

Abstract
Simultaneous elucidation of the glycan structure and the glycosylation site are needed to reveal the biological function of protein glycosylation. In this study, we employed a recent type of fragmentation termed higher energy collisional dissociation (HCD) to examine fragmentation patterns of intact glycopeptides generated from a mixture of standard glycosylated proteins. The normalized collisional energy (NCE) value for HCD was varied from 30 to 60% to evaluate the optimal conditions for the fragmentation of peptide backbones and glycoconjugates. Our results indicated that HCD with lower NCE values preferentially fragmented the sugar chains attached to the peptides to generate a ladder of neutral loss of monosaccharides, thereby enabling the putative glycan structure characterization. In addition, detection of the oxonium ions enabled unambiguous differentiation of glycopeptides from non-glycopeptides. In contrast, HCD with higher NCE values preferentially fragmented the peptide backbone and, thus, provided information needed for confident peptide identification. We evaluated the HCD approach with alternating NCE parameters for confident characterization of intact N- and O-linked glycopeptides in a single liquid chromatography–tandem mass spectrometry (LC–MS/MS) analysis. In addition, we applied a novel data analysis pipeline, so-called GlycoFinder, to form a basis for automated data analysis. Overall, 38 unique intact glycopeptides corresponding to eight glycosylation sites (six N-linked and two O-linked sites) were confidently identified from a standard protein mixture. This approach provided concurrent characterization of both the peptide and the glycan, thereby enabling comprehensive structural characterization of glycoproteins in a single LC–MS/MS analysis.
Keywords: Glycosylation, Glycopeptides, LC–MS/MS, HCD, NCE, Automated identification
Glycosylation is one of the most prominent protein posttranslational modifications and is linked to various structural and functional roles in many biological processes, including fertilization, immune defense, viral replication, parasitic infection, cell growth, cell–cell adhesion, inflammation, and signal transduction [1–5]. Glycans can be either covalently bound to the amide group of asparagine (i.e., N-linked glycosylation), or attached to the hydroxyl group of serine or threonine (i.e., O-linked glycosylation).
Despite the great biological and medical importance, glycopeptide characterization still presents significant analytical challenges. Glycopeptides are often difficult to detect without enrichment because of their low abundance. In addition, the ionization efficiency of glycopeptides is typically low, and the signal is often suppressed by the overwhelming majority of coexistent non-glycosylated peptides [6–8]. To tackle abundance and ionization challenges, various enrichment methods, such as zwitterionic hydrophilic interaction chromatography (ZIC–HILIC)1 [7,9] and lectin [10], porous graphitic carbon [11,12], and hydrazine [13,14] chemistries, are typically used. In addition, the often highly complex nature of the attached glycan makes the sequencing and identification very difficult. The most commonly applied strategy is enzymatic deglycosylation of peptides prior to liquid chromatography– mass spectrometry (LC–MS) analysis. Although this method allows for comprehensive characterization of glycoproteins and their glycosylation sites, it has obvious drawbacks in that it does not provide oligosaccharide composition and structure information critical for deciphering the function. Furthermore, none of the available glycosidases is universal. For example, the most commonly used endoglycosidase, PNGase F, did not perform well when applied to the bacterium Campylobacter jejuni, likely because of its unique monosaccharide composition [15]; in addition, there is no known enzyme that can cleave O-linked glycans as efficiently as PNGase processes N-linked glycans. Optimization of the existing analytical approaches for intact glycopeptide characterization [15,16], therefore, is critically needed to facilitate simultaneous characterization of the glycan structure and the corresponding peptide sequence.
Multiple tandem MS (MS/MS) strategies [15–19] and various bioinformatics tools [17,20,21] have been applied to characterize intact glycopeptides. Collisionally induced dissociation (CID) is known to preferentially cleave the glycosidic linkages and produce limited peptide backbone fragment ions [18]. An interesting approach to alleviate this issue and enable concurrent peptide and glycosylation site identification is adding one more fragmentation stage (MS3) [18]. However, this approach is limited by the requirement of sufficient parent ion intensity to generate useful information from the MS3 experiment. Besides, there currently is no available software to automatically interpret and combine information from different fragmentation stages, further limiting the applicability of the MS3 approach, particularly for more complex samples.
Alternatively, electron transfer dissociation (ETD) can selectively fragment the peptide backbone while leaving the labile glycosidic bond intact. ETD is often combined with CID to comprehensively elucidate glycopeptide structure. Higher energy collisional dissociation (HCD) [22] has proven to be effective and is widely applied for characterization of various modifications [15], including glycosylation. Due to the ability of HCD to overcome the signal disruption caused by labile glycan fragmentation and to provide peptide sequencing, it can be combined with the CID spectra derived from glycoconjugate fragmentation to better determine the composition of the glycan moiety and the site of glycosylation [15]. However, many of the low mass-to-charge ratio (m/z) diagnostic glycan oxonium ions [16,23] might not be detectable in CID spectra due to the “low mass cutoff”, so-called “one third” rule, and this may result in less glycopeptide identification.
Here, we describe a novel LC–MS/MS strategy for comprehensive characterization of N- and O-linked glycopeptides using HCD with alternating normalized collisional energy (NCE) parameters. By evaluating a series of HCD parameters, we found that HCD with lower NCE values preferentially cleaves the more fragile glycan structure, whereas HCD with higher NCE values preferentially cleaves the peptide backbone. Finally, we propose an automated data analysis strategy, based on the principles of de novo peptide sequencing [24], which facilitates localization of glycosylation sites and determination of glycan structures in a high-throughput manner.
Materials and methods
Glycopeptide enrichment by ZIC–HILIC column
Albumin, fetuin, α1-acid glycoprotein, and ribonuclease B mixture (1 mg each, Sigma–Aldrich, Milwaukee, WI, USA), and the secreted proteins (10 mg) from Aspergillus niger prepared according to the previous work [25], followed by the addition of urea (8 M), were reduced with 10 mM dithiothreitol (DTT, Sigma–Aldrich) for 30 min at 60 °C, and then an appropriate amount of iodoacetamide (IAA, Sigma–Aldrich) was added in a sample to reach a concentration of 40 mM. The sample was shaken at 800 rpm at 37 °C in the dark for 1 h to alkylate. Following incubation, protein samples were digested for 3 h at 37 °C by using porcine sequencing-grade trypsin (Promega, Madison, WI, USA) at a substrate/enzyme mass ratio of 50:1.
The peptide mixture digested from albumin, fetuin, α1-acid glycoprotein, and ribonuclease B was separated by a capillary ZIC–HILIC column (10 cm length, 530 μm inner diameter [i.d.], 3.5 μm, 200 Å, Merck SeQuant, Umeå, Sweden) using an in-house-designed LC system with a flow rate of 6 μl/min (Fig. 1). The mobile phases consisted of 0.1% formic acid (FA) in acetonitrile (ACN) (A) and 0.1% FA in H2O (B). Samples were diluted to double their original volume by mobile phase A, and insoluble material was removed by centrifugation at 10,000 rpm for 1 min. Sample concentration was approximately 3 μg/μl, and a 20-μl sample was loaded onto the column. Following loading, the column was first washed twice by 99% mobile phase A for 20 min, followed by 20% mobile phase B for 10 min. Bound peptides were eluted by 80% mobile phase B for 10 min and 100% mobile phase B for 20 min. Eluted peptides were concentrated using SpeedVac.
Fig. 1.
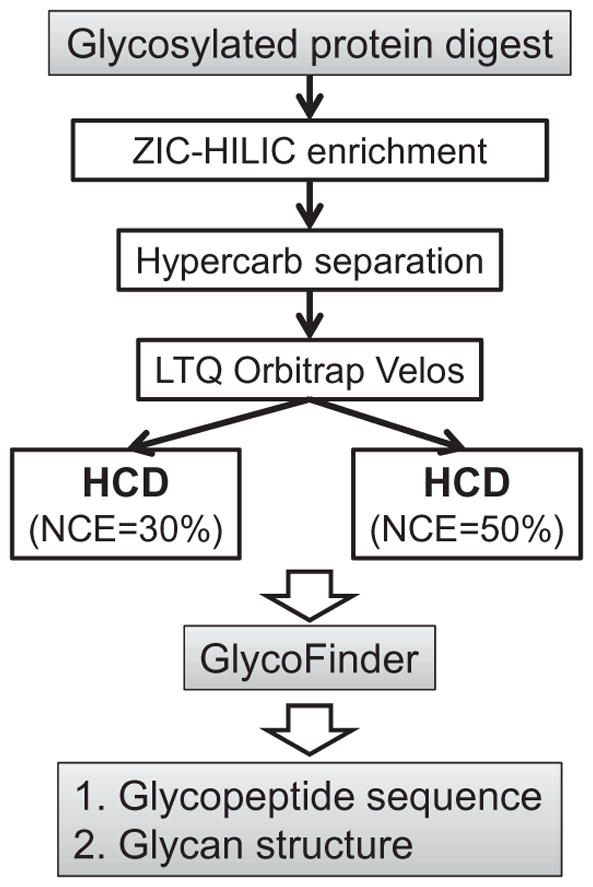
Schematic representation of the experimental workflow.
The tryptic peptides from A. niger secreted proteins were diluted to 0.535 ml with mobile phase B and loaded onto the ZIC–HILIC column (150 × 4.6 mm, 3.5 μm, 200 Å, Nest Group, cat. no. Q150449S32-155) at a flow rate of 0.6 ml/ min for 10 min with 99% mobile phase A. A linear gradient of 0 to 20% mobile phase B was then used to wash the column for 10 min, followed by a second wash of a linear gradient of 20 to 80% mobile phase B for 15 min. The enriched peptides were eluted using a linear gradient of 80 to 100% mobile phase B for 15 min, lyophilized overnight, and dialyzed for 4 h and then overnight with nanopure water using a 2-ml, 1-kDa MWCO (molecular weight cutoff) filter (GE Healthcare, cat. no. 80-6483-94) at 4 °C. The dialyzed eluent was then concentrated to dryness using SpeedVac.
Hypercarb LC–HCD MS/MS
Approximately 1.2 μg of glycopeptide mixture was reconstituted in 4 μl of nanopure water and further separated using a 30-cm-long, 75-μm-i.d. Hypercarb (porous graphitic carbon) column (Thermo Scientific, San Jose, CA, USA) and a nanoACQUITY UltraPerformance LC system (Waters, Manchester, UK). The mobile phases were the same as for ZIC–HILIC enrichment described above except that mobile phases A and B were switched. The glycopeptides were eluted using a gradient from 5 to 50% mobile phase B (0.1% FA in ACN) over 58 min at 400 nl/min directly into a Thermo LTQ Orbitrap Velos mass spectrometer (Thermo Scientific). The LTQ Orbitrap Velos was operated in a data-dependent MS/MS mode with the top eight ions (intensity) per duty cycle selected for HCD. In the initial experiments, a series of four NCEs (30, 40, 50, and 60%), for HCD were tested to determine optimal collisional energy (i.e., ideal HCD spectra). In the following experiments, the LTQ Orbitrap Velos was also operated in a data-dependent mode with the top eight ions and the HCD acquisitions automatically switching between NCEs of 30 and 50%.
Data processing
To automate and streamline the data processing for LC–MS/MS datasets with alternating HCD NCE parameters, we propose a new data analysis pipeline, GlycoFinder, which allows for high-throughput characterization of glycopeptides. First, a characteristic glycan oxonium ion list was generated using previously published data, including N-acetylhexosamine (HexNAc) (m/z = 138.05 and 204.09), hexose (Hex) (m/z = 163.06), N-acetylneuraminic acid or sialic acid (NeuAc, m/z = 274.09 and 292.10), Hex + HexNAc (m/z = 366.14), and NeuAc + Hex + HexNAc (m/z = 657.24) [16,23]. Using in-house-developed software, all tandem mass spectra were searched against the compiled list of oxonium ions with high mass accuracy (±10 ppm), and the candidate glycopeptide spectra were selected based on matched diagnostic oxonium ions. To minimize false positives derived from random matches with diagnostic ions, we applied heuristic rules (e.g., coexistence of Hex oxonium ion and HexNAc oxonium ion as the diagnostic ions of NeuAc + Hex + HexNAc). To assign possible monosaccharide composition for the candidate glycopeptides, the HCD spectra obtained with low NCE values (i.e., 30%) were analyzed in a manner similar to de novo sequencing of amino acid residues: de-isotoped spectra were analyzed based on the difference between fragment masses that can be fit within the specified mass measurement accuracy (e.g., 10 ppm) to the mass of known monosaccharide residue. HCD spectra obtained with high NCE values (i.e., 50%) were analyzed by an approach similar to USTags [24] and manually validated. The resulting data were integrated for a final glycopeptide assignment.
Results and discussion
Two-dimensional enrichment and separation of glycopeptides
Comprehensive glycopeptide characterization requires a selective and reproducible method to separate glycopeptides from non-glycopeptides. Here, we applied an offline two-dimensional (2D) LC approach to enrich and separate glycopeptides (Fig. 1). We used a ZIC–HILIC column in the first dimension to enrich glycopeptides from the complex glycoprotein digestion mixture offline, using a fast step-washing method. ZIC–HILIC resin has been demonstrated to provide highly selective and specific glycoprotein and glycopeptide enrichment [7,26,27], based mainly on the hydrophilicity difference between glycosylated and non-glycosylated protein and peptide [28]. As a second dimension of separation, we used a graphitized carbon column and coupled it directly to the mass spectrometer. Contrary to ZIC–HILIC, the separation mechanism of graphitized carbon is based mainly on hydrophobic interaction. Compared with reversed-phase columns on which glycopeptides are barely retained, a graphitized carbon column can effectively bind glycopeptides, making it ideal for online coupling to MS for glycopeptide separation. Overall, 43% of the tandem mass spectra acquired in a single LC–MS/MS analysis were assigned to glycopeptides based on oxonium ion signatures. These results indicate that relatively straightforward stepwise ZIC–HILIC enrichment can efficiently enrich glycopeptides for subsequent MS analysis.
HCD fragmentation
Fig. 2 depicts the example HCD spectra for an N-linked glycopeptide and an O-linked glycopeptide released from fetuin protein under different NCE settings. Fig. 2A (NCE = 30%) and Fig. 2B (NCE = 50%) show tandem mass spectra of an N-glycopeptide NLTK, featuring a high mannose-type glycan attached to the asparagine. Fig. 2C (NCE = 30%) and Fig. 2D (NCE = 50%) show tandem mass spectra of an O-glycopeptide VVVGPSVVAVPLPLHR, with sialylated glycans attached to the serine. In each HCD spectrum, the glycan oxonium ions were detected with high intensity, confirming the glycopeptide nature of these precursors. A series of ions from Y1 to Y7 corresponding to a laddered pattern of glycan cleavage are shown clearly in Fig. 2A with HCD under NCE = 30%, providing crucial information for reconstruction of monosaccharide residue structure of the glycan moiety. In contrast, HCD under NCE = 50% (Fig. 2B) yielded a rich series of y and b fragment ions, thereby enabling peptide sequence assignment. Similar behavior was observed for the O-linked glycopeptides. For instance, HCD with NCE = 30% of a triply charged O-linked glycopeptide (VVVGPSVVA VPLPLHR) produced mainly Y ions, with very few peptide backbone fragment ions. The presence of NeuAc at the terminal position was suggested by the presence of the oxonium ions at m/z = 274.09 and 292.10 and was also confirmed by the mass difference between the precursor ion (766.09+3) and the Y2 ion (1003.8+2), which corresponded to the mass of the assigned glycan. Extensive backbone fragments were detected when HCD with NCE = 50% was applied, and the glycopeptide sequence was validated by the y and b ions, as shown in Fig. 2D.
Fig. 2.

Examples of N-linked glycopeptide (A,B) and O-linked glycopeptide (C,D) tandem mass spectra acquired using HCD collisional energy: NCE = 30% (A,C) and NCE = 50% (B,D). B ions and Y ions (uppercase) are glycan fragmentation ions, and the designations follow the rule described in the previous publication [30]; b ions and y ions (lowercase) are peptide fragmentation ions. Monosaccharides are labeled according to the nomenclature outlined by the Consortium for Functional Glycomics (http://glycomics.scripps.edu/CFGnomenclature.pdf); squares, circles, and diamonds represent HexNAc, Hex, and NeuAc, respectively. Glycan is represented by the total number of each type of monosaccharide (i.e., 2* □ represents two HexNAc in the detected glycan). Mass-to-charge ratio (m/z) and charge state (CS) of the selected precursor ion are listed on the top of each spectrum.
Fragments from both the peptide backbone and the glycan are necessary for confident glycopeptide characterization. We tested four NCE settings (30, 40, 50, and 60%). Although the NCE setting is not distinct for the favorable fragmentation of the sugar chain or peptide chain, the general trend is obvious that glycan was preferentially fragmented at lower NCE values, whereas the peptide backbone was preferentially fragmented at higher NCE values (Fig. 3). Hence, using these complementary fragmentation regimes, one can obtain both peptide and glycan fragments needed for structural characterization of glycopeptides.
Fig. 3.

Normalized intensity of N- and O-linked glycopeptide fragment ions plotted for different HCD collisional energies: (A) peptide plus monosaccharide ions for N-linked glycopeptides; (B) peptide plus glycopeptide ions for O-linked glycopeptides; (C) singly charged peptide and y ions derived from the N-linked glycopeptides; (D) doubly charged peptide and y ions corresponding to the O-linked glycopeptide.
Normalized intensities of different fragment ions were plotted over a range of HCD NCE values (Fig. 3) according to the published method [23]. Fig. 3A and C show averaged fragment intensities of three N-linked glycopeptides. All three N-linked glycopeptides have the same amino acid backbone sequence (NLTK) derived from RNase B, but with different glycans attached: 2 HexNAc + 7 Hex, 2 HexNAc + 6 Hex, and 2 HexNAc + 5 Hex. The intensities of the same fragment ion from each N-linked glycopeptides were averaged. Then, the averaged values of different fragment ions were normalized using the highest fragment ion intensity as 100%. Fig. 3B and D show averaged fragment intensities of two O-linked glycopeptides (VVVGPSVVAVPLPLHR and IVGQPSIPGGPVR) derived from fetuin. For N-linked glycopeptides, at lower NCE values (i.e., 30%), no peptide ions or backbone fragment ions were detected (Fig. 3A). With increased NCE, the relative intensities of peptide ion or backbone fragment ions were also increased, reaching a maximum value at NCE = 50%; at even higher NCE (i.e., 60%), their relative intensities were either comparable or slightly decreased. Similar observations were made for O-linked glycopeptides (Fig. 3B). Conversely, the relative intensities of Y series ions were at their highest value for NCE = 30%. The Y ions disappeared when NCE reached 50%. These findings suggest that the use of alternating HCD NCE values provides the optimal solution for comprehensive structural characterization of glycopeptides; lower NCE values such as 30% can be chosen to majorly fragment glycans, and higher NCE values such as 50% can be chosen to majorly fragment glycopeptide backbones. Although the NCE parameters for favorable fragmentation of glycans or peptides are not distinct, they fall in a close range.
LC–MS/MS analysis for glycopeptides using alternating HCD NCE parameters
The LC–MS/MS analyses were performed on the glycopeptides enriched from a standard protein mixture using alternating HCD collisional energies, NCE = 30 and 50%. A data analysis pipeline, GlycoFinder, was developed in-house to enable automated glycopeptide identification and characterization. The spectra resulting from HCD with low NCE values provided rich information in the form of oxonium ions and monosaccharide neutral loss and were used to generate a list of candidate glycopeptides. Accurate mass information from HCD spectra increased the confidence of the assignment. In addition, based on the detected y-ion ladders, GlycoFinder calculated the peptide mass by subtracting the mass of a glycan from the mass of a glycopeptide. This information was then combined with peptide sequence information derived from the NCE = 50% HCD spectra.
In this study, 38 N- and O-linked glycopeptides (eight glycosylation sites) derived from fetuin, α-1 acid glycoprotein, and RNase B were confidently identified (see Supplementary Table 1 in online supplementary material). Interestingly, three N-linked glycopeptides (one glycosylation site) derived from bovine α2-macroglobulin were also identified. α2-Macroglobulin was reported as the mitogenic component of crude fetuin preparations and most likely existed as a small contamination in the purchased bovine fetuin standard [29]. No glycopeptides from ovalbumin were identified, likely because of missed cleavage. Ovalbumin contains only one potential N-glycosylation site at amino acid residue 293. If the tryptic cleavage site is missed in the neighborhood of the glycosylation site, or if trypsin fails to act efficiently on highly posttranslationally modified protein, the resulting glycopeptide could be too large for efficient LC–MS detection. The identified glycosylation sites are all confirmed in UniProt (http://www.uniprot.org), and glycan compositions are all confirmed in GlycoSuiteDB (http://glycosuitedb.expasy.org/glycosuite/glycodb).
Application to the biological complex sample secretome from A. niger
Preliminary data suggest that HCD with alternating NCE parameters has also been successfully applied to the complex biology sample, ZIC–HILIC-enriched secretome from A. niger. Fig. 4 shows the MS/MS spectrum of the N-linked glycopeptide TFADGFVSIVETHAASN*GSMSEQYDK from glucan 1,4-α-glucosidase. The enzyme is responsible for successive hydrolysis of α-(1→4)-linked glucose residue from nonreducing ends of the glycan chain. Under low NCE settings, the glycan structure was determined to be 8 Hex + 2 HexNAc based on the mass difference between the precursor ion (4492.8401) and peptide (2790.2442), observable characteristic oxonium ions for HexNAc (m/z = 204.09) and Hex+HexNAc (m/z = 366.14) [16], and ladder peaks in the mass spectra for Hex and HexNAc (Fig. 4A). Under high NCE settings, rich fragment ladder ions from peptide backbone sequence were identified and the detected glycopeptide was further confirmed as TFADGFVSIVETHAASNGSMSEQYDK (Fig. 4B). The glycosylation site was localized at Asn419, the sole Asn where N-linked glycosylation can occur within the peptide sequence. The glycosylation site at Asn419 was also reported in previous work [25], but the glycan structure is identified only here.
Fig. 4.

Identification of N-linked glycopeptide from A. niger secretome (AMYG_ASPNG glucoamylase) using HCD with alternating NCE parameters: (A) HCD analysis under low NCE settings; (B) HCD analysis under high NCE settings.
The details for high-throughput analysis of glycoproteins from the secretome of A. niger and the complete glycoprotein list will be published in a separate article. In addition, our future work will also incorporate the evaluation of false discovery rate for the identified glycopeptides.
Conclusions
We have demonstrated that HCD with alternating NCE parameters can effectively generate fragment ions of the glycan chain and the peptide backbone. Mass of the peptide and monosaccharide composition of the glycan can be derived from HCD tandem mass spectra with lower NCE values, whereas the peptide sequence can be derived from HCD tandem mass spectra with higher NCE. These results suggest that the characterization of the glycopeptides using the described approach provides a unique platform for rapid and comprehensive analysis of site-specific glycosylation in complex mixtures.
Supplementary Material
Acknowledgments
This work was supported by funds from Environmental Molecular Science Laboratory (EMSL) intramural research projects and EMSL capability development projects, the U.S. Department of Energy (DOE) Office of Biological and Environmental Research, and the National Institutes of Health’s National Center for Research Resources (grant RR018522). Portions of this research were supported by the Department of Energy Office of Biological and Environmental Research Genome Sciences Program under the Panomics project. We thank T. Clauss for mass spec instrument calibration. This research was performed in the W. R. Wiley EMSL, a national scientific user facility sponsored by the U.S. Department of Energy’s Office of Biological and Environmental Research and located at the Pacific Northwest National Laboratory. The Pacific Northwest National Laboratory is operated by the Battelle Memorial Institute for the U.S. Department of Energy under contract DE-AC05-76RLO-1830.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.ab.2014.01.003.
Footnotes
Abbreviations used: ZIC–HILIC, zwitterionic hydrophilic interaction chromatography; LC, liquid chromatography; MS, mass spectrometry; MS/MS, tandem MS; CID, collisionally induced dissociation; ETD, electron transfer dissociation; HCD, higher energy collisional dissociation; m/z, mass-to-charge ratio; NCE, normalized collisional energy; DTT, dithiothreitol; IAA, iodoacetamide; i.d., inner diameter; FA, formic acid; ACN, acetonitrile; HexNAc, N-acetylhexosamine; Hex, hexose; NeuAc, N-acetylneuraminic acid or sialic acid.
References
- 1.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 2.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 3.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: Direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci USA. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 5.Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Glycosylation and the immune system. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]
- 6.Dell A, Morris HR. Glycoprotein structure determination mass spectrometry. Science. 2001;291:2351–2356. doi: 10.1126/science.1058890. [DOI] [PubMed] [Google Scholar]
- 7.Mysling S, Palmisano G, Hojrup P, Thaysen-Andersen M. Utilizing ion-pairing hydrophilic interaction chromatography solid phase extraction for efficient glycopeptide enrichment in glycoproteomics. Anal Chem. 2010;82:5598–5609. doi: 10.1021/ac100530w. [DOI] [PubMed] [Google Scholar]
- 8.Peterman SM, Mulholland JJ. A novel approach for identification and characterization of glycoproteins using a hybrid linear ion trap/FT–ICR mass spectrometer. J Am Soc Mass Spectrom. 2006;17:168–179. doi: 10.1016/j.jasms.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Neue K, Mormann M, Peter-Katalinic J, Pohlentz G. Elucidation of glycoprotein structures by unspecific proteolysis and direct nanoESI mass spectrometric analysis of ZIC–HILIC-enriched glycopeptides. J Proteome Res. 2011;10:2248–2260. doi: 10.1021/pr101082c. [DOI] [PubMed] [Google Scholar]
- 10.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 11.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Anal Chem. 2003;75:5628–5637. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 12.Larsen MR, Cordwell SJ, Roepstorff P. Graphite powder as an alternative or supplement to reversed-phase material for desalting and concentration of peptide mixtures prior to matrix-assisted laser desorption/ionization–mass spectrometry. Proteomics. 2002;2:1277–1287. doi: 10.1002/1615-9861(200209)2:9<1277::AID-PROT1277>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 13.Wang L, Aryal UK, Dai ZY, Mason AC, Monroe ME, Tian ZX, Zhou JY, Su D, Weitz KK, Liu T, Camp DG, Smith RD, Baker SE, Qian WJ. Mapping N-linked glycosylation sites in the secretome and whole cells of Aspergillus niger using hydrazide chemistry and mass spectrometry. J Proteome Res. 2012;11:143–156. doi: 10.1021/pr200916k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling, and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 15.Scott NE, Parker BL, Connolly AM, Paulech J, Edwards AVG, Crossett B, Falconer L, Kolarich D, Djordjevic SP, Hojrup P, Packer NH, Larsen MR, Cordwell SJ. Simultaneous glycan–peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the Nlinked glycoproteome of Campylobacter jejuni. Mol Cell Proteomics. 2011;10:M000031–MCP201. doi: 10.1074/mcp.M000031-MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mayampurath AM, Wu Y, Segu ZM, Mechref Y, Tang HX. Improving confidence in detection and characterization of protein N-glycosylation sites and microheterogeneity. Rapid Commun Mass Spectrom. 2011;25:2007–2019. doi: 10.1002/rcm.5059. [DOI] [PubMed] [Google Scholar]
- 17.Yin X, Bern M, Xing Q, Ho J, Viner R, Mayr M. Glycoproteomic analysis of the secretome of human endothelial cells. Mol Cell Proteomics. 2013;12:956–978. doi: 10.1074/mcp.M112.024018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wuhrer M, Catalina MI, Deelder AM, Hokke CH. Glycoproteomics based on tandem mass spectrometry of glycopeptides. J Chromatogr B. 2007;849:115–128. doi: 10.1016/j.jchromb.2006.09.041. [DOI] [PubMed] [Google Scholar]
- 19.Leymarie N, Zaia J. Effective use of mass spectrometry for glycan and glycopeptide structural analysis. Anal Chem. 2012;84:3040–3048. doi: 10.1021/ac3000573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woodin CL, Hua D, Maxon M, Rebecchi KR, Go EP, Desaire H. GlycoPep Grader: a Web-based utility for assigning the composition of N-linked glycopeptides. Anal Chem. 2012;84:4821–4829. doi: 10.1021/ac300393t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lohmann KK, von der Lieth CW. GlycoFragment and GlycoSearchMS: Web tools to support the interpretation of mass spectra of complex carbohydrates. Nucleic Acids Res. 2004;32:W261–W266. doi: 10.1093/nar/gkh392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-energy C-trap dissociation for peptide modification analysis. Nat Methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 23.Segu ZM, Mechref Y. Characterizing protein glycosylation sites through higher-energy C-trap dissociation. Rapid Commun Mass Spectrom. 2010;24:1217–1225. doi: 10.1002/rcm.4485. [DOI] [PubMed] [Google Scholar]
- 24.Shen YF, Tolic N, Hixson KK, Purvine SO, Anderson GA, Smith RD. De novo sequencing of unique sequence tags for discovery of post-translational modifications of proteins. Anal Chem. 2008;80:7742–7754. doi: 10.1021/ac801123p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang L, KAryal U, Dai Z, Mason AC, Monroe ME, Tian ZX, Zhou JY, Su D, Weitz KK, Liu T, Camp DG, Smith RD, Baker SE, Qian WJ. Mapping N-linked glycosylation sites in the secretome and whole cells of Aspergillus niger using hydrazide chemistry and mass spectrometry. J ProteomeRes. 2012;11:143–156. doi: 10.1021/pr200916k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wohlgemuth J, Karas M, Jiang W, Hendriks R, Andrecht S. Enhanced glycoprofiling by specific glycopeptide enrichment and complementary monolithic nano-LC (ZIC–HILIC/RP18e)/ESI–MS analysis. J Sep Sci. 2010;33:880–890. doi: 10.1002/jssc.200900771. [DOI] [PubMed] [Google Scholar]
- 27.Calvano CD, Zambonin CG, Jensen ON. Assessment of lectin and HILIC based enrichment protocols for characterization of serum glycoproteins by mass spectrometry. J Proteomics. 2008;71:304–317. doi: 10.1016/j.jprot.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 28.Alpert AJ. Hydrophilic-interaction chromatography for the separation of peptides, nucleic-acids, and other polar compounds. J Chromatogr. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 29.Libby P, Raines EW, Cullinane PM, Ross R. Analysis of the mitogenic effect of fetuin preparations on arterial smooth-muscle cells: the role of contaminant platelet-derived growth-factor. J Cell Physiol. 1985;125:357–366. doi: 10.1002/jcp.1041250302. [DOI] [PubMed] [Google Scholar]
- 30.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in Fab–MS/MS spectra of glycoconjugates. Glycoconj J. 1988;5:397–409. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.