6–72.2 | |||||
|
| ||||||||
| Tobacco Use | 0.519 | NS | ||||||
|
| ||||||||
| Yes | 46 | 52.2 | 41.4–63.0 | |||||
| No | 70 | 52.2 | 37.9–66.5 | |||||
HERG blockers decreased sphere formation in high hERG expressing GBM cell lines
Based on hERG expression levels found in GSC-derived xenografts, we wanted to determine whether hERG blockers could decrease proliferation rates in high hERG expressing GPDC lines more than low expressing hERG GPDC lines. GPDCs were dissociated into single cells and 100 cells were seeded into a 96-well plate. E-4031, a known hERG-specific blocker, was added at increasing drug concentrations. (25),. After two weeks, spheres were counted and normalized to vehicle control. E-4031 decreased sphere formation at increasing drug concentrations, with a statistically significant difference in sphere formation seen at drug concentrations 5 uM.(Fig. 3). This suggests that hERG blockers have the ability to decrease GPDC sphere formation and proliferation in a hERG-specific manner, and hERG inhibition might be a potentially useful GBM therapeutic strategy.
Fig. 3. Sphere formation is reduced with hERG blockers.
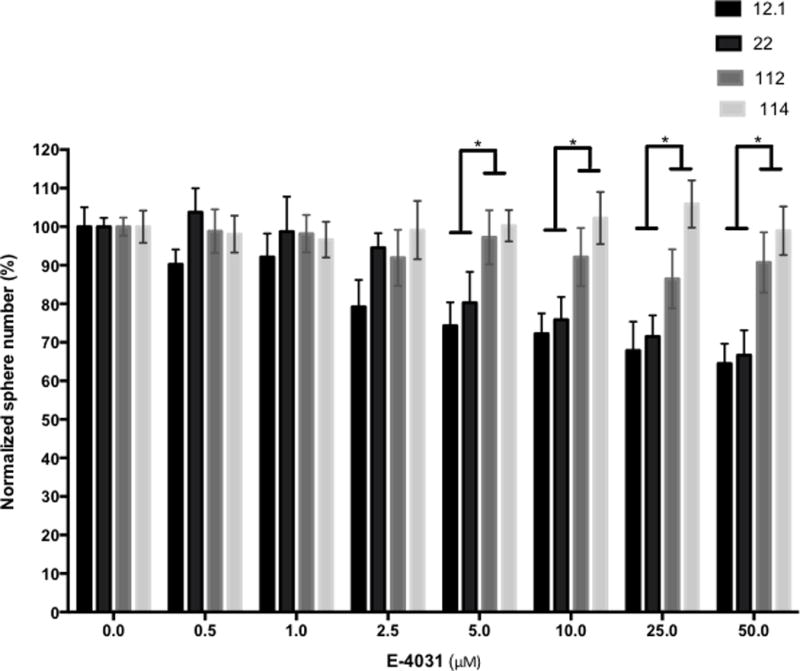
HERG specific blockerbE-4031 was used in increasing concentrations on 12.1, 22, 112, and 114 GPDCs. After two weeks of drug treatment, the number of spheres were counted and normalized to vehicle control. (*= p-value <0.05)
Patients who received hERG blockers had a better overall survival rate
To determine whether hERG blockers affected patient survival, we examined whether patients represented on the GBM tissue microarray received commonly-prescribed hERG blockers after their GBM diagnosis (26). Of the 110 patients analyzed, 66 patients received at least one of the following drugs known to exhibit off-target hERG block: phenytoin, haloperidol, fluoxetine, tamoxifen, amitriptyline, and ketoconazole (Supp. Fig 1). Patients prescribed at least one hERG blocker exhibited significantly enhanced survival compared to those who did not receive a hERG blocker, based on the log-rank test (p=0.0015) with a median survival of 69.6 weeks (N=66) and 43.5 weeks (N=44), respectively (Fig. 4). Patients who did not receive a hERG blocker had a 64% greater chance of dying before patients who did receive a hERG blocker (log-rank hazard ratio of 1.790). This data suggest that hERG blockers may increase patient survival. Interestingly, of the 66 patients who received at least one hERG blocker, 7 patients received two or more hERG blockers and had a median survival of 73.9 weeks, also a statistically significant improvement in survival compared to those patients who did not receive a hERG blocker (p=0.0117). Patients who did not receive a hERG blocker were 71% more likely to die before patients who received two or more hERG blockers (log-rank hazard ratio of 2.344). This suggests a patient survival benefit with greater hERG blockade.
Fig. 4. Receipt of hERG blockers correlate with a better GBM patient survival.

Patients who received more than one hERG blocker were compared to patients that had not received any hERG blockers and there was a statistically significant difference in their survival (p=0.0015).
hERG blockers enhance survival specifically for patients with high hERG expression levels
Interestingly, the correlation of hERG blockers with survival was not statistically significant for patients on the tissue microarray with low hERG expressing GBM (p=0.4136) (Fig. 5A). However, patients with high hERG expressing GBM who received a hERG blocker fared better than those who did not (p=0.0458) (Fig. 5B), with a median survival of 56.5 weeks (N=15) compared to 34.8 weeks (N=16), respectively. Patients with high hERG expressing GBM who did not receive hERG blockers had a 66% greater chance of dying first (a log-rank hazard ratio of 1.906) compared to patients with high hERG expressing GBM who received a hERG blocker. This suggests that hERG blockers are beneficial for patients with high hERG expressing GBMs, and supports the idea that hERG blockers improve survival times via a direct action on hERG channels, rather than other targets for these drugs. Moreover, the findings suggest the potential of tailoring GBM treatments according to hERG expression level stratification.
Fig. 5. Patient hERG expression levels correlate with benefit of receiving a hERG.

Patients were stratified based on whether or not they had high hERG or low hERG expression levels. In each group, patients were further stratified based on whether they had received a hERG blocker. (A)There was not a statistically significant difference in survival between patients who had low hERG expression levels (p=0.4136); however, (B) there was a statistically significant difference in survival of patients with high hERG expression levels based on whether or not they received a hERG blocker (p=0.0458)
Discussion
In this study, we found that upregulation of hERG expression correlated with greater tumor proliferation in heterogeneous GPDC-derived mouse xenografts and a worse patient survival rate. We found that the hERG blocker E-4031 decreased proliferation as assayed by sphere formation in a GPDC line exhibiting high levels of hERG expression, whereas blocking hERG in low hERG expressing lines does not decrease proliferation to the same extent. Survival was enhanced in patients receiving one or more hERG blockers, but only when their tumors exhibited high levels of hERG expression. These findings suggest that hERG may be a useful biomarker for GBM survival, and that non-torsadogenic hERG blockers may be potentially repurposed for adjuvant GBM therapy.
GBMs are heterogeneous and a variety of subtypes have been described (24, 27). Research to characterize differences in GBMs has led to a better understanding of GBM progression and new therapeutic targets (28, 29). For example, the efficacy of temozolomide is affected by the methylation status of MGMT, an enzyme that repairs the DNA damage resulting from temozolomide’s chemotherapeutic action (30). Methylation of the MGMT promoter silences protein production, preventing repair of DNA damage and leads to tumor cell death (31). MGMT status is now commonly tested in patients and it is well documented that GBM with MGMT promoter methylation are associated with higher median survival (30). Although since the current study was performed with a tissue microarray of GBM patients treated before the current standard of adjuvant temozomolide chemoradiation, and markers such as MGMT methylation and IDH mutation status were not known, we are working to confirm our findings with an updated GBM TMA. Other researchers have discussed the role of hERG as a novel biomarker for several other cancers and our work further supports this approach in GBM (32, 33). If validated, hERG expression may be used in the future to stratify GBM patients for possibly hERG inhibitor adjuvant therapy.
Many drugs inhibit hERG channels off-target, but some of these lack the pro-arrhythmic risk associated with hERG block. Phenytoin, prescribed to control seizures for most patients in this study, poses little risk for torsades de pointes arrhythmia. However, other troubling side effects have led to substitution of other anti-epileptic drugs such as levetiracetam, which does not block hERG channels (34). Whether this trend will affect survival of patients, especially those with tumors expressing high hERG levels, is unknown. Our study provides the rationale for clinical trials to determine whether other drugs targeting hERG may provide benefit as adjuvant therapies, by repurposing drugs such as fluoxetine (Prozac®), also a hERG blocker, to reduce tumor proliferation in a subset of patients with the added benefit of also treating depression.
While many FDA approved hERG blockers exist, some of them have the potential to cause arrhythmias due to cardiac hERG blockade especially under conditions of polypharmacy (35). One way to avoid off-target effects of hERG blockade might be combination with tumor targeting agents, such as alkylphosphocholine (APC) analogs (36, 37). Tumor cells are selectively targeted by long term APC retention, whereas normal tissues metabolize APCs. APC analogs can be modified by addition other moieties, such as radioactive iodine or optical fluorescence for diagnostic or therapeutic purposes (27, 28). Based on our findings, hERG blockers that are already FDA approved for other purposes may be repurposed as new therapeutic options for GBM patients. Studies in other cancers, such as leukemia, have shown that specific hERG blockers can have therapeutic effects without causing cardiac toxicities (38). Prospective clinical trials will be needed to test and validate hERG as a GBM biomarker, and potentially stratify GBM patients for hERG inhibitor adjuvant therapy.
Supplementary Material
Translational Relevance.
Glioblastoma (GBM) is the most malignant and deadly primary brain tumor, with a median survival of less than two years despite maximal therapy. Human ether à go-go-related-gene (hERG) is a voltage-dependent K+ channel found overexpressed in GBM cell lines, and linked to aberrant proliferation in other cancers. We analyzed hERG expression in glioblastoma patient-derived cells (GPDCs) and a clinically annotated human GBM tissue microarray (TMA) to determine correlation with patient survival. Since all FDA-approved drugs undergo cardiotoxicity profiling that includes hERG inhibition, GBM patient survival was also analyzed after subgrouping based on GBM hERG expression and whether they received hERG inhibitory drugs (phenytoin, haloperidol, fluoxetine, tamoxifen, amitriptyline, and/or ketoconazole). Our research demonstrates that hERG is a potential GBM biomarker, and that non-torsadogenic hERG blockers may be repurposed as potential adjuvant therapies for high hERG-expressing GBM.
Acknowledgments
We thank David Jones, Fang Liu, Ray Zhang, and Will Lyon for their project advice and technical assistance.
Funding: This work was supported in part by NIH grants R01NS75995 (KBP, JSK), T32 GM008692 (KBP), UL1RR025011 and P30CA014520 (UW Carbone Cancer Center), R01CA15880, Headrush Brain Tumor Research Professorship, Roger Loff Memorial Fund “Farming against Brain Cancer” (JSK), R01HL081780 (GAR) and R01NS081320 (GAR).
Footnotes
Competing interest: The authors declare no conflicts of interest.
Author contributions: Conceived and designed the experiments (KBP, KWE, GAR, JSK), Performed the experiments (KBP and PAC), Analyzed the data (KBP,PAC, KWE, GAR, JSK), contributed reagents/materials/analysis tools (MSS,GAR, JSK), Wrote the manuscript (KBP, PAC, KWE, MSS,GAR, JSK).
References
- 1.Society AC. Cancer Facts and Figures 2009. American Cancer Society; 2009. [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Zhu W, Begum G, Pointer K, Clark PA, Yang SS, Lin SH, et al. WNK1-OSR1 kinase-mediated phospho-activation of Na+-K+-2Cl− cotransporter facilitates glioma migration. Mol Cancer. 2014;13:31. doi: 10.1186/1476-4598-13-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou W, Guo S, Xiong Z, Liu M. Oncogenic role and therapeutic target of transient receptor potential melastatin 7 channel in malignancy. Expert Opin Ther Targets. 2014:1–20. doi: 10.1517/14728222.2014.940894. [DOI] [PubMed] [Google Scholar]
- 5.Huang X, Jan LY. Targeting potassium channels in cancer. J Cell Biol. 2014;206:151–62. doi: 10.1083/jcb.201404136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garzon-Muvdi T, Schiapparelli P, ap Rhys C, Guerrero-Cazares H, Smith C, Kim DH, et al. Regulation of brain tumor dispersal by NKCC1 through a novel role in focal adhesion regulation. PLoS Biol. 2012;10:e1001320. doi: 10.1371/journal.pbio.1001320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner KL, Honasoge A, Robert SM, McFerrin MM, Sontheimer H. A proinvasive role for the Ca(2+) -activated K(+) channel KCa3.1 in malignant glioma. Glia. 2014;62:971–81. doi: 10.1002/glia.22655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Masi A, Becchetti A, Restano-Cassulini R, Polvani S, Hofmann G, Buccoliero AM, et al. hERG1 channels are overexpressed in glioblastoma multiforme and modulate VEGF secretion in glioblastoma cell lines. Br J Cancer. 2005;93:781–92. doi: 10.1038/sj.bjc.6602775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- 10.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- 11.Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006;440:463–9. doi: 10.1038/nature04710. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Liu L, Guo T, Zhang J, Li X, Du W, et al. Expression and functional role of HERG1, K+ channels in leukemic cells and leukemic stem cells. J Huazhong Univ Sci Technolog Med Sci. 2007;27:257–60. doi: 10.1007/s11596-007-0310-2. [DOI] [PubMed] [Google Scholar]
- 13.Shao XD, Wu KC, Guo XZ, Xie MJ, Zhang J, Fan DM. Expression and significance of HERG protein in gastric cancer. Cancer Biol Ther. 2008;7:45–50. doi: 10.4161/cbt.7.1.5126. [DOI] [PubMed] [Google Scholar]
- 14.Crociani O, Zanieri F, Pillozzi S, Lastraioli E, Stefanini M, Fiore A, et al. hERG1 channels modulate integrin signaling to trigger angiogenesis and tumor progression in colorectal cancer. Sci Rep. 2013;3:3308. doi: 10.1038/srep03308. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Crociani O, Lastraioli E, Boni L, Pillozzi S, Romoli MR, D'Amico M, et al. hERG1 channels regulate VEGF-A secretion in human gastric cancer: clinicopathological correlations and therapeutical implications. Clin Cancer Res. 2014;20:1502–12. doi: 10.1158/1078-0432.CCR-13-2633. [DOI] [PubMed] [Google Scholar]
- 16.Jehle J, Schweizer PA, Katus HA, Thomas D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011;2:e193. doi: 10.1038/cddis.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Staudacher I, Jehle J, Staudacher K, Pledl HW, Lemke D, Schweizer PA, et al. HERG K+ channel-dependent apoptosis and cell cycle arrest in human glioblastoma cells. PLoS One. 2014;9:e88164. doi: 10.1371/journal.pone.0088164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nat Neurosci. 2015;18:1236–46. doi: 10.1038/nn.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee J, Kotliarova S, Kotliarov Y, Li A, Su Q, Donin NM, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 20.Pointer KB, Clark PA, Zorniak M, Alrfaei BM, Kuo JS. Glioblastoma cancer stem cells: Biomarker and therapeutic advances. Neurochem Int. 2014;71:1–7. doi: 10.1016/j.neuint.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ignatova TN, Kukekov VG, Laywell ED, Suslov ON, Vrionis FD, Steindler DA. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia. 2002;39:193–206. doi: 10.1002/glia.10094. [DOI] [PubMed] [Google Scholar]
- 22.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 23.Svendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S, Ostenfeld T, et al. A new method for the rapid and long term growth of human neural precursor cells. J Neurosci Methods. 1998;85:141–52. doi: 10.1016/s0165-0270(98)00126-5. [DOI] [PubMed] [Google Scholar]
- 24.Zorniak M, Clark PA, Leeper HE, Tipping MD, Francis DM, Kozak KR, et al. Differential expression of 2',3'-cyclic-nucleotide 3'-phosphodiesterase and neural lineage markers correlate with glioblastoma xenograft infiltration and patient survival. Clin Cancer Res. 2012;18:3628–36. doi: 10.1158/1078-0432.CCR-12-0339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanguinetti MC, Jurkiewicz NK, Scott A, Siegl PK. Isoproterenol antagonizes prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea pig myocytes. Mechanism of action. Circ Res. 1991;68:77–84. doi: 10.1161/01.res.68.1.77. [DOI] [PubMed] [Google Scholar]
- 26.van Noord C, Sturkenboom MC, Straus SM, Witteman JC, Stricker BH. Non-cardiovascular drugs that inhibit hERG-encoded potassium channels and risk of sudden cardiac death. Heart. 2011;97:215–20. doi: 10.1136/hrt.2009.188367. [DOI] [PubMed] [Google Scholar]
- 27.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene. 2008;27:2897–909. doi: 10.1038/sj.onc.1210949. [DOI] [PubMed] [Google Scholar]
- 29.Fine HA. Glioma stem cells: not all created equal. Cancer Cell. 2009;15:247–9. doi: 10.1016/j.ccr.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 31.Zhang J, Stevens MF, Bradshaw TD. Temozolomide: mechanisms of action, repair and resistance. Curr Mol Pharmacol. 2012;5:102–14. doi: 10.2174/1874467211205010102. [DOI] [PubMed] [Google Scholar]
- 32.Lastraioli E, Lottini T, Bencini L, Bernini M, Arcangeli A. hERG1 Potassium Channels: Novel Biomarkers in Human Solid Cancers. Biomed Res Int. 2015;2015:896432. doi: 10.1155/2015/896432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arcangeli A, Becchetti A. Novel perspectives in cancer therapy: Targeting ion channels. Drug Resist Updat. 2015;21–22:11–9. doi: 10.1016/j.drup.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 34.Fuller KL, Wang YY, Cook MJ, Murphy MA, D'Souza WJ. Tolerability, safety, and side effects of levetiracetam versus phenytoin in intravenous and total prophylactic regimen among craniotomy patients: a prospective randomized study. Epilepsia. 2013;54:45–57. doi: 10.1111/j.1528-1167.2012.03563.x. [DOI] [PubMed] [Google Scholar]
- 35.Pollard CE, Valentin JP, Hammond TG. Strategies to reduce the risk of drug-induced QT interval prolongation: a pharmaceutical company perspective. Br J Pharmacol. 2008;154:1538–43. doi: 10.1038/bjp.2008.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swanson KI, Clark PA, Zhang RR, Kandela IK, Farhoud M, Weichert JP, et al. Fluorescent cancer-selective alkylphosphocholine analogs for intraoperative glioma detection. Neurosurgery. 2015;76:115–23. doi: 10.1227/NEU.0000000000000622. discussion 23–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weichert JP, Clark PA, Kandela IK, Vaccaro AM, Clarke W, Longino MA, et al. Alkylphosphocholine analogs for broad-spectrum cancer imaging and therapy. Sci Transl Med. 2014;6:240ra75. doi: 10.1126/scitranslmed.3007646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gasparoli L, D'Amico M, Masselli M, Pillozzi S, Caves R, Khuwaileh R, et al. New pyrimido-indole compound CD-160130 preferentially inhibits the KV11.1B isoform and produces antileukemic effects without cardiotoxicity. Mol Pharmacol. 2015;87:183–96. doi: 10.1124/mol.114.094920. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.