

Abstract
Two immortalized human juvenile chondrocyte cell lines, T/C28a2 and C28/I2, were employed to determine the extent to which recombinant human (rh) IL-6, a known cytokine activator of the Janus kinase/signal transducers and activators of transcription (JAK/STAT) pathway in many cell types, caused STAT proteins to be phosphorylated. The results showed that STAT3 was constitutively phosphorylated in the absence of rhIL-6 in T/C28a2 chondrocytes. However, C28/I2 chondrocytes treated with rhIL-6 caused STAT1, STAT3, and STAT5 to be phosphorylated without altering total unphosphorylated STAT proteins. STAT3 phosphorylation in response to rhIL-6 in T/C28a and C28/I2 chondrocytes was efficiently blocked by the JAK3-selective inhibitor WHI-P131 (Janex-1) and by soluble IL-6 receptor-α (sIL-6R). However, the combination of rhIL-6 and ruxolitinib, a JAK1/JAK2-selective inhibitor, was a less effective inhibitor of STAT protein activation. These findings showed that rhIL-6 activated STAT proteins in the C28/I2 chondrocyte cell line. STAT protein phosphorylation could be blocked by a JAK3-selective inhibitor or by the combination of rhIL-6 and sIL-6R.
Keywords: chondrocyte cell lines, cytokine, human, signal transduction
Introduction
Several proinflammatory cytokines, including IL-6, activate the Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling pathway where phosphorylation of STAT proteins is critical for maintaining normal cellular homeostasis. Seven STAT proteins, STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6, have been identified and localized to the cytoplasm,1 with STAT1, STAT3, and STAT5 proteins being the most prominent in various musculoskeletal tissues.
STAT proteins contain Src homology-2 regions which are the sites for STAT protein phosphorylation by cytokines or growth factors.2 In the first step in this cascade, one of several JAK isoforms, JAK1, JAK2, JAK3, JAK5A, JAK5B, or JAK6, is phosphorylated (ie, activated) following the binding of cytokine(s) and/or growth factor(s) to their specific receptors.3–5 After this step is completed, activated JAK phosphorylates U-STAT proteins. Once STAT proteins are phosphorylated, they form p-STAT homodimers and/or p-STAT heterodimers. These STAT dimers undergo nuclear translocation, with p-STAT proteins acting as potent transcription factors by interacting with gene promoter regions containing STAT-responsive DNA motifs.6
The repertoire of human chondrocyte STAT proteins undergoing phosphorylation in response to rhIL-6 or other proinflammatory cytokines has not been extensively studied. Therefore, to achieve this objective, the extent to which rhIL-6 activated STAT1, STAT3, and STAT5 was assessed in two cell lines of immortalized human juvenile chondrocytes. The results of this analysis with rhIL-6 were compared to STAT proteins which were phosphorylated in response to another proinflammatory cytokine, rhTNF-α.
We employed the immortalized human juvenile T/C28a2 and C28/I2 chondrocyte lines for these analyses. These immortalized human chondrocyte lines have proven to be useful cell culture models for human articular chondrocyte responses to proinflammatory cytokines.7 Furthermore and most importantly, these chondrocyte lines were also previously shown to express cartilage-specific extracellular matrix protein genes7,8 as well as several other molecular characteristics of authentic human chondrocytes, most notably the molecular signature SOX9 gene, considered to be the “master” transcriptional regulator of several cartilage-specific genes, including type II collagen (COL2A1) and aggrecan (AGRN).7–9
Recently, JAK1, JAK2, and JAK3 have been identified as targets for pharmacologic intervention in rheumatoid arthritis (RA) primarily because several of the cytokines, growth factors, and other soluble mediators of inflammation known to activate these JAK isoforms were significantly increased in the sera and/or synovial fluid of RA patients.10 On this basis, the proinflammatory cytokines that were considered to be most relevant to RA and that also activate the JAK/STAT pathway included INF-γ, IL-7, IL-17, and IL-23,11 but most prominently, members of the IL-6 cytokine family, IL-6, OSM, ciliary neutrotrophic factor, leukemia inhibitory factor, and cardiotrophin-1.12
STAT1 and STAT3 were reported to be preferentially phosphorylated by the classical or “canonical” pathways involving IL-6/IL-6Rα/gp130-receptor-mediated signaling.12 However, additional evidence also indicated that activation of STAT1 by IL-6 could occur via membrane-bound IL-6R (mIL-6R) as well as through the IL-6 trans-signaling pathway involving soluble IL-6R (sIL-6R). The analysis of STAT5 phosphorylation in response to IL-6 has generally been limited to that involved with T-cell activation with little or no focus on human articular chondrocytes. For example, with respect to cells of the immune system, Tormo et al13 showed that the treatment of CD4+ and CD8+ T cells as well as natural killer cells with rhIL-6 in vitro caused activation of STAT5.
In the present study, we also probed the extent to which neutralizing the interaction between IL-6 and the soluble IL-6 receptor-α (sIL-6Rα)14 or with JAK-selective small-molecule inhibitors (SMIs) altered STAT protein phosphorylation in response to rhIL-6 or rhTNF-α.
Materials and methods
Materials
Human chondrocyte cell lines
The immortalized human juvenile chondrocyte cell lines T/C28a2 and C28/I2 were obtained from the laboratory of Professor Mary B Goldring (Hospital for Special Surgery/Weill Medical College of Cornell University, New York, NY, USA).
U-STAT, p-STAT, and β-actin antibodies
Antibodies that are reactive specifically with U-STAT1, U-STAT3, and U-STAT5; p-STAT1, p-STAT3, and p-STAT5 proteins; or β-actin were employed. Human STAT1 antibody (monoclonal mouse IgG2B clone #655210) and human/mouse p-STAT1 (antigen affinity-purified polyclonal rabbit IgG; Y701); anti-human/mouse/rat STAT3 antibody and human p-STAT3 (antigen affinity-purified polyclonal rabbit IgG; Y705); and monoclonal anti-STAT5B antibody and human p-STAT5A/B antigen affinity-purified polyclonal rabbit IgG; Y694/Y699) were all purchased from R&D Systems (Minneapolis, MN, USA). The β-actin antibody was purchased from Cell Signaling Technology (Danvers, MA, USA).
JAK inhibitors
WHI-P131 (4-[(6,7-dimethoxy-4-quinazolinyl) amino]-phenol; Janex-1), a JAK-SMI, was purchased from Cayman Chemicals (Ann Arbor, MI, USA) or EMD4 Biosciences (Temecula, CA, USA). Janex-1 was reported to selectively inhibit JAK3 (half-maximal inhibitory concentration [IC50] =78 μM) without affecting JAK1, JAK2, or other protein tyrosine kinases (IC50 >350 μM).15 The JAK1/2-SMI, ruxolitinib, also known as INCB018424, was purchased from Selleck Chemicals (Houston, TX, USA). Ruxolitinib is reported to be a selective JAK1-/JAK2-SMI with a >130-fold selectivity for JAK1/JAK2 compared to JAK3.16
Recombinant human (rh) IL-6, rhTNF-α, sIL-6Rα, and protease/phosphatase inhibitor “cocktail”
The cytokines, recombinant human (rh) IL-6, and rhTNF-α as well as soluble IL-6Rα (sIL-6R) were purchased from various commercial vendors. A protease/phosphatase inhibitor “cocktail” was purchased from Cell Signaling Technology.
Methods
Human T/C28a2 and C28/I2 chondrocytes
T/C28a2 or C28/I2 chondrocytes were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)–F12 medium (1:1 ratio) supplemented with 10% (v/v) fetal bovine serum (FBS). For the measurement of total U-STAT and p-STAT proteins, T/C28a2 or C28/I2 chondrocytes were subcultured into 60 mm culture dishes containing 3 mL of DMEM–F12– 10% FBS (v/v) at an initial cell density of 3×105 cells. The culture medium was changed every 2–3 days until the cells became confluent, generally within 3–5 days after subpassage. At that time, the medium was aspirated, chondrocytes washed once with phosphate-buffered saline (PBS), and DMEM–F12 supplemented with 0.1% FBS (v/v) added to each culture dish.
Analysis of total U-STAT and p-STAT proteins
To analyze total U-STAT and p-STAT proteins, T/C28a2 or C28/I2 chondrocytes were maintained in DMEM–F12 (1:1) containing 0.1% FBS at 25°C (ie, “no additions,” [N/A], 0 min) in order to determine the level of baseline constitutive p-STAT proteins. Alternatively, chondrocytes were incubated in DMEM–F12 (1:1) containing 0.1% FBS and rhIL-6 (50 ng/mL) or rhTNF-α (20 ng/mL), or with rhIL-6 plus sIL-6Rα. (30 ng/mL), or these various treatments plus Janex-1 (100 μM), or ruxolitinib, generally at 50 or 100 μM, for 30 or 60 min at 37°C.
The 30- and 60-min time points were employed for the Western blot analyses of U-STAT and p-STAT proteins based on our previously published Western blot results,17 which showed that p-STAT3 protein accumulated in human chondrocyte protein lysates in response to rhTNF-α for up to 30 min after which, at 60 min, the anti-p-STAT3 antibody reactive band was undetectable on Western blots.
Thus, at baseline (0 min) and after 30 and 60 min, the culture medium was aspirated; cells were washed once with ice-cold PBS, and then scraped into fresh ice-cold PBS or ice-cold PBS containing a protease/phosphatase inhibitor “cocktail”. The cell pellet was collected by centrifugation and a cell lysate produced by sonication of the cell pellets. Cell lysates were stored at −80°C until used for the various assays.
Western blots
Western blots were produced essentially as previously described17 with one essential modification, which was required for quantifying the Western blots. Thus, for this purpose, we employed a sequential antibody-probing/antibody-stripping technique to localize either U-STAT or p-STAT proteins on the Western blots after which these Western blots were restripped and blotted with the β-actin antibody. The various antibody-reactive bands corresponding to the migration rate (Mr) of authentic U-STAT or p-STAT proteins were identified. These antibody-reactive bands were assigned a relative molecular size consistent with the Mr of standard proteins on SDS-PAGE.
However, for the initial qualitative assessment of STAT3 activation, the protein band reactive with the anti-U-STAT3 or anti-p-STAT3 antibody was identified by their respective Mr. Then the ratio of p-STAT3 to U-STAT3 was used to determine the extent to which U-STAT3 was phosphorylated in response to the various incubation conditions.
For all of the subsequent quantitative assessments of total U-STAT1, U-STAT3, or U-STAT5, and STAT1, STAT3, or STAT5 phosphorylation, anti-U-STAT antibody or P-STAT antibody-reactive bands were identified on Western blots. The average signal intensity of U-STAT and p-STAT proteins, as well as their associated anti-β-actin antibody signal intensity, was determined from computer-scanned images of autoradiographs using Metamorph software (MetaMorph Inc., Nashville, TN, USA). The average intensity values from each β-actin Western blot were normalized to the highest value measured in control and treatment groups. These values were then multiplied by the relative intensity values obtained for antibody-reactive bands in their respective Western blot (ie, blots probed with U-STAT or p-STAT antibodies) in order to normalize each antibody-reactive band to its corresponding antibody-reactive band probed with the anti-β-actin antibody. Finally, each of these calculated values were normalized to the experimental treatment group (ie, generally normalized to either lane 1 or lane 2 of each Western blot) to obtain the relative intensity value. Bar graphs were produced using Excel.
A “no additions” control group was employed for comparing all of the other incubation conditions except those treatment groups which contained Janex-1 or ruxolitinib. For those treatment groups, a control group containing dimethyl sulfoxide (DMSO; ie, generally lane 2 of the Western blots) was employed for comparison purposes because Janex-1 or ruxolitinib was initially dissolved in DMSO, and a Janex-1 or ruxolitinib stock solution was produced after diluting the stock solution 1:1 with DMEM–F12 (1:1). Thus, treatment groups containing Janex-1 or ruxolitinib had the same concentration of DMSO as did the corresponding DMSO control group.
Data analysis
The relative differences in antibody-reactive band intensities between control and various treatment groups on individual Western blots were expressed as ±Δ%. The significance of these single-point values were analyzed by χ2 analysis, with the expected difference between control and treated groups set at Δ = ±20%, where p<0.05 was considered significant.
Results
Treatment with rhIL-6 results in the phosphorylation of STAT3 by T/C28a2 and C28/I2 human chondrocytes
Confluent cultures of T/C28a or C28/I2 chondrocytes were treated with “no additions” plus DMSO, rhIL-6 (50 ng/mL), or rhIL-6 plus Janex-1 (100 μM) and held for ~10 min at 25° (“no incubation”) or incubated at 37°C for 30 min with either “no additions” or “no additions” plus DMSO rhIL-6 (50 ng/mL), or rhIL-6 plus sIL-6R (30 ng/mL), or rhIL-6 plus Janex-1 (100 μM). The Western blot, as can be seen in Figure 1, showed that STAT3 was constitutively phosphorylated in T/C28a2 chondrocytes in both the “no additions” control group and the “no additions” plus DMSO control group (Figure 1A). However, incubation of T/C28a2 chondrocytes with rhIL-6 at 37°C for 30 min increased the intensity of the anti-p-STAT3 antibody-reactive band without altering total U-STAT3 (Figure 1A). Importantly, the p-STAT3 band was not detected in C28/I2 chondrocytes in the “no additions” or “no additions” plus DMSO group under the “no incubation” condition or after 30 min at 37°C (Figure 1B), indicating that STAT3 was not constitutively phosphorylated in the C28/I2 chondrocyte line. Of note, as with the T/C28a2 chondrocyte line, the incubation of C28/I2 chondrocytes with rhIL-6 increased the signal intensity of p-STAT3 without altering total U-STAT3 (Figure 1B). The combination of rhIL-6 and Janex-1 resulted in a complete loss of the anti-p-STAT3 antibody-reactive band when compared to the “no additions” plus DMSO control group and, in addition, without altering the signal intensity of the anti-U-STAT3 antibody-reactive band in both T/C28a2 (Figure 1A) and C28/I2 chondrocytes (Figure 1B). The appearance of U-STAT3 antibody-reactive bands in all of the lanes of this Western blot provided a good indication that each lane was loaded with the same amount of protein lysate. With this in mind, the result of this experiment provided ample evidence that Janex-1 or the combination of rhIL-6 and sIL-6R dampened the phosphorylation of STAT3 in both of these immortalized human chondrocyte lines (Figure 1A and B).
Figure 1.

Western blot analysis of STAT3 phosphorylation by T/C28a2 and C28/I2 chondrocytes.
Notes: T/C28a2 (A) or C28/I2 (B) chondrocytes were held at 25°C (no incubation) or incubated at 37°C for 30 min with “no additions,” “no additions” plus DMSO, rhIL-6 (50 ng/mL), sIL-6Rα (30 ng/mL), Janex-1 (100 μM), or various combinations thereof. Western blots were produced with either an anti-U-STAT3 (STAT3) antibody or an anti-p-STAT3 antibody.
Abbreviations: DMSO, dimethyl sulfoxide; sIL-6Rα, soluble IL-6 receptor α; STAT, signal transducers and activators of transcription.
We also determined whether or not JAK1/JAK2 activation in response to rhTNF-α or rhIL-6 contributed to STAT3 phosphorylation by C28/I2 chondrocytes. However, for this analysis, we employed a quantitative Western blot analysis to determine the effect of the JAK1/JAK2-selective SMI, ruxolitinib, on STAT3 activation (Figure 2). In general, none of the incubation conditions altered the ratio of total U-STAT3 to β-actin, with the one exception of C28/I2 chondrocytes treated with rhTNF-α (20 ng/mL) for 30 min (Figure 2A, lane 7) where a statistically significant reduction in U-STAT3 was found (χ2=26.0; p<0.05). However, rhTNF-α had no effect on the ratio of p-STAT3 to β-actin (χ2=0.85; p=0.36) (Figure 2B, lane 7), nor did the combination of rhTNF-α plus ruxolitinib alter this result (χ2=1.28; p=0.26) (Figure 2B, lane 8). Although rhIL-6 when combined with ruxolitinib (Figure 2, lane 6) reduced the ratio of p-STAT3 to β-actin compared to rhIL-6, this change was not statistically significant (χ2=2.3; p=0.13).
Figure 2.
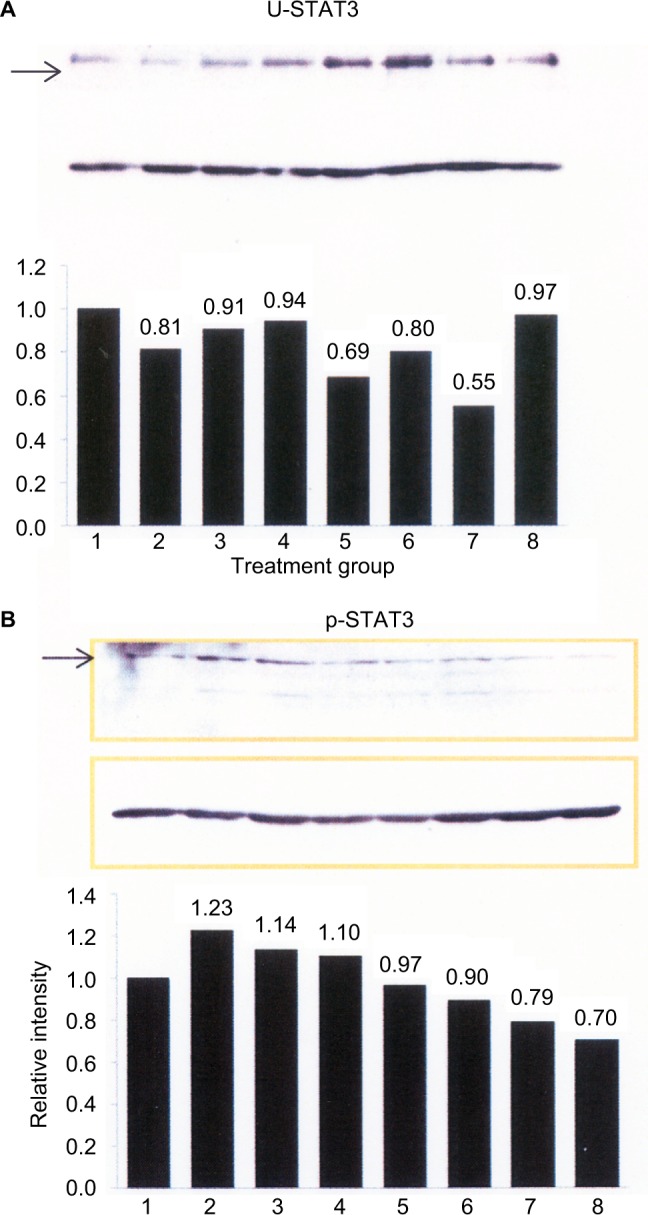
Quantitative Western blot analysis of STAT3 phosphorylation by C28/I2 chondrocytes.
Notes: C28/I2 chondrocytes were incubated for 30 min under the following conditions: lane 1, control (no additions); lane 2, control 1 – DMSO, 133 μL; lane 3, control 2 – DMSO, 66 μL: lane 4, rhIL-6 (50 ng/mL); lane 5, rhIL-6 + ruxolitinib (100 μM); lane 6, rhIL-6 + ruxolitinib (50 μM); lane 7, rhTNF-α (20 ng/mL); lane 8, rhTNF-α plus ruxolitinib (100 μM). control 1 – DMSO was the comparator for ruxolitinib, 100 μM; control 2 – DMSO for ruxolitinib, 50 μM. Western blots were produced with an anti-STAT3 (U-STAT3) antibody or anti-P-STAT3 antibody (A) or with the β-actin antibody (B). The bar graphs were produced by Excel and provide the ratio of U-STAT3 to β-actin (A) or p-STAT3 to β-actin (B) as determined by Metamorph® software. The upper rectangular box in (A) and (B) (arrows) shows U-STAT3 and p-STAT3, respectively, whereas the lower rectangular box in (A) and (B) shows the β-actin immunoblots.
Abbreviation: DMSO, dimethyl sulfoxide; rh, recombinant human; STAT, signal transducers and activators of transcription.
When the quantitative Western blot results of STAT3 in Figure 2 were considered in conjunction with the results shown in Figure 1, we saw that the combination of rhIL-6 plus sIL-6R reduced the ratio of p-STAT3 to β-actin (Figure 2). This result, in turn, supported the results of our initial qualitative analysis on the effect of sIL-6R on rhIL-6-induced p-STAT3 (Figure 1).
Next, we examined the profile of STAT1 and STAT5 proteins produced by C28/I2 chondrocytes. The Western blot for this analysis is shown in Figure 3. With respect to STAT1 and p-STAT1, 3 bands were identified that reacted with the STAT1 or p-STAT1 antibody. One band, identified as STAT1A (Figure 3B) or p-STAT1A (Figure 3B), comigrated with authentic STAT1A, whereas the bands provisionally labeled STAT1B, STAT1C (Figure 3B) or p-STAT1B, p-STAT1C (Figure 3D) migrated faster than STAT1A/p-STAT1A. Thus, the ratio of STAT1A or p-STAT1A to β-actin was only performed on these immunoreactive bands (Figure 3).
Figure 3.

Quantitative Western blot analysis of STAT1 and STAT5 phosphorylation.
Notes: C28/I2 chondrocytes were incubated for 30 (A, B, E, F) or 60 min (C, D, G, H) under the following conditions: lane 1, control (no additions); lane 2, control 1 – DMSO, 133 μL; lane 3, control 2 – DMSO, 66 μL: lane 4, rhIL-6 (50 ng/mL); lane 5, rhIL-6 + ruxolitinib (100 μM; DMSO – 133 μL); lane 6, rhIL-6 + ruxolitinib (50 μM; DMSO – 66 μL); lane 7, rhTNF-α (20 ng/mL); lane 8, rhTNF-α plus ruxolitinib (100 μM). Western blots were produced with anti-STAT1, anti-STAT5 antibody or anti-p-STAT1, anti-p-STAT5 antibody or anti-β-actin antibody. The bar graphs were produced by Excel and provide the quantitative ratio of STAT1A (U-STAT1A; A and E) or p-STAT5A (C and G) to β-actin or the ratio of p-STAT1A (B and D) or p-STAT5B (F and H) to β-actin as determined by Metamorph® software.
Abbreviations: DMSO, dimethyl sulfoxide, rh, recombinant human; STAT, signal transducers and activators of transcription.
A single anti-STAT5 antibody-reactive band (arrow) which migrated with authentic STAT5A was also identified by its reactivity with the anti-STAT5 antibody (Figure 3E and G). Similar patterns of STAT1 (Figure 3A and C) and STAT5 (Figure 3E and G) were identified at 30 and 60 min.
To determine whether or not the faster migrating anti-STAT antibody-reactive bands resulted from STAT protein degradation, C28/I2 chondrocyte protein lysates were prepared in PBS containing a protease/phosphatase inhibitor “cocktail” used at the concentration recommended by the manufacturer. The results from Western blots employing this alternative protein lysate preparatory method indicated that the faster migrating anti-STAT antibody-reactive protein bands remained detectable, thus making it less likely that the faster migrating anti-STAT antibody-reactive bands were products of STAT protein degradation (data not shown).
Phosphorylation of STAT1 and STAT5 proteins by rhIL-6 or rhTNF-α
We then examined the extent to which the U-STAT1 and U-STAT5 proteins identified in these Western blots were phosphorylated in response to rhIL-6 or rhTNF-α at 30 and 60 min (Figure 3). Three forms of p-STAT1 corresponding to p-STAT1A (arrow), p-STAT1B (arrow), and most likely p-STAT1C (arrow) (Figure 3B and D) were detected. Two forms of p-STAT5 corresponding to p-STAT5A (arrow) and p-STAT5B (arrow) were identified at 30 min (Figure 3F), but only p-STAT5A was detected at 60 min (Figure 3H). We assigned the detectable p-STAT5 band at this time point to be p-STAT5A because this STAT5A/5B antibody-reactive band reflected the intensity of the anti-p-STAT5A/B antibody band migrating at ~90 kDa in all of the Western blots, whereas the p-STAT5B band was expected to migrate to ~70 kDa. However, at 60 min, p-STAT5A was detected in only 1 of the DMSO control groups (Figure 3H, lane 3) but more importantly in the rhIL-6 treatment group (Figure 3H, lane 4).
Ruxolitinib did not alter total U-STAT1 or inhibit phosphorylation of STAT1 (p-STAT1) in response to rhIL-6 or rhTNF-α
As shown in Figure 3, ruxolitinib employed at either 50 or 100 μM did not alter total U-STAT1A, U-STAT1B, or U-STAT5A. In addition, ruxolitinib combined with rhIL-6 or rhTNF-α also generally failed to inhibit p-STAT1A, p-STAT1B, or p-STAT1C at either 30 or 60 min (Figure 3B, D, and F, lanes 5, 6, and 8).
Effect of ruxolitinib on U-STAT5A/U-STAT5 and p-STAT5A/p-STAT5B
Unexpectedly, when C28/I2 chondrocytes were incubated with rhIL-6 and the effect on p-STAT5A/p-STAT5B was assessed at 60 min (Figure 3H, lane 4), we found a significant increase in the ratio of p-STAT5A to β-actin (χ2=22.36; p=2×10–6). This result could not be mimicked by rhTNF-α (Figure 3H, lane 7). Importantly, the anti-p-STAT5A antibody-reactive band was completely undetectable when ruxolitinib was combined with either rhIL-6 (Figure 3H, lanes 5 and 6) or rhTNF-α (Figure 3H, lane 8).
Discussion
The principal objective of this study was to determine the extent to which rhIL-6 activated the JAK/STAT pathway in the T/C28a2 and C28/I2 human immortalized chondrocyte cell lines. Although the IL-6/IL-6Rα/gp130 complex is the principal JAK/STAT signaling mechanism activated by IL-6,1 the findings in the present study also indicated that rhIL-6 in combination with sIL-6R activated STAT3. This suggested that the “noncanonical” IL-6 trans-signaling pathway was likely to be active in the C28/I2 chondrocyte response to rhIL-6, as we previously reported for rhIL-6-mediated phosphorylation of STAT1 in the T/C28a2 chondrocyte line.18 Thus, our initial experiments showed that STAT3 was constitutively phosphorylated in the T/C28a2 line of immortalized human chondrocytes. This preliminary finding prompted us to employ the C28/I2 chondrocyte line for most of the remainder of these studies, which examined the extent to which rhIL-6 or rhIL-6 plus sIL-6R activated STAT1, STAT3, and STAT5 proteins.
It may be a critical issue going forward to more precisely determine the extent to which alternative IL-6 pathways, ie, induction by either IL-6 trans-signaling or mIL-6R, absent gp130, are differentially activated by rhIL-6. Although tocilizumab is a potent inhibitor of IL-6-mediated JAK/STAT activation,14 it is unlikely that tocilizumab would be a suitable inhibitor for this purpose since it was reportedly unable to selectively inhibit the IL-6/IL-6Rα/gp130 pathway.19
To summarize the results of these Western blots, we showed that rhIL-6 or rhIL-6 plus sIL-6R activated STAT3, which was inhibited by the JAK3-selective SMI, Janex-1. However, the results of the quantitative Western blot analyses of C28/I2 chondrocytes from the various treatment groups treated with the JAK1-/JAK2-SMI, ruxolitinib, were less consistent. Thus, based on these results, only a weak case can be made at this juncture for the involvement of JAK1 and/or JAK2 in rhIL-6-mediated changes in STAT activation in these human chondrocyte lines. Additionally, we have also come to recognize that the concentrations of ruxolitinib which were employed in these studies were higher than the reported IC50 for ruxolitinib, with these IC50 values having been calculated from an analysis of the ruxolitinib-inhibitory effect on JAK1/JAK2 in other cell types.16
What was most intriguing based on the Western blot analyses shown in Figure 3 was that the anti-STAT5A and anti-p-STAT5B antibody-reactive bands in the rhIL-6 plus ruxolitinib-treatment group as well as in the rhTNF-α-treatment group were undetectable at 60 min, despite ample evidence of equal gel loading based on the results in the anti-U-STAT5 antibody and anti-β-actin-antibody lanes. However, we did detect an anti-p-STAT5A antibody-reactive band in one of the DMSO control groups as well as an even stronger anti-p-STAT5A antibody-reactive band in the rhIL-6-treatment group at 60 min. Although the overall significance of these findings remains to be completely elucidated, it appears for the present that STAT5A continued to be phosphorylated after 60 min in the DMSO control group, but also most impressively in the presence of rhIL-6.
It should be pointed out that the results of another study indicated that DMSO employed at a concentration in DMEM–F12 (1:1) plus 0.5% FBS used in the present study increased the frequency of 4,6-diamidine-2-phenylindole dihydrochloride/Terminal deoxynucleotidyl transferase dUTP nick end labeling (DAPI/TUNEL)-positive C28/I2 chondrocytes, as a measure of apoptosis, compared to C28/I2 chondrocytes maintained in either DMEM–F12 (1:1) plus 0.5% FBS or DMEM–F12 (1:1) plus 10% FBS.20 However, no difference was found in the frequency of DAPI/TUNEL-positive chondrocytes when medium containing 0.5% FBS was compared to medium containing 10% FBS. Thus, it must be considered that DMSO has a weak cytotoxic effect on C28/I2 chondrocytes, but this does not help to explain the effect of DMSO on phosphorylation of STAT5 at 60 min (Figure 3H).
Finally, these human chondrocyte cell lines, especially, C28/I2, which were employed in this study, appear to be very useful for determining chondrocyte responsiveness to rhIL-6 and rhTNF-α, as has been previously shown to be the case for IL-1β.7 This is particularly germane for studying human chondrocyte homeostasis in health and disease because these human chondrocyte lines can be continuously maintained in high-density culture without losing their chondrogenic phenotype.
Acknowledgments
This study was supported by a contract between Genentech/Roche Group (Principal Investigator, Charles J. Malemud, Ph.D.) and Case Western Reserve University (CWRU), and by the CWRU Visual Sciences Core Center Grant (NEI P30-EY11373) (Co-principal Investigators, Eric Pearlman and Irina Pikuleva). We thank Scott Howell, Ph.D. for his support in performing the quantitative Western blot analyses.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Malemud CJ, Pearlman E. Targeting JAK/STAT signaling pathway in inflammatory diseases. Curr Signal Transduct Ther. 2009;4:201–221. [Google Scholar]
- 2.Malemud CJ. Differential activation of JAK enzymes in rheumatoid arthritis and autoimmune disorders by proinflammatory cytokines – potential drug targets. Int J Interferon Cytokine Mediator Res. 2010;2:97–111. [Google Scholar]
- 3.Ivashkiv LB, Hu X. Signaling by STATs. Arthritis Res Ther. 2004;6(4):159–168. doi: 10.1186/ar1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray PJ. The JAK-STAT signaling pathway: input and output integration. J Immunol. 2007;178(5):1650–1653. doi: 10.4049/jimmunol.178.5.2623. [DOI] [PubMed] [Google Scholar]
- 5.Malemud CJ. Intracellular signaling pathways in rheumatoid arthritis. J Clin Cell Immunol. 2013;4:160. doi: 10.4172/2155-9899.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malemud CJ. Suppression of pro-inflammatory cytokines via targeting of STAT-responsive genes. In: El-Shemy H, editor. Drug Discovery. Rijeka, Croatia: InTech Publishing; 2013. pp. 373–411. [Google Scholar]
- 7.Goldring MB. Culture of immortalized chondrocytes and their use as models for chondrocyte function. Methods Mol Med. 2004;100:37–52. doi: 10.1385/1-59259-810-2:037. [DOI] [PubMed] [Google Scholar]
- 8.Koyenyesi R, Tan L, Robbins JR, Goldring MB. Proteoglycan production by immortalized human chondrocyte cell lines cultured under conditions that promote the expression of the differentiated state. Arch Biochem Biophys. 2000;383(1):79–90. doi: 10.1006/abbi.2000.2044. [DOI] [PubMed] [Google Scholar]
- 9.Finger F, Schörle C, Zien A, et al. Molecular phenotyping of human chondrocyte cell lines T/C-28a2, T/C-28a4 and C-28/I2. Arthritis Rheum. 2003;48(12):3395–3403. doi: 10.1002/art.11341. [DOI] [PubMed] [Google Scholar]
- 10.Malemud CJ, Blumenthal DE. Protein kinase small molecule inhibitors for rheumatoid arthritis: medicinal chemistry/clinical perspectives. World J Orthopedics. 2014;5(4):496–503. doi: 10.5312/wjo.v5.i4.496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imboden JB. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol. 2009;4:417–434. doi: 10.1146/annurev.pathol.4.110807.092254. [DOI] [PubMed] [Google Scholar]
- 12.Malemud CJ. Recent advances in neutralizing the IL-6 pathway in arthritis. Open Access Rheumatol Res Rev. 2009;1:133–150. doi: 10.2147/oarrr.s6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tormo AJ, Letellier MC, Sharma M, et al. IL-6 activates STAT5 in T cells. Cytokine. 2012;60(2):575–582. doi: 10.1016/j.cyto.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Shetty A, Hanson R, Korsten P, et al. Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther. 2014;8:349–364. doi: 10.2147/DDDT.S41437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sudbeck EA, Liu XP, Naria NK, et al. Structure-based design of specific inhibitors of Janus kinase 3 as apoptosis anti-leukemia agents. Clin Cancer Res. 1999;5(6):1569–1582. [PubMed] [Google Scholar]
- 16.Quintás-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective Jak1/2 inhibitor, INCB018424: therapeutic implications for the treatment of myeloproliferative diseases. Blood. 2010;115(15):3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malemud CJ, Sun Y, Pearlman E, et al. Monosodium urate and tumor necrosis factor-α increase apoptosis in human chondrocyte cultures. Rheumatology (Sunnyvale) 2012;2:113. doi: 10.4172/2161-1149.1000113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meszaros EC, Malemud CJ. STAT1 is constitutively phosphorylated in the T/C28a2 immortalized juvenile human chondrocyte cell line and stimulated by IL-6 plus soluble IL-6 receptor. J Clin Cell Immunol. 2015;6(2) doi: 10.4172/2155-9899.1000307. pii, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rose-John S, Waetzig GH, Scheller J, et al. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin Ther Targets. 2009;11(5):613–624. doi: 10.1517/14728222.11.5.613. [DOI] [PubMed] [Google Scholar]
- 20.Malemud CJ, Lewis AC, Meszaros EC, Wylie MA, Skomorovska-Prokvolit Y, Mesiano S. Inhibition of ERK1/2 phosphorylation by U0126 increases tumor necrosis factor-α-induced apoptosis, but not interleukin-6-induced apoptosis in C28/I2 human chondrocytes. J Autoimmun Disord. 2015;1:1. doi: 10.21767/2471-8153.100004. [DOI] [PMC free article] [PubMed] [Google Scholar]