

Supplemental Digital Content is available in the text
Keywords: CNGB3, cone dystrophy, homozygous by descent, protein modeling, whole exome sequencing
Abstract
Rationale:
Genetic elucidation of cone-dominated retinal dystrophies in Indian subcontinent is much needed to identify and catalog underlying genetic defects. In this context, the present study recruited a consanguineous Indian family affected with autosomal recessive cone dystrophy (CD). Considering the huge genetic heterogeneity and recessive inheritance of the disease, we chose to dissect out causal variant in this family by whole exome sequencing (WES).
Patient concerns:
In the recruited family, three of the six siblings had complaints of poor visual acuity, photophobia, and disturbed colour vision since early childhood. Fundus examination disclosed vascular attenuation and macular retinal pigment epithelium (RPE) changes in all the affected siblings, signifying degeneration of photoreceptor cells.
Diagnosis:
Complete clinical investigation and electroretinography studies led to the diagnosis of cone dystrophy in three siblings of the family.
Interventions:
Detailed ophthalmic examination, including family history, visual function testing, and retinal imaging, was performed. We captured and sequenced exomes of 2 affected siblings and their mother using SureSelect Human All Exon V5 Kit on Illumina HiSeq 2000/2500 platform with 100 bp paired-end sequencing method. Candidates after data analysis were screened by segregation analysis and Sanger sequencing. Considering recessive inheritance and consanguinity in the pedigree, we attempted to map large loci homozygous by descent in the genome of patients using exome sequencing variants. Extensive protein modeling was carried out to assess possible consequences of the identified variant on the 3-dimensional structure of the protein.
Outcomes:
WES generated more than 65,000 variants for each individual. Assuming recessive inheritance, 13,026 variants were selected. Further filtering on the basis of their position in gene, class, and minor allele frequency constricted the huge list to 12 rare variants. Finally, we ascertained a single base deletion c.1148delC (p.Thr383fs) in the gene CNGB3 as the causal variant. This is a recurrent frameshift mutation resulting in truncated CNGB3 protein. We mapped a large 15-Mb stretch of homozygous markers spanning the causal variant in the proband. The gene CNGB3 encodes modulatory subunit of cyclic nucleotide-gated channels in cone photoreceptors. Protein modeling reveals loss of 2 transmembrane helices and conserved CAP_ED domain in truncated CNGB3, which eventually is predicted to form nonfunctional channels and hamper phototransduction.
Lessons:
We have identified a recurrent mutation c.1148delC (p.Thr383fs) in CNGB3 for autosomal recessive CD. The present report provides the first description of CNGB3 mutation from India. It is also the foremost investigation of familial CD in Indian patients; therefore, it presents the primary genetic etiology of CD in India.
1. Introduction
Cone dystrophies (CDs) cover a prominent class of inherited retinal degenerations (RDs) that principally involve deterioration of cone photoreceptors. Poor visual acuity and abnormal color vision, generally arising in the first 2 decades of life, are the primary complaints in CD, which later also involves nystagmus, photosensitivity, and central scotoma.[1] In addition to these typical symptoms, ophthalmoscopic abnormalities in the macula and retinal pigment epithelium (RPE) might also be observed.[2] Considerable amount of genetic variability in CD further complicates the disease management. Therefore, an efficient therapeutic strategy in this disease demands not only the right clinical interpretation but also the precise recognition of underlying genetic defect. Attempts to identify such genetic defects have revealed existence of all forms of Mendelian inheritance in CD, namely X-linked (XL), autosomal dominant (AD), and autosomal recessive (AR). Much of the genetics behind XL and AD forms has been explained. However, 75% of AR-CD cases are still unanswered despite its highest prevalence among all Mendelian forms.[1]
In the present study, we recruited a north Indian consanguineous Muslim family affected with AR-CD (Fig. 1A). CD is a rare disease with a prevalence of 1 in 30,000 to 40,000 individuals[3]; therefore, families affected with CD are an ideal source to expedite disease gene identification. To date, there are at least 30 genes implicated in cone-dominated retinal dystrophies (RetNet - Retinal Information Network). Several of them such as ABCA4, ADAM9, CACNA1F, GUCY2D, CDHR1, CNGB3, PDE6C etc, have large number of protein-coding exonic regions. Therefore, Sanger sequencing based targeted screening of all the candidate genes is expensive and laborious. In this context, whole exome sequencing is a time-saving, cost-effective, and promising technology, which can dissect out causal variants for Mendelian as well as complex diseases even in simplex familial cases.[4–6] In comparison to complex, AD or XL forms, AR familial cases are easier to solve with WES, as one needs to focus only on the variants that are consistently homozygous in all the affected members.[7] Here, consanguineous pedigrees offer added benefit because the disease gene is supposed to be located in a large locus homozygous by descent.[8,9] In such cases, it becomes important to map large blocks of homozygous markers in the genome of patients. Subsequently, majority of the WES generated variants that lie outside these homozygous blocks can be filtered out. In the present study, taking into consideration the genetic heterogeneity and recessive inheritance of CD and consanguinity in the recruited family, we chose to identify the causal mutation by whole exome sequencing. Our next objective was to map the loci homozygous by descent in the genome of the affected siblings. Finally, we evaluated plausible consequences of the identified variant by protein structural modeling.
Figure 1.
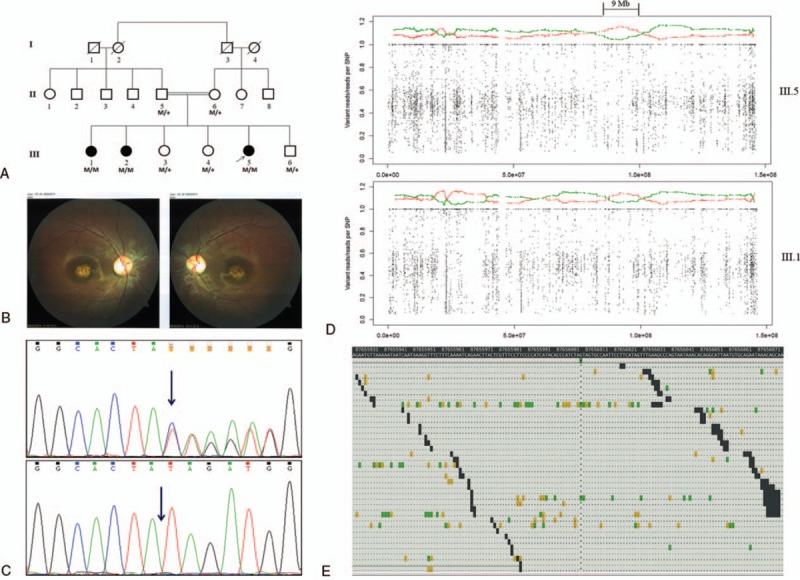
Genetic examination of the family affected with cone dystrophy. (A) Pedigree of the consanguineous Indian family affected with AR cone dystrophy. Squares present males, and circles denote females. Shaded shapes indicate individuals affected in the family. M: Mutation NG_016980.1:c.1148delC, M/M: Homozygous for the deletion, M/+: Heterozygous for the deletion. (B) Fundus photograph of III.1. (C) Chromatogram of Sanger sequencing demonstrating heterozygosity for c.1148delC in parents and healthy siblings (upper) and homozygosity for the deletion in affected siblings (lower). (D) Mapping of the regions of shared homozygosity on eighth chromosome in III.5 (upper) and III.1 (lower). Each dot stands for a single nucleotide variant used in mapping. X-axis denotes genomic position along chromosome 8, and Y-axis shows ratio of number of reads containing nonreference (variant) allele to the total number of reads. Each point in this graph represents relative frequency of a variant over a span of 500 flanking variants (250 left and 250 right side). Red color denotes relative frequency of homozygous variants (≥95%) out of total (homozygous and heterozygous), and green color shows the same for heterozygous variants. The regions where frequency of homozygous markers (red) is 2.5 times higher than that of heterozygous markers (green) has been considered under autozygous locus. A 9 Mb long autozygous locus common in III.5 and III.1 has been indicated. (E) Genomic location depicting “AG/A” at position 87656008 within CNGB3 (-).
2. Methods
2.1. Clinical assessment and genetic analysis
The north Indian Muslim family (Fig. 1A) residing in Jaunpur, Uttar Pradesh, India was recruited in our ongoing genetic study on RD approved by the ethical committee of Faculty of Science (currently known as Institute of Science), Banaras Hindu University, Varanasi, India. Complete ophthalmologic evaluation, including family history, visual function testing, and retinal imaging, was carried out. The patients were referred to All India Institute of Medical Sciences (AIIMS), New Delhi, India for electroretinography (ERG) examination. After procuring written informed consent, peripheral blood samples were taken from 8 members including both parents (II.5 and II.6) and 6 siblings (III.1 to III.6). We used standard salting out protocol to extract genomic DNA from peripheral blood. Initially, 3 family members including mother (II.6) and 2 affected sisters (III.1 and III.5) were chosen for the genetic investigation by whole exome sequencing. Exome enrichment was carried out with 3 μg of genomic DNA using Agilent SureSelect Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA) according to manufacturer's protocol on the Illumina HiSeq 2000/2500 platform (Illumina, San Diego, CA). Each sample was sequenced to at least 100X raw target depth, with 100 bp paired-end sequencing method.
During bioinformatics analysis, we checked several parameters from fastq files, including base quality score distribution, sequence quality score distribution, average base content per read, GC distribution in the reads, PCR amplification issue, over-represented sequences, and adapter trimming. On the basis of the quality of fastq files, sequence reads were trimmed where necessary to retain only high-quality sequence for further analysis. In addition, low-quality sequence reads were excluded from further investigation. Adapter trimming was performed using fastq-mcf program (version - 1.04.676). Paired-end reads were aligned to the reference human genome primary assembly hg19 downloaded from UCSC database. Alignment was performed using BWA program (version - bwa-0.7.8). While running BWA, trimming was performed using –q = 20. Reads after alignment with mapping quality ≥20 and with insert size ≥100 bp and ≤1000 bp were taken further for analysis. Paired reads that mapped to 2 different chromosomes were totally discarded from the analysis. Aligned reads were first sorted by Picard tool (picard-tools - 1.115) using SortSam command, and read duplicates were removed using Picard Mark Duplicates command. After removing the duplicates, reads were realigned around the known indels provided by GATK group. This was followed by base recalibration step. After recalibration, quality score of each base was more accurate. Known variant positions were taken into account to recalibrate the quality score. After realignment, we used GenomeAnalysisTKLite-2.3–9 toolkit Unified Genotyper (https://www.broadinstitute.org/) to identify single nucleotide variants (SNVs) and short Indels. We further filtered variants in order to retain good quality (depth and variant score) variants. Identified variants were annotated using in-house program (VariMAT). The gene model used for annotation was downloaded from Ensembl database Release 75. Variant class prediction was done using VeP Release 75 (http://www.ensembl.org) and internal annotation pipeline (VariMAT).
2.2. Mapping the regions of shared homozygosity
We used a modified version of an earlier reported algorithm[10] and attempted to map blocks of homozygous markers in the genome of diseased family members employing exome variant data. For the purpose of mapping, known as well as novel single nucleotide variants (SNVs) having ≥10 reads were used. It included exonic, intronic, intergenic, splice site, synonymous, nonsynonymous, and UTR variants. Next, we sorted out SNVs called with variant reads ≥95% as homozygous and SNVs with variant reads ≥30% to ≤70% as heterozygous ones. Reads with variants ≤5% were removed from the account. Rest of the variant calls (5% to <30%, >70% to <95%) were considered ambiguous, and they were not used for plotting. Chromosome wise plotting of the data was carried out using R-project.
2.3. Protein structural modeling
In order to understand the possible consequences of the frameshift deletion on the 3-dimensional structure of the protein, we generated protein model of the wild-type and mutant CNGB3 protein. Because of the low sequence identity between CNGB3 and template PDBID: 5K7L (20%), we used secondary structure-based threading server PHYRE[11] to generate the CNGB3 model, taking the Eag1 crystal structure (PDB ID: 5K7L chain A) as a template. All sequence similarity searches were carried out in the MPI Bioinformatics Toolkit using HHpred[12] with default settings. HHpred searches for template were performed against a database comprising PDB70 (protein databank structures, as available on January 19, 2017) clustered at 70% sequence identity. Chimera tool was used for visualization and analysis of the modeled protein structure.[13]
3. Results
3.1. Clinical assessment
In the recruited family (Fig. 1A), 3 of the 6 siblings had complaints of poor visual acuity, photophobia, and disturbed color vision since early childhood. Fundus examination disclosed vascular attenuation and macular RPE changes in all the affected siblings, signifying degeneration of photoreceptor cells (Fig. 1B). The index patient III.5 was investigated initially at the age of 9 years, with complaints of poor vision and photophobia since early childhood. Her best corrected visual acuity was 0.6 OD/0.6 OS with refraction (spherical equivalent in diopters) -3.25 OD/-1.25 OS. She was also diagnosed to have jerk nystagmus without any null, divergent squint 30 prism diopters (PD), and abnormal color vision. Fundus examination revealed vascular attenuation, macular RPE changes, and mid-peripheral RPE changes. Two of her sisters (III.1 aged 18 years and III.2 aged 14 years) presented with similar complaints of poor vision and photophobia. The eldest sibling III.1 had best corrected visual acuity 0.7 OD/0.7 OS with refraction -3.75 OD/ -2 OS. The sibling III.2 was noted with best corrected visual acuity 0.7 OD/0.7 OS and refraction -4.25 OD/ -4.00 OS. Both these siblings (III.1 and III.2) had jerk nystagmus without null and abnormal color vision. The patient III.1 manifested intermittent divergent squint 20 PD, whereas the sibling III.2 had intermittent divergent squint 15 PD. On fundus evaluation, both III.1 and III.2 exhibited temporal pallor of disc and vascular attenuation along with macular atrophy. Fundus of III.1 had a typical macular metallic sheen, whereas III.2 had only macular RPE changes. As per the ERG reports from AIIMS, New Delhi, all the 3 affected siblings demonstrated normal rod response but abnormal cone responses. Therefore, the clinical features and ERG studies led to the diagnosis of cone dystrophy.
3.2. WES and mapping the regions of shared homozygosity
Whole exome sequencing generated around 22 Gigabases (Gb) of mappable sequence data in the proband, 19 Gb in her affected sister, and 22 Gb in their mother. More than 99% of these reads were mapped. Percentage of targets with 10X coverage in III.5, III.1, and II.6 was 97.13%, 95.91%, and 97.15%, respectively, whereas corresponding mean target coverage was 93X, 79X, and 95X. WES provided us 66,803, 65,364, and 67,929 variants in the 3 members III.5, III.1, and II.6 individually, while the coding variants after quality filtering (>5 variant reads, >15%variation) in them were 19,836, 19,566, and 20,129, respectively. The causal mutation was supposed to be one among the homozygous variants in the affected sisters. Therefore, taking into consideration the AR mode of inheritance, we chose variants that were consistently homozygous (>80% variation reads) in both the affected sisters but heterozygous in mother. This rendered us only with 13,026 variants. We further filtered them on the basis of their position in gene (including exonic-CDS, exonic-PC_ncRNA, intronic 3’-splice site, intronic 5’-splice site), class (all except synonymous variants), and minor allele frequency (1000 genome minor allele frequency less than or equal to 0.01 and seen in less than 2 individuals within the in-house database). As a result, we obtained a final list of 12 rare variants (Supplementary Figure 1). We checked these variants in RetNet - Retinal Information Network database and DAVID tool. Among all these variants, CNGB3 (NG_016980.1) was found to be the only gene having expression in retina and association with retinal dystrophy. The variant c.1148delC (p.Thr383fs) identified in this gene was further confirmed by Sanger sequencing in both the parents and all 6 siblings (Fig. 1C). Parents and healthy siblings were carrier of this deletion, while the affected siblings were homozygous. We also checked 25 unrelated healthy individuals (50 chromosomes) from the same north Indian population and found them negative for this deletion. Therefore, the variant NG_016980.1:c.1148delC was ascertained as the causal variant for CD in the family.
Further, in an attempt to map large regions of shared homozygosity in the genome of affected siblings (Fig. 1D), we identified a large 15-Mb stretch of homozygous markers, which spanned the mutation on chromosome 8 in the proband (III.5). We confirmed the presence of these autozygous regions in the affected siblings (III.5 and III.1) using AgileVCFMapper (Supplementary Figure 2A and B). Interestingly, we observed that a long block of homozygous markers in the proband covers a locus from 81263715 (81.2 Mb) to 96598761 (96.5 Mb), whereas such a block in III.1 is present from 87666251 (87.6 Mb) to 96598761 (96.5 Mb). Therefore, a region of shared homozygosity in both the siblings extends from 87666251 (87.6 Mb) to 96598761 (96.5 Mb) and is 9 Mb long. It may also be noted that the CNGB3 variant present at position 87656008 is slightly outside the block of homozygous markers in the second affected sibling (III.1). Figure 1E also demonstrates genomic location of the single base deletion on chromosome 8.
3.3. Protein structural modeling
The target protein CNGB3 showed significant similarity with recently solved structure of a mammalian potassium voltage-gated channel subfamily protein: metal transport-calcium binding protein complex (PDBID 5K7L_chain A),[14] with 427 aligned columns, 20% identity, and E-value 3.4e-52. However, the match between the template and target proteins was within the region of 205–641. The N-terminus and C-terminus sequences of CNGB3 did not match with any available template in PDB. Therefore, it needed a hybrid approach with homology modeling and ab-initio approach of modeling. Understanding the domain composition of the lost structural region, especially from 383 amino acid residue onwards was important in our analysis. Structural analysis of CNGB3 obtained using PHYRE2 server revealed that the target protein was composed of alpha helix (67%), beta strand (7%), and transmembrane (TM) helix (18%). Seven TM helices were observed in the region 217–445 (Fig. 2A). Importantly, in mutant form, the last 2 TM helices were lost due to truncation (Fig. 2B). Also, conserved domain analysis of NCBI revealed that the truncated region had a CAP_ED domain, which is an effector domain of the CAP family of transcription factors (Fig. 2C, D).
Figure 2.
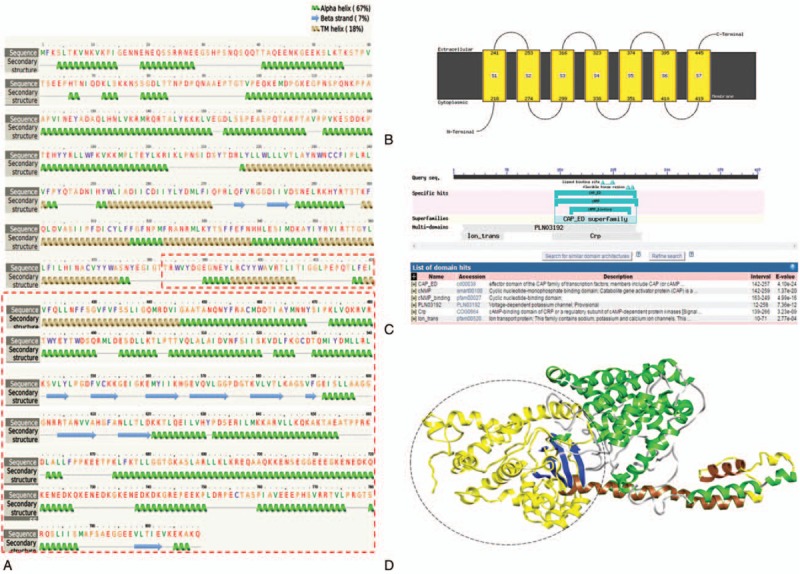
Secondary structure analysis and protein modeling of CNGB3. (A) Predicted secondary structure of full-length CNGB3 using PHYRE2 server. The transmembrane helices 6 and 7 are lost in the truncated form of protein. (B) Transmembrane region showing arrangement of TM-helices. (C) NCBI-CDD analysis of truncated domain region. CAP_ED domain is lost in truncated CNGB3 protein. (D) Model structure of CNBG3 (full-length) using PHYRE server. Helices are shown in green and yellow ribbon, and strands are in blue ribbon. Part of TM-helix region, helices 6 and 7 (shown in brown color ribbon), are lost in truncated protein. CAP_ED domain is located in the truncated region, encircled and shown in yellow ribbon.
3.4. Discussion and conclusion
A detailed catalogue of causal variants for inherited RD from north Indian patients is much needed, as there is limited understanding on its genetic epidemiology from this geographical region. In this context, a north Indian family affected with a rare recessive cone dystrophy was investigated in the current study. This was a small family with only 3 affected siblings in a single generation. Linkage-based screening cannot be employed in small families with a limited number of affected members. Moreover, because of a large number of candidate genes implicated in CD, Sanger sequencing based targeted screening was an expensive and tedious option. We elected WES as an efficient approach for this familial case. Consanguinity in the family was an additional advantage in terms of data analysis, as it allowed us to focus only on the variants that were present within the IBD locus in the affected siblings. Our study also favors earlier ideas that the next-generation sequencing (NGS) based technologies have better potential than targeted Sanger sequencing or array-based screening for genetic diagnosis of heterogeneous diseases such as RD.[15] WES is one of such NGS-based techniques that can track known as well as novel disease-causing mutations efficiently in heterogeneous diseases. It is equally capable of identifying common as well as private variants and works even in the cases when limited number of family members are available for diagnosis. WES-based diagnosis in our study has successfully revealed a recurrent frameshift deletion in CNGB3 gene as the causal variant for CD in the affected family.
The CNGB3 gene was identified within ACHM3 locus on chromosome 8q21 in an attempt to identify the underlying gene for achromatopsia in pedigrees negative for earlier known CNGA3 mutations, and the 1 base deletion c.1148delC (p.Thr383fs) was observed in homozygous and compound heterozygous state in several patients from different geographic origins.[16] It is estimated that the CNGB3 mutations are responsible for 50% of achromatopsia cases, and the single base deletion accounts for 70% of the CNGB3 mutant alleles.[17] The same deletion in compound heterozygous state was later shown to be associated with cone dystrophy.[3] Concordant with this report, we also observe nystagmus, macular changes, and abnormal cone but normal rod responses in the affected individuals. Although the clinical presentations of CD in the present study are similar to those described earlier, the patients are homozygous for the deletion. Ours is also the first report of this deletion in Indian patients.
The recruited Indian–Muslim family presented a case of consanguineous marriage, which is common in several societies of India including all Muslims communities.[18,19] In the family, the parents (II.5 and II.6) were first cousins, and the children (III.1, III.2, and III.5) were affected with a rare AR disease. This encouraged us to look for large stretches of homozygous markers in the affected siblings. In our attempt to map such IBD loci using exome variant data, we identified a 15-Mb stretch of homozygous markers spanning the CNGB3 variant in the proband. However, such a locus in another affected sibling (III.1) was only 9 Mb long and did not embrace the identified mutation. The deletion in the second sibling was slightly separate from the 9-Mb locus. Apparently, exome-wide sequence variants are not as efficient as genome-wide SNP markers are to map IBD loci in the children of a consanguineous marriage. Microarray SNP genotyping data are better to map large autozygous regions precisely, as it offers superior marker density and distribution.[20] Becker et al[10] who previously proposed mapping the regions of shared homozygosity using exome variants located large homozygous blocks only in the index patient. Whereas, we tried mapping autozygous regions in 2 affected siblings whose exomes were sequenced. Although consistent length of autozygous regions could not be located in both siblings, we identified the CNGB3 variant independently by exome sequencing.
Our detailed protein modeling also supports the damaging consequences of this deletion on CNGB3 protein. The deletion results in truncated protein of just 394 residues instead of the 809 residues long wild-type protein. There are 7 TM helices in full length protein as demonstrated in the secondary structure of CNGB3. The TM helices appear to contribute in the architecture of channels by forming a hydrophobic core through the lipid bilayer. Therefore, loss of 2 of these helices might lead to formation of nonfunctional channels. In addition, we also demonstrate loss of CAP_ED domain in the truncated protein. The cyclic nucleotide-gated (CNG) ion channels are activated by binding of cytoplasmic cyclic nucleotides such as cGMP or cAMP. It is the CAP_ED domain that facilitates this binding. Therefore, the truncation resulting in loss of TM helices and CAP_ED domain not only disrupts the architecture of the channels but also hinders its activation. The CNG channels comprise of 2 structurally related subunits A and B. The A subunit is accountable for ion conduction through the channel, whereas the B subunit has modulatory function.[21,22] While CNGA1 and CNGB1 subunits are present in rod photoreceptors, CNGA3 and CNGB3 constitute the channels in cone photoreceptors. To answer, how CNGA3 and CNGB3 mutations are responsible for CDs, in vivo studies have been carried out. Mutations in the gene CNGA3 appear to disrupt channels’ cellular trafficking and cell membrane targeting,[23] whereas those in CNGB3 have been demonstrated to cause cone dystrophy by downregulating CNGA3 biosynthesis.[17]
In summary, whole exome sequencing in the current study has revealed the mutation NG_016980.1:c.1148delC for cone dystrophy in an Indian family. This is the first report of CNGB3 mutation from India. This mutation results in truncation of the protein leading to loss of TM helices and CAP_ED domain. Therefore, the truncation not only disrupts fundamental function of the channel but also deteriorates downstream signaling responsible for channel activation. Our study also supports promising potential of WES in genetic diagnosis of RD. To the best of our knowledge, there are no reports on genetic analysis of cone-dominated RD from India. Ours is the first investigation of familial CD in Indian patients; hence, it presents the primary genetic etiology of CD in India.
Supplementary Material
Acknowledgments
We sincerely acknowledge the kind participation and cooperation of the affected family throughout analysis. We are also grateful for fellowship from Council of Scientific and Industrial Research, India to SG and financial support from Department of Biotechnology, India to MM.
Footnotes
Abbreviations: CD = cone dystrophy, CNG = cyclic nucleotide-gated, ERG = electroretinography, IBD = identical by descent, RD = retinal dystrophy, RPE = retinal pigment epithelium, SNP = single nucleotide polymorphism, TM = transmembrane, WES = whole exome sequencing.
The authors report no conflicts of interest.
Supplemental Digital Content is available for this article.
References
- [1].Roosing S, Thiadens AA, Hoyng CB, et al. Causes and consequences of inherited cone disorders. Prog Retin Eye Res 2016;42:1–26. [DOI] [PubMed] [Google Scholar]
- [2].Michaelides M, Hardcastle AJ, Hunt DM, et al. Progressive cone and cone-rod dystrophies: phenotypes and underlying molecular genetic basis. Surv Ophthalmol 2006;51:232–58. [DOI] [PubMed] [Google Scholar]
- [3].Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol 2004;88:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet 2014;59:5–15. [DOI] [PubMed] [Google Scholar]
- [5].Priya RR, Rajasimha HK, Brooks MJ, et al. Retinal Development: Methods and Protocols Methods in Molecular Biology. 2012;New York, NY: Springer Science Business Media, 335–51. [Google Scholar]
- [6].Majewski J, Schwartzentruber J, Lalonde E, et al. What can exome sequencing do for you? J Med Genet 2011;48:580–9. [DOI] [PubMed] [Google Scholar]
- [7].Gilissen C, Hoischen A, Brunner HG, et al. Disease gene identification strategies for exome sequencing. Eur J Hum Genet 2012;20:490–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987;236:1567–70. [DOI] [PubMed] [Google Scholar]
- [9].Lazar CH, Kimchi A, Namburi P, et al. Nonsyndromic early-onset cone-rod dystrophy and limb-girdle muscular dystrophy in a consanguineous Israeli family are caused by two independent yet linked mutations in ALMS1 and DYSF. Hum Mutat 2015;36:836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Becker J, Semler O, Gilissen C, et al. Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta. Am J Hum Genet 2011;88:362–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat protoc 2015;10:845–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Söding J, Biegert A, Lupas AN. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 2005;33(Suppl 2):W244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- [14].Whicher JR, MacKinnon R. Structure of the voltage-gated K+ channel Eag1 reveals an alternative voltage sensing mechanism. Science 2016;353:644–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Glöckle N, Kohl S, Mohr J, et al. Panel-based next generation sequencing as a reliable and efficient technique to detect mutations in unselected patients with retinal dystrophies. Eur J Hum Genet 2014;22:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the β-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 2000;9:2107–16. [DOI] [PubMed] [Google Scholar]
- [17].Ding XQ, Harry CS, Umino Y, et al. Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum Mol Genet 2009;18:4770–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bittles AH. The impact of consanguinity on the Indian population. Indian J Hum Genet 2002;8:45–51. [Google Scholar]
- [19].Bittles AH, Hussain R. An analysis of consanguineous marriage in the Muslim population of India at regional and state levels. Ann Hum Biol 2000;27:163–71. [DOI] [PubMed] [Google Scholar]
- [20].Watson CM, Crinnion LA, Gurgel-Gianetti J, et al. Rapid detection of rare deleterious variants by next generation sequencing with optional microarray SNP genotype data. Hum Mutat 2015;36:823–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zagotta WN, Siegelbaum SA. Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci 1996;19:235–63. [DOI] [PubMed] [Google Scholar]
- [22].Gerstner A, Zong X, Hofmann F, et al. Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J Neurosci 2000;20:1324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ding XQ, Fitzgerald JB, Quiambao AB, et al. Molecular pathogenesis of achromatopsia associated with mutations in the cone cyclic nucleotide-gated channel CNGA3 subunit. Adv Exp Med Biol 2010;664:245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.