

Abstract
Aberrant glycosylation has been observed for decades in essentially all types of cancer, and is now well established as an indicator of carcinogenesis. Mining the glycome for biomarkers, however, requires analytical methods that can rapidly separate, identify, and quantify isomeric glycans. We have developed a rapid-throughput method for chromatographic glycan profiling using microfluidic chip-based nanoflow liquid chromatography (nano-LC)/mass spectrometry. To demonstrate the utility of this method, we analyzed and compared serum samples from epithelial ovarian cancer cases (n = 46) and healthy control individuals (n = 48). Over 250 N-linked glycan compound peaks with over 100 distinct N-linked glycan compositions were identified. Statistical testing identified 26 potential glycan biomarkers based on both compositional and structure-specific analyses. Using these results, an optimized model was created incorporating the combined abundances of seven potential glycan biomarkers. The receiver operating characteristic (ROC) curve of this optimized model had an area under the curve (AUC) of 0.96, indicating robust discrimination between cancer cases and healthy controls. Rapid-throughput chromatographic glycan profiling was found to be an effective platform for structure-specific biomarker discovery.
Keywords: Chip nano-LC/MS, Cancer biomarker, Glycan isomer separation, Rapid-throughput, Structure-specific profiling, Serum glycan
1. Introduction
Glycosylation is an important determinant of protein function, yet it is highly sensitive to its biochemical environment. Major biological changes such as cancer have been repeatedly associated with aberrant glycosylation in humans [1,2]. These alterations in turn modulate many cancer-related processes, including apoptosis [3,4], angiogenesis [5,6], growth factor receptor binding [7,8], integrin–cadherin function [9,10], etc. The glycome, thus, serves as a rich source of potential biomarkers for cancer and other diseases [11].
Global profiling of human serum glycans has already identified potential biomarkers for several types of cancer [12–24]. However, many of these studies focus on compositional glycan profiling. In contrast, structure-specific glycan profiling has the potential to uncover more robust glycan biomarkers with higher specificity than compositional profiling alone. For example, changes in the glycosidic linkages of single monosaccharide residues have been associated with Alzheimer’s disease and pancreatic cancer [25,26]. Additionally, since each glycan composition can comprise multiple glycan structures, structure-specific glycan profiling provides a significantly larger set of potential biomarkers [27,28].
In order to gain structure-specific information about the glycome, analytical methods for isomer differentiation and characterization must be applied. Tandem mass spectrometry has historically been used to differentiate between certain targeted glycan linkages [23,29,30]; however, this typically requires extra derivatization steps (commonly, permethylation) to render glycans amenable to analysis and furthermore provides only partial information about possible isomers. More recently, ion mobility spectroscopy has been used to attain partial separation of glycan isomers [22,31]. Chromatographic separation, however, has been the most universally successful method of isomer-specific glycan profiling to date [19–21,32–34].
Chromatographic glycan profiling utilizes isomer-sensitive stationary phases to chromatographically separate complex glycan mixtures. In contrast to tandem MS-centric methods, chromatographic glycan profiling can be performed on native glycans with minimal sample processing [19,35]. In particular, MS/MS structural elucidation as well as linkage-specific glycosidase digestions have previously shown chip-based porous graphitized carbon (PGC) nano-LC to be highly effective at separating isomeric oligosaccharides, glycans, and glycopeptides with high chromatographic resolution and retention time reproducibility [36–40]. LC provides a second dimension of separation that complements our previously-developed strategy for compositional glycan profiling by high-resolution MS. By coupling together reproducible isomer-sensitive LC with accurate mass MS, glycan isomers can be separated and rapidly identified according to both retention time and exact mass [33,34,36–38,41,42].
Chip-based nano-LC/MS has proven extremely effective for the global separation of serum glycans [19,43]. Nano-LC/MS offers significantly improved sensitivity over conventional LC/MS or MALDI-MS [16,17]. In addition, nano-ESI generally produces lower energy ions and therefore yields less in-source fragmentation than MALDI. Integration of these features within a microfluidic chip vastly simplifies analysis while providing unparalleled retention time reproducibility [19,44]. Coupling chip-based nano-LC with a time-of-flight (TOF) MS detector imparts the added benefits of high mass accuracy and dynamic range of detection [45–48].
Some groups have previously reported that porous graphitized carbon can separate not only glycan isomers, but also alpha and beta anomers [34]. One common sample preparation technique is to chemically reduce native N-glycans, removing the possibility of anomerization and simplifying analysis of the resulting chromatogram [33,34,41,43]. However, the high chromatographic resolution and retention time reproducibility exhibited by the chip-based nano-LC format enable a technological rather than chemical solution to this problem – using a combination of accurate mass and retention time, native glycan isomer peaks, including anomers as well as regioisomers, may be easily compared and quantified across many chromatographic runs [19]. Additionally, analysis of native glycans sidesteps the additional sample handling, extended sample cleanup, and inevitable chemical artifacts associated with chemical derivatization strategies. As a result, native glycans can be isolated and analyzed with less sample processing and greater quantitative precision than reduced or otherwise-derivatized glycans, making them the ideal analyte for biomarker applications.
We have developed a rapid-throughput method for comprehensive, isomer-specific chromatographic profiling of native serum glycans and applied it to glycan biomarker discovery. Chip-based porous graphitized carbon nano-LC/MS was used to quickly separate and quantify native, underivatized N-glycans. Serum samples from epithelial ovarian cancer cases and healthy control individuals were profiled and compared both by overall compositional abundance and in relation to specific isomers. Statistical tests were performed to detect significant differences in cancer cases vs. healthy controls as well as determine the discriminatory power of potential glycan biomarkers. Rapid-throughput chromatographic glycan profiling was shown to be a powerful platform for glycan biomarker discovery, providing rapid yet detailed characterization and quantitation of large sample sets.
2. Materials and methods
2.1. Acquisition of human sera
Human sera were acquired from (a) Sigma–Aldrich, for method development and reproducibility studies; and (b) the Gynecological Oncology Group (GOG) tissue bank, for cancer biomarker studies. All GOG sera, including both cancer cases and healthy controls, were collected using a standardized protocol approved by the institutional review board (IRB) of each participating institution.
GOG sera originated from females that had been diagnosed with epithelial ovarian cancer (cancer cases, n = 46) as well as normal, healthy females (controls, n = 48). There were four samples from early stage cancer cases (Stage I, n = 1; Stage II, n = 3) and 42 from late stage cancer cases (Stage III, n = 34; Stage IV, n = 8). As the number of early stage cancer cases was very limited, analysis by stage was not performed in this biomarker discovery set. To minimize potential confounding effects, epithelial ovarian cancer histology was kept uniform (serous and papillary serous), and cases and controls were age-matched by 5-year blocks (40–45, 46–50, 51–55, 56–60, and 61–65 years). Samples were blinded for processing, analysis, and data extraction.
2.2. Enzymatic release of N-glycans
N-glycan release and associated processing steps were performed according to previously published rapid-throughput procedures developed by Kronewitter et al. [49]. To denature the serum proteins and facilitate enzymatic N-glycan release, 50 μL of serum were added to an equal volume of aqueous 200 mM ammonium bicarbonate and 10 mM dithiothreitol solution. The mixture was thermally denatured by alternating between a 100 °C and 25 °C water bath for six cycles of 20 s each. Next, 2.0 μL (or 1000 U) of peptide N-glycosidase F (New England Biolabs) were added and the mixture was incubated in a microwave reactor (CEM Corporation) for 10 min at 20 watts. Finally, 400 μL of cold ethanol were added and the mixture was chilled at −80 °C for 1 h in order to precipitate out the deglycosylated proteins. Following centrifugation, released N-glycans were isolated in the supernatant fraction and dried in vacuo.
2.3. N-glycan enrichment with graphitized carbon SPE
N-glycan enrichment was performed according to previously published rapid-throughput procedures developed by Kronewitter et al. [49]. Released N-glycans were purified by graphitized carbon solid-phase extraction using an automated GX-274 ASPEC liquid handler (Gilson). Graphitized carbon cartridges (150 mg, 4.0 mL, Grace Davison) were washed with 3.0 mL of 80% acetonitrile and 0.10% trifluoroacetic acid (v/v) in water, followed by conditioning with 6.0 mL of pure water. Aqueous N-glycan solutions (200 μL) were loaded onto the cartridge and then washed with 7.0 mL of pure water at a flow rate of approximately 1 mL/min in order to remove salts and buffer. Serum N-glycans were eluted with 8.0 mL of 40% acetonitrile and 0.05% trifluoroacetic acid (v/v) in water. Samples were dried in vacuo.
2.4. Chromatographic separation and MS analysis of the serum N-glycome
Samples were reconstituted in water and analyzed using an Agilent HPLC-Chip/Time-of-Flight (Chip/TOF) MS system comprising an autosampler (maintained at 6 °C), capillary pump, nano pump, HPLC-Chip/MS interface, and the Agilent 6210 TOF MS detector. The chip used consisted of a 9 × 0.075 mm i.d. enrichment column and a 43 × 0.075 mm i.d. analytical column, both packed with 5 μm porous graphitized carbon as the stationary phase, with an integrated nano-ESI spray tip. For each sample, 1.0 μL (corresponding to 200 nL of serum) was loaded onto the enrichment column and washed with a solution of 3.0% acetonitrile and 0.1% formic acid (v/v) in water at 4.0 μL/min. A rapid glycan elution gradient was delivered at 0.4 μL/min using solutions of (A) 3.0% acetonitrile and 0.1% formic acid (v/v) in water, and (B) 90.0% acetonitrile and 0.1% formic acid (v/v) in water, at the following proportions and time points: 5% to 32.8% B, 0 min to 13.3 min; and 32.8% to 35.9% B, 13.3 min to 16.5 min. Remaining non-glycan compounds were flushed out with 100% B at 0.8 μL/min for 5 min. Finally, the analytical column was re-equilibrated with 5% B at 0.8 μL/min for 10 min, while the enrichment column was re-equilibrated with 0% B at 8 μL/min for 10 min. The drying gas temperature was set at 325 °C with a flow rate of 4 L/min (2 L of filtered nitrogen gas and 2L of filtered dry compressed air).
MS spectra were acquired in positive ionization mode over a mass range of m/z 600–2000 with an acquisition time of 1.5 s per spectrum. Mass correction was enabled using reference masses of m/z 622.029, 922.010, 1221.991, and 1521.971 (ESI-TOF Calibrant Mix G1969-85000, Agilent Technologies).
MS/MS spectra were acquired in the positive ionization mode over a mass range of m/z 100–3000 with an acquisition time of 1.5 s per spectrum. Following an MS scan, precursor compounds were automatically selected for MS/MS analysis by the acquisition software based on ion abundance and charge state (z = 2, 3, or 4) and isolated in the quadrupole with a mass bandpass FWHM (full width at half maximum) of 1.3 m/z. Collision energies for CID fragmentation were calculated for each precursor compound based on the following formula:
where Vcollision is the potential difference across the collision cell.
Data for all 94 samples were acquired over a continuous period of 55 h, at a rate of 35 min per run. To minimize possible bias due to injection order and/or instrumental drift, samples were blinded and injected in randomized order, using the same solvents, over the course of a single instrument session.
3. Results and discussion
3.1. Nano-LC/MS method reproducibility
To supplement previous studies on the reproducibility of the serum processing steps [49], the reproducibility of the nano-LC/MS analysis was tested. N-glycans were released from a commercially-bought serum standard. Fig. 1 shows overlaid total ion chromatograms (TICs) of ten replicate injections from the same serum N-glycan sample. In order to quantify the run-to run reproducibility, TICs were analyzed by a computer algorithm in which every single X–Y coordinate (n = 628) of the TIC function was compared to its corresponding X–Y coordinate in every other TIC function (n = 10), and the distance between each was recorded. From this data, average errors in the X axis (retention time) and the Y axis (intensity) were calculated. Average retention time error was only 1.67 s ± 0.06 s, while average peak intensity error was 2.43% ± 0.12%, representing a significant improvement over previous methods for chromatographic separation of N-glycans [19]. The propagated error resulting from combined uncertainties due to serum processing [49] and nano-LC/MS analysis was calculated for statistically significant glycans (Table 1) and found to be much lower (on average, 6.85%) than detected biological differences (i.e. fold change) between cancer cases and healthy controls. Thus, the method was deemed appropriate for glycan profiling and biomarker discovery applications.
Fig. 1.
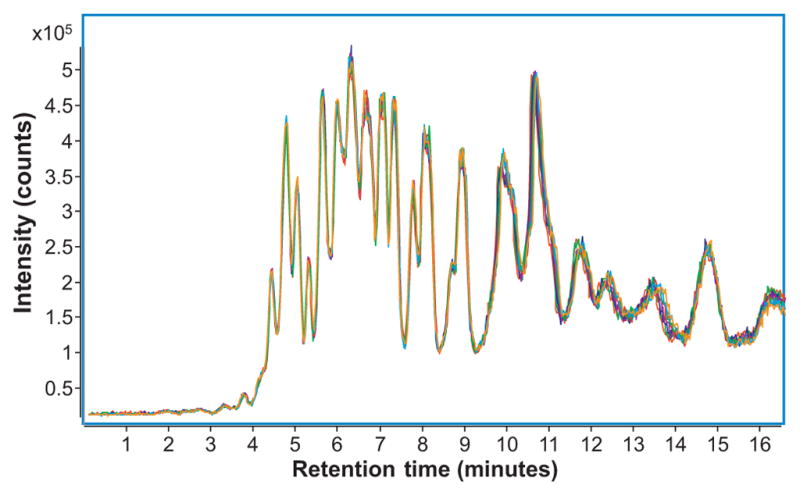
Overlaid total ion chromatograms (TICs) for ten replicate injections of serum N-glycans isolated from commercially-bought serum. Each injection corresponded to 200 nL of serum.
Table 1.
Serum N-glycans that are differentially expressed in epithelial ovarian cancer cases vs. healthy control individuals.
| Monosaccharide composition | Relative abundance (%) | Fold change | T-test | ||||
|---|---|---|---|---|---|---|---|
|
|
|
||||||
| Hex | HexNAc | Fuc | NeuAc | Control | Cancer | (Control to cancer) | (p-Values) |
| High mannose (and core) | |||||||
| 3 | 2 | 0.521 | 0.319 | −1.63 | 1.18 × 10−8 | ||
| 4 | 2 | 0.695 | 0.563 | −1.23 | 6.60 × 10−6 | ||
| 5 | 2 | 1.10 | 0.889 | −1.24 | 1.02 × 10−4 | ||
| 6 | 2 | 1.29 | 1.06 | −1.21 | 2.05 × 10−4 | ||
| 7 | 2 | 0.630 | 0.504 | −1.25 | 5.63 × 10−5 | ||
| 8 | 2 | 1.33 | 1.10 | −1.20 | 7.47 × 10−5 | ||
| Complex/hybrid (undecorated) | |||||||
| 5 | 3 | 0.246 | 0.175 | −1.41 | 2.59 × 10−5 | ||
| 4 | 4 | 0.707 | 0.515 | −1.37 | 6.19 × 10−6 | ||
| 6 | 3 | 0.222 | 0.144 | −1.55 | 3.09 × 10−11 | ||
| 5 | 5 | 0.769 | 0.535 | −1.44 | 1.52 × 10−10 | ||
| Complex/hybrid (fucosylated and/or sialylated) | |||||||
| 4 | 3 | 1 | 0.355 | 0.278 | −1.28 | 3.24 × 10−4 | |
| 4 | 4 | 1 | 3.17 | 2.41 | −1.31 | 4.34 × 10−5 | |
| 6 | 3 | 1 | 0.0156 | 0.00556 | −2.80 | 1.17 × 10−6 | |
| 4 | 4 | 1 | 0.637 | 0.558 | −1.14 | 2.46 × 10−4 | |
| 5 | 4 | 1 | 2.03 | 1.29 | −1.58 | 6.22 × 10−8 | |
| 4 | 5 | 1 | 0.875 | 0.660 | −1.32 | 9.84 × 10−6 | |
| 5 | 5 | 1 | 0.807 | 0.605 | −1.33 | 3.42 × 10−5 | |
| 5 | 4 | 1 | 1 | 4.05 | 3.40 | −1.19 | 2.99 × 10−5 |
| 6 | 6 | 1 | 0.00857 | 0.00234 | −3.66 | 7.62 × 10−5 | |
| 5 | 5 | 2 | 1 | 0.517 | 0.210 | −2.46 | 5.56 × 10−11 |
| 5 | 6 | 4 | 0.0372 | 0.00434 | −8.56 | 4.47 × 10−12 | |
| 6 | 5 | 1 | 2 | 0.808 | 2.05 | 2.54 | 3.77 × 10−8 |
| 7 | 6 | 1 | 1 | 0.0365 | 0.0826 | 2.27 | 6.02 × 10−5 |
| 7 | 6 | 1 | 2 | 0.103 | 0.346 | 3.36 | 1.15 × 10−9 |
All glycans listed show statistically significant differences in abundance (p < 6.33 × 10−4). Glycan compositions were assigned based on accurate mass. Abundance was calculated relative to the total glycan abundance. Fold change is a measure of the change in glycan abundances between healthy controls and cancer cases. Positive fold changes denote the ratio of cancer to control, whereas negative fold changes denote the ratio of control to cancer.
3.2. Detection and identification of N-glycans
In order to obtain N-glycan profiles of each serum sample, commercially-available computerized algorithms first extracted a generalized list of compound peaks in the sample and then identified the N-glycan compositions by accurate mass.
Raw LC/MS data was filtered with a signal-to-noise ratio of 5.0 and parsed into a series of extracted ion chromatograms (XICs) using the Molecular Feature Extractor algorithm included in the MassHunter Qualitative Analysis software (Version B.03.01, Agilent Technologies). Using expected isotopic distribution and charge state information, XICs were combined to create extracted compound chromatograms (ECCs) representing the summed signal from all ion species associated with a single compound (e.g. the doubly protonated ion, the triply protonated ion, and all associated isotopologues). Thus, each individual ECC peak could be taken to represent the total ion count associated with a single distinct compound.
Each ECC peak was matched by accurate mass to a glycan library. Our laboratory has developed theoretical glycan mass libraries that cover all possible complex, hybrid, and high-mannose glycan compositions based on known biological synthesis pathways and glycosylation patterns [50]. Deconvoluted masses of each ECC peak were compared against theoretical glycan masses using a mass error tolerance of 20 ppm. As the sample set originated from human serum, only glycan compositions containing hexose (Hex), N-acetylhexosamine (HexNAc), fucose (Fuc), and N-acetylneuraminic acid (NeuAc) were considered. These parameters confined the number of potential glycan masses to a finite list of possibilities which, combined with the high resolution and mass accuracy of the TOF MS, enabled automated detection of glycans with a false discovery rate of 0.6% (calculated as the proportion of possible glycan masses, ±20 ppm, within the total MS mass range).
On average, our nano-LC/MS method was able to identify over 250 N-linked glycan compound peaks with over 100 distinct N-linked glycan compositions. Each of the identified compositions included two or more peaks corresponding to either structural and/or linkage isomers (regioisomers) or, in some cases, anomeric isomers. Fig. 2 shows ECCs of the serum N-glycans identified in a commercially-bought serum standard. Separation of the different N-glycan types may be easily observed – truncated complex/hybrid glycans (blue) eluted first, followed by high mannose (green) and fucosylated complex/hybrid glycans (red). Complex/hybrid glycans exhibiting sialylation (purple) or sialylation/fucosylation (orange) eluted last. Highly-sialylated glycans with up to three or four sialic acid residues were detected by MS and confirmed by MS/MS; however, they were excluded from analysis due to extremely poor peak shapes and chromatographic resolution. Previous research [51] as well as our own experiments with the PGC chip indicate that this issue may be resolved by modification of solvent pH or ionic strength with minimal effect on MS sensitivity. Since tri- and tetra-sialylated species have previously been associated with cancer [52–55], improved separation of highly sialylated glycan species will be a significant focus for future refinement of the present method.
Fig. 2.

(a) Extracted compound chromatograms (ECCs) of N-glycans found in a commercially-bought serum; and (b) a magnified view of a short segment of the glycan elution profile, showing the high sensitivity and resolution achieved by nano-LC separation. Colors denote different glycan classes – high mannose (high Man); complex/hybrid undecorated (C/H); complex/hybrid fucosylated but not sialylated (C/H Fuc); complex/hybrid sialylated but not fucosylated (C/H Sia); and complex/hybrid fucosylated and sialylated (C/H Sia & Fuc). Structures shown are merely putative assignments based on previously published structural characterization of common serum glycans [40]. (For interpretation of the references to color in the artwork, the reader is referred to the web version of the article.)
A cursory examination of Fig. 2a shows that approximately three-quarters of the glycan signal originated from a set of only about 30 high-abundance glycans. However, a closer look at the zoomed-in view provided by Fig. 2b reveals that hundreds of low-abundance glycans were also detected simultaneously. Despite the extreme dynamic range of serum glycans, our nano-LC/MS method was able to detect, identify, and precisely quantify glycans with chromatographic peak abundances spanning five orders of magnitude.
3.3. Glycosylation changes in epithelial ovarian cancer
Overarching changes in the glycosylation machinery were detected by grouping together glycans of similar composition or structure and analyzing them as a correlated set. Groupings were based on three criteria: (1) N-glycan type; (2) fucosylation and/or sialylation; and (3) degree of branching. In order to minimize the effects of any potential sample processing or injection errors, abundances were calculated relative to the total ion abundance of all N-glycans in a particular nano-LC run; i.e. relative abundance. In order to determine whether differences between cancer cases and healthy controls were significant, standard unpaired, two-tailed T-tests were performed [56].
To compare different N-glycan types, glycans were grouped into complex, hybrid, and high mannose types. Fig. 3a shows the average relative abundances and standard errors associated with each glycan type in cancer cases vs. healthy controls. When compared to controls, sera from cancer cases contained significantly higher abundances of complex type glycans (p = 4.09 × 10−6), significantly lower abundances of hybrid type glycans (p = 4.80 × 10−4), and significantly lower abundances of high mannose type glycans (p = 5.13 × 10−6).
Fig. 3.

(a) Average relative abundances and standard errors associated with complex, hybrid, and high mannose type N-glycans; (b) average relative abundances and standard errors associated with the sialylated, fucosylated, and undecorated (complex type) glycans; and (c) average relative abundances and standard errors associated with mono-, bi-, tri-, tetra-, and penta-antennary complex type glycans in cancer cases (striped pink, n = 48) and controls (checkered blue, n = 46). For statistically significant differences between cancer cases and controls, T-test p-values are shown. Note that error bars are present for all data, though extremely small. (For interpretation of the references to color in the artwork, the reader is referred to the web version of the article.)
Terminal glycosyltransferases (such as sialyltransferases or fucosyltransferases) are often overexpressed in cancerous cells, leading to differential expression of fucose and sialic acid [57–61]. Thus, global sialylation and fucosylation were compared in cancer cases vs. healthy controls. Fig. 3b shows the average relative abundances and standard errors associated with the sialylated, fucosylated, and undecorated (complex type) glycans in cancer cases vs. healthy controls. Since some glycans were both sialylated and fucosylated, the total of these relative abundances is greater than 100%. When compared to controls, sera from cancer cases contained significantly higher abundances of sialylated glycans (p = 7.56 × 10−4) and significantly lower abundances of undecorated glycans (p = 8.03 × 10−7). These results are consistent with previous research and support evidence that sialyltransferases are upregulated with epithelial ovarian cancer [57,58].
Increased glycan branching is another hallmark of cancer, particularly β(1,6)-GlcNAc branching [62,63]. The degrees of glycan branching in cancer cases and healthy controls are compared in Fig. 3c. Average relative abundances and standard errors associated with mono-, bi-, tri-, tetra-, and penta-antennary complex type glycans are shown. When compared to controls, sera from cancer cases contained significantly higher abundances of biantennary (p = 0.0302) and tetraantennary (p = 0.0304) complex type glycans. The relatively low significance of these p-values is likely influenced by statistical interference from other factors such as sialylation/fucosylation (or lack thereof).
3.4. Glycan mass profiling for compositional biomarker discovery
While grouped analysis of the serum N-glycome provides clues about biochemical changes to the glycosylation pathway, more detailed information is typically required for sensitive and specific detection of disease states. For example, previous studies have linked ovarian cancer to significant increases in certain fucosylated serum glycoproteins [64,65]. Serum N-glycome profiling could uncover cancer-related changes in specific fucosylated glycans associated with these glycoproteins. Conversely, association of specific serum glycans with ovarian cancer might suggest the involvement of specific glycoproteins that are known to display these glycans.
Specific changes to the serum N-glycan profile were uncovered by separately considering each individual glycan composition. As before, T-tests were performed on each individual composition in order to determine whether differences between cancer cases and healthy controls were significant. Not all glycans were detected above the limit of quantitation in all samples; thus, glycans were only considered for statistical testing if they were above the limit of quantitation in more than one third of the samples from at least one of the two groups. Adjusting for multiple comparisons (n = 79) using a simple Bonferroni correction, the critical p-value at a 5% significance level was set at 6.33 × 10−4, below which the difference between cancer cases and healthy controls was taken to be significant.
Fig. 4 shows the average relative abundances and standard errors associated with the top ten most abundant glycan compositions in cancer cases vs. healthy controls. When compared to controls, sera from cancer cases contained significantly lower abundances of Hex5HexNAc4FucNeuAc (p = 2.99 × 10−5) and Hex4HexNAc4Fuc (p = 4.34 × 10−5) but significantly higher abundances of Hex6HexNAc5FucNeuAc2 (p = 3.77 × 10−8). Statistically significant differences between cancer cases and healthy controls were also evident for less-abundant glycan compositions. When compared to controls, sera from cancer cases showed significantly different abundances (p < 6.33 × 10−4) of 24 glycan compositions, including high mannose, complex, and hybrid type glycans. These statistically significant glycans have been listed in Table 1.
Fig. 4.
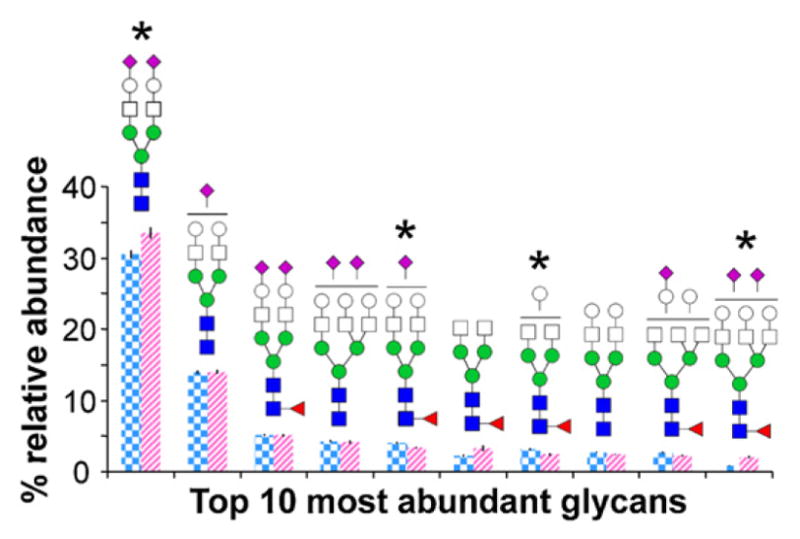
Average relative abundances and standard errors associated with the top ten most abundant N-glycan compositions in cancer cases (striped pink, n = 48) and controls (checkered blue, n = 46). Note that error bars are present for all data, though extremely small. *Denotes a statistically significant difference between cancer cases and controls. (For interpretation of the references to color in the artwork, the reader is referred to the web version of the article.)
3.5. Chromatographic glycan profiling for structure-specific biomarker discovery
The isomer separation capabilities of porous graphitized carbon have been well-established by both MS/MS and exoglycosidase-based structural studies, particularly with the chip-based nano-LC platform [36–39]. Chromatographic profiling, thus, provides structure-specific information about glycans that can be used to uncover more robust biomarkers. Identification of glycan structural isomers that exhibit statistically significant differences in abundance may provide a window into the up- and down-regulation of glycosyltransferase activity under changing biological conditions.
Our nano-LC method was able to easily separate and baseline resolve up to 7 different glycan structural isomers for each composition. Commercially-available computerized algorithms (see Section 3.2) parsed the MS chromatograms and calculated the abundances of each isomer relative to the total glycan abundance. Due to the reproducibility of the method, equivalent isomers in different samples were easily matched by retention time, enabling separate consideration of each glycan structural isomer. As before, T-tests were performed on each individual isomer in order to determine whether differences between cancer cases and healthy controls were significant. Eligibility for statistical testing was determined by the same criteria previously described, i.e. whether the isomer was detected above the limit of quantitation in more than one third of the samples from at least one of the two groups. Since there were many more glycan structures (n ~ 250) than compositions, the Bonferroni correction was recalculated. Adjusting for multiple comparisons with n = 250, the critical p-value at a 5% significance level was set at 2.00 × 10−4, below which the difference between cancer cases and healthy controls was taken to be significant.
Chromatographic glycan profiling was found in many instances to increase the specificity and power of statistical comparisons between cancer cases and healthy controls. Each glycan composition is made up of two or more glycan isomers, but not all of these isomers show statistically significant differences in cancer cases vs. healthy controls. By considering only the relevant glycan structures, chromatographic profiling enables a more focused view of the changes in the serum glycome.
For example, Fig. 5a shows the average relative abundances and standard errors associated with five structural isomers of Hex5HexNAc4FucNeuAc for cancer cases vs. healthy controls. Separate T-tests comparing the relative abundances in cancer cases vs. healthy controls of each isomer revealed that different isomers had different p-values. For the change in abundance of the specific structural isomer eluting at 10.19 min, the p-value was just 2.72 × 10−8. In contrast, for the overall composition, the p-value was 2.99 × 10−5. Thus, when focusing on specific isomers, the differences between cancer cases and healthy controls were more pronounced.
Fig. 5.
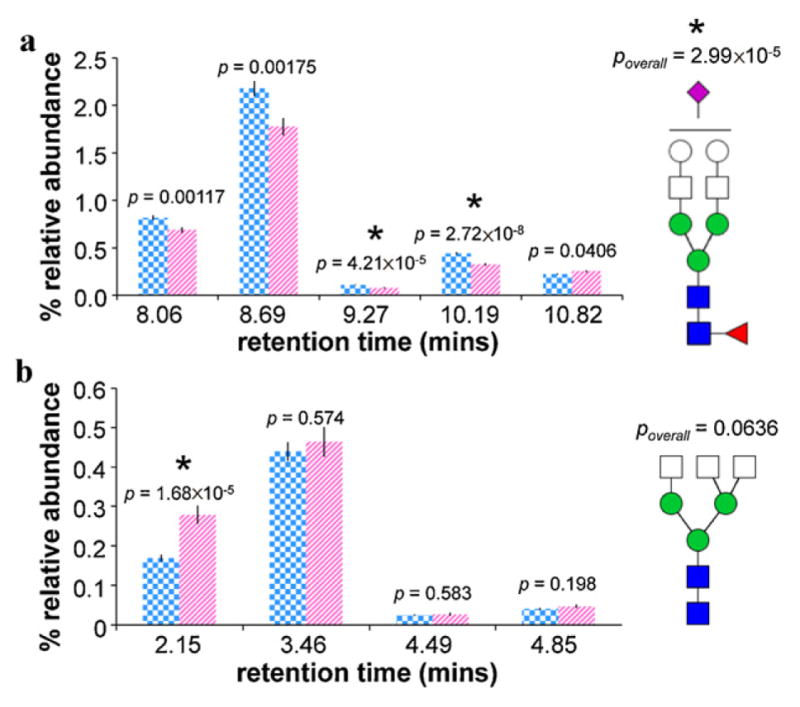
(a) Average relative abundances and standard errors associated with five structural isomers of Hex5 HexNAc4 FucNeuAc; and (b) average relative abundances and standard errors associated with four structural isomers of Hex3 HexNAc5 in cancer cases (striped pink, n = 48) and controls (checkered blue, n = 46). T-test p-values are shown for each isomer as well as for the overall composition. Note that error bars are present for all data, though extremely small. *Denotes a statistically significant difference between cancer cases and controls. (For interpretation of the references to color in the artwork, the reader is referred to the web version of the article.)
Fig. 5b shows a similar application of chromatographic glycan profiling. Average relative abundances and standard errors associated with four structural isomers of Hex3HexNAc5 are shown for cancer cases and healthy controls. For the change in abundance of the specific structural isomer eluting at 2.15 min, the p-value was just 1.68 × 10−5. In contrast, for the overall composition, the p-value was 0.0636, which is not significant. In this way, isomer-specific analysis was able to uncover a statistically significant difference between cancer cases and healthy controls that was overlooked by compositional profiling.
It should be noted that, in some instances, the glycan isomers separated by porous graphitized carbon may include alpha and beta anomers [34]. Given the exceptionally high retention time reproducibility demonstrated by this nano-LC method, detection and identification of any potential anomer peaks may easily be addressed in the future with the use of retention time and tandem MS libraries [36–38]. For instance, Fig. 6 shows the isomer-specific MS/MS spectra for two isomers of Hex5HexNAc4NeuAc eluting at 7.5 min (top) and 8.0 min (bottom). Stark differences can be seen between the two spectra – for example, m/z 274.09 (NeuAc-H2O), 292.10 (NeuAc), and 657.24 (HexHexNAcNeuAc) are relatively more abundant in the bottom spectra, while m/z 528.19 (Hex2HexNAc) and 1567.56 (Hex4HexNAc3NeuAc) are relatively more abundant in the top spectrum. While these differences do not permit de novo identification of the isomer, the unique fragmentation patterns do serve as an identifier. Future tandem MS libraries that match these mass spectral fingerprints with specific glycan structures may eventually assist in the rapid identification of glycan isomers.
Fig. 6.
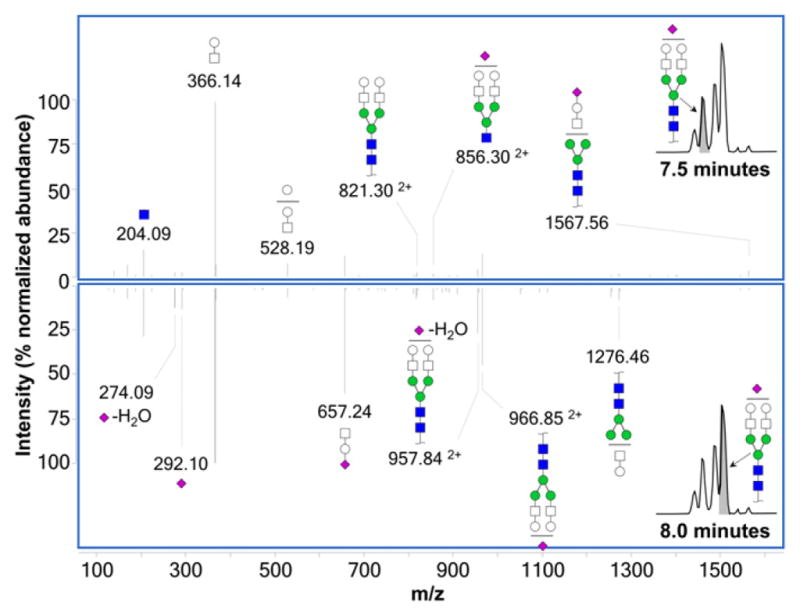
Isomer-specific MS/MS spectra (taken in positive mode) for two isomers of biantennary monosialylated glycan Hex5 HexNAc4 NeuAc eluting at 7.5 min (top) and 8.0 min (bottom), showing unique fragmentation patterns for each. All ions are singly protonated unless marked otherwise.
Table 2 summarizes the instances in which chromatographic glycan profiling was found to be more effective than compositional profiling alone. In all, 12 instances were found where differences between cancer cases and healthy controls were more pronounced with specific glycan structural isomers than with glycan compositions. Of these, nine statistically significant differences between cancer cases and healthy controls were best described by a single isomer, whereas the other three statistically significant differences were best described by the summed abundance of two related isomers (identified in Table 2 by retention times). In two instances, chromatographic profiling uncovered a statistically significant difference that compositional profiling did not have the power to detect.
Table 2.
Comparison of T-test p-values associated with compositional vs. structure-specific profiling of selected serum N-glycans.
| Monosaccharide composition | Compositional | Structure-specific | Ret. time | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| Hex | HexNAc | Fuc | NeuAc | Fold change | T-test | Fold change | T-test | (min) |
| 3 | 5 | 1.20 | 6.37 × 10−2 | 1.65 | 1.68 × 10−5 | 2.21 | ||
| 5 | 5 | −1.37 | 6.19 × 10−6 | −1.49 | 2.81 × 10−6 | 3.68 | ||
| 7 | 2 | −1.25 | 5.63 × 10−5 | −1.24 | 1.37 × 10−5 | 4.24 and 4.77 | ||
| 8 | 2 | −1.20 | 7.47 × 10−5 | −1.20 | 3.64 × 10−5 | 4.28 and 4.85 | ||
| 6 | 2 | −1.21 | 2.05 × 10−4 | −1.27 | 7.57 × 10−5 | 4.60 | ||
| 5 | 3 | −1.41 | 2.60 × 10−5 | −1.50 | 1.75 × 10−6 | 4.61 | ||
| 6 | 3 | −1.55 | 3.09 × 10−11 | −1.56 | 7.21 × 10−12 | 5.20 and 5.82 | ||
| 5 | 2 | −1.24 | 1.01 × 10−4 | −1.40 | 2.55 × 10−5 | 5.86 | ||
| 4 | 4 | 1 | −1.31 | 4.34 × 10−5 | −1.36 | 1.42 × 10−5 | 6.04 | |
| 6 | 5 | 1 | 1 | 1.32 | 1.20 × 10−3 | 1.39 | 1.83 × 10−4 | 8.58 |
| 5 | 6 | 4 | −8.56 | 4.47 × 10−12 | −8.74 | 2.51 × 10−12 | 9.55 | |
| 5 | 4 | 1 | 1 | −1.20 | 2.99 × 10−5 | −1.36 | 2.72 × 10−8 | 10.22 |
All glycans listed show statistically significant differences in the abundance of at least one structural isomer (p < 2.00 × 10−4). Glycan compositions were assigned based on accurate mass. For most compositions, relative abundances in cancer and control cases were given previously in Table 1. Retention times identify specific glycan structural isomers that were found to be most statistically significant. Fold change is a measure of the change in glycan abundances between healthy controls and cancer cases. Positive fold changes denote the ratio of cancer to control, whereas negative fold changes denote the ratio of control to cancer.
3.6. Statistical evaluation of potential glycan biomarkers
Although statistically significant correlations can provide valuable insights into the biological role of glycosylation in epithelial ovarian cancer, they are not necessarily indicative of clinical utility [66–68]. In order for a compound (or combination of compounds) to be useful as a diagnostic indicator, it (or they) must exhibit both sensitivity (i.e. a low false negative rate) and specificity (i.e. a low false positive rate).
Receiver operating characteristic (ROC) curves graphically plot the sensitivity vs. specificity of a binary diagnostic test as discrimination threshold is varied. The area under the curve (AUC) of a test’s ROC curve is used as an indicator of the accuracy of the test, with an AUC of 1.0 corresponding to a perfectly accurate test while an AUC of 0.5 corresponds to a completely uninformative test [69]. In order to evaluate the usefulness of the most statistically significant glycans (Table 1) as epithelial ovarian cancer biomarkers, ROC curves were generated for each glycan, and AUCs calculated for each ROC curve [70].
Table 3 lists the AUCs of the ten glycans with the greatest statistical significances when comparing cancer cases vs. healthy controls (i.e. those with the ten lowest p-values). The AUCs ranged between 0.77 and 0.87, indicating that (for this sample set) any of the ten individual glycans listed could be applied to diagnostic discrimination of cancer cases and healthy controls.
Table 3.
Receiver operating characteristic (ROC) curve areas and T-test p-values for the ten serum N-glycans considered for inclusion in an optimized composite score for diagnostic discrimination of cancer cases and controls.
| Monosaccharide composition | Fold change | T-test | ROC | Inclusion in composite | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Hex | HexNAc | Fuc | NeuAc | (Control to cancer) | (p-Values) | (AUC) | |
| 5 | 6 | 4 | −8.56 | 4.47 × 10−12 | 0.87 | ✓ | |
| 6 | 3 | −1.55 | 3.09 × 10−11 | 0.85 | ✓ | ||
| 5 | 5 | 2 | 1 | −2.46 | 5.56 × 10−11 | 0.87 | ✓ |
| 5 | 5 | −1.44 | 1.52 × 10−10 | 0.77 | ✓ | ||
| 7 | 6 | 1 | 2 | 3.36 | 1.15 × 10−9 | 0.85 | ✓ |
| 3 | 2 | −1.63 | 1.18 × 10−8 | 0.83 | |||
| 6 | 5 | 1 | 2 | 2.54 | 3.77 × 10−8 | 0.82 | ✓ |
| 5 | 4 | 1 | −1.58 | 6.22 × 10−8 | 0.82 | ||
| 6 | 3 | 1 | −2.80 | 1.17 × 10−6 | 0.79 | ✓ | |
| 4 | 4 | −1.37 | 6.19 × 10−6 | 0.86 | |||
| Composite index: | −2410 | 2.46 × 10−20 | 0.96 | ||||
Glycan compositions were assigned based on accurate mass. Fold change is a measure of the change in glycan abundances between healthy controls and cancer cases. Positive fold changes denote the ratio of cancer to control, whereas negative fold changes denote the ratio of control to cancer. AUC denotes area under the ROC curve (out of a maximum of 1.0000).
To assess the complementary performance of the potential glycan biomarkers in Table 3, a composite score was derived using multivariate logistic regression [71–73]. First, the abundance of each glycan in each sample was normalized so that low- and high-abundance glycans would be weighted equally. To calculate the normalized abundance, the following formula was used:
where ni,j is the normalized abundance of glycan j in sample i, ri,j is the relative abundance of glycan j in sample i, r̄cancer,j is the mean abundance of glycan j among all cancer cases, and r̄cancer,j is the mean abundance of glycan j among all controls. Then, the composite score was calculated according to the following formula:
where ci is the composite score of sample i, Aj is the regression coefficient for glycan j, S is the set of all glycans in Table 3, and ni,j is (as before) the normalized abundance of glycan j in sample i. To select an optimal set of glycans that would maximize the discrimination between cancer cases and healthy controls while minimizing overfitting, a simplified logistic regression was used in which relevant glycans were each given equal weight by confining the regression coefficient Aj to either 1 (indicating that glycan j was beneficial to the index) or 0 (indicating that glycan j was detrimental or irrelevant to the index). The results of the logistic regression determined which glycans were included in or removed from the composite score.
The optimized composite score incorporated the normalized abundances of seven different glycans with ROC curve AUCs between 0.77 and 0.87 (Fig. 7(a)–(g)). However, the ROC curve of the optimized composite score had an AUC of 0.96 (Fig. 7h), demonstrating the dramatic improvement in diagnostic power provided by combining multiple glycan tests.
Fig. 7.

Individual ROC curves for (a) Hex6 HexNAc3 ; (b) Hex5 HexNAc6 Fuc4 ; (c) Hex5 HexNAc5 Fuc2 NeuAc; (d) Hex7 HexNAc6 FucNeuAc2 ; (e) Hex6 HexNAc5 FucNeuAc2 ; (f) Hex6 HexNAc3 Fuc; (g) Hex5 HexNAc5 ; and (h) the combined ROC curve for the optimized seven-glycan composite score, based on their relative abundances in cancer cases vs. controls.
The seven glycans that contributed to the optimized composite score included complex and hybrid type glycans. Five were fucosylated, three were sialylated, and two were undecorated. The remaining three glycans (out of the ten most significant) were found to be unhelpful for distinguishing between cancer cases and healthy controls and were thus excluded from the composite score. The three excluded glycans were (in order of decreasing abundance): Hex5HexNAc4Fuc, Hex4HexNAc4, and Hex3HexNAc2. Interestingly, the first two excluded glycans are associated primarily with serum immunoglobulins, while the third is simply the unmodified N-glycan core [38].
4. Conclusion
We have developed a rapid-throughput method for comprehensive, isomer-specific chromatographic profiling of serum glycans. Chip-based porous graphitized carbon nano-LC/MS was used to quickly separate and quantify native, underivatized N-glycans. In order to accommodate a biomarker discovery workflow, nano-LC gradients were optimized to minimize run time yet preserve sensitivity, reproducibility, and isomer specificity. Over 250 N-linked glycan compound peaks with over 100 distinct N-linked glycan compositions were identified from the equivalent of only 200 nL of serum. Retention times varied from run to run by only a few seconds, facilitating isomer-specific quantitation of selected glycans across all sample injections.
Application of this method to serum samples from epithelial ovarian cancer patients (n = 46) and healthy control individuals (n = 48) revealed altered glycosylation trends, while compositional and structure-specific glycan profiling each identified a number of potential biomarkers. Based on an optimized model incorporating the combined abundances of seven potential biomarkers, a ROC curve was generated with an AUC of 0.96, indicating exceptionally robust discrimination between cancer cases and healthy controls.
While compelling, it must be emphasized that these are preliminary findings that will require clinical validation in blinded, unsupervised studies before they can be developed into a clinically-applicable diagnostic test. In particular, stratification of future sample sets into early and advanced stage epithelial ovarian cancer cases will help determine the early stage detection capabilities of glycan-based biomarkers. Additionally, further structural glycan analysis will be necessary to confirm the exact structures of the significant glycan isomers identified by chromatographic profiling. Complete structural annotation of the human serum glycome is ongoing, enabling rapid future identification of significant glycan structures according to mass, retention time, and MS/MS fragmentation pattern [38].
However, for now, it seems clear that rapid-throughput chromatographic glycan profiling is an immensely powerful platform for biomarker discovery. The increased power of structure-specific glycan profiling over compositional profiling highlights the highly specific nature of the glycosylation changes that take place during carcinogenesis. Using this new methodology, we will be able to identify specific glycan structures that vary with disease, thus aiding the development of effective diagnostic tools for the detection of cancer.
Acknowledgments
Funding was provided by the National Institutes of Health (RO1GM049077 for C. B. Lebrilla), the Converging Research Center Program through the Ministry of Education, Science and Technology (2012K001505 for H. J. An), and the Ovarian Cancer Research Fund (for G. S. Leiserowitz)
References
- 1.Dube DH, Bertozzi CR. Nat Rev Drug Discov. 2005;4:477. doi: 10.1038/nrd1751. [DOI] [PubMed] [Google Scholar]
- 2.Fuster MM, Esko JD. Nat Rev Cancer. 2005;5:526. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 3.Hauptmann P, Riel C, Kunz-Schughart LA, Fröhlich KU, Madeo F, Lehle L. Mol Microbiol. 2006;59:765. doi: 10.1111/j.1365-2958.2005.04981.x. [DOI] [PubMed] [Google Scholar]
- 4.Rapoport E, Pendu J. Glycobiology. 1999;9:1337. doi: 10.1093/glycob/9.12.1337. [DOI] [PubMed] [Google Scholar]
- 5.Saito T, Miyoshi E, Sasai K, Nakano N, Eguchi H, Honke K, Taniguchi N. J Biol Chem. 2002;277:17002. doi: 10.1074/jbc.M200521200. [DOI] [PubMed] [Google Scholar]
- 6.Pili R, Chang J, Partis RA, Mueller RA, Chrest FJ, Passaniti A. Cancer Res. 1995;55:2920. [PubMed] [Google Scholar]
- 7.Duchesne L, Tissot B, Rudd TR, Dell A, Fernig DG. J Biol Chem. 2006;281:27178. doi: 10.1074/jbc.M601248200. [DOI] [PubMed] [Google Scholar]
- 8.Triantis V, Saeland E, Bijl N, Oude-Elferink RP, Jansen PLM. Hepatology. 2010;9999 doi: 10.1002/hep.23708. NA. [DOI] [PubMed] [Google Scholar]
- 9.Guo HB, Lee I, Kamar M, Akiyama SK, Pierce M. Cancer Res. 2002;62:6837. [PubMed] [Google Scholar]
- 10.Nita-Lazar M, Noonan V, Rebustini I, Walker J, Menko AS, Kukuruzinska MA. Cancer Res. 2009;69:5673. doi: 10.1158/0008-5472.CAN-08-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Drake PM, Cho W, Li B, Prakobphol A, Johansen E, Anderson NL, Regnier FE, Gibson BW, Fisher SJ. Clin Chem. 2010;56:223. doi: 10.1373/clinchem.2009.136333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.An HJ, Miyamoto S, Lancaster KS, Kirmiz C, Li B, Lam KS, Leiserowitz GS, Lebrilla CB. J Proteome Res. 2006;5:1626. doi: 10.1021/pr060010k. [DOI] [PubMed] [Google Scholar]
- 13.de Leoz MLA, An HJ, Kronewitter S, Kim J, Beecroft S, Vinall R, Miyamoto S, de Vere White R, Lam KS, Lebrilla C. Dis Markers. 2008;25:243. doi: 10.1155/2008/515318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leiserowitz GS, Lebrilla C, Miyamoto S, An HJ, Duong H, Kirmiz C, Li B, Liu H, Lam KS. Int J Gynecol Cancer. 2008;18:470. doi: 10.1111/j.1525-1438.2007.01028.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirmiz C, Li B, An HJ, Clowers BH, Chew HK, Lam KS, Ferrige A, Alecio R, Borowsky AD, Sulaimon S, Lebrilla CB, Miyamoto S. Mol Cell Proteomics. 2007;6:43. doi: 10.1074/mcp.M600171-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Bereman MS, Williams TI, Muddiman DC. Anal Chem. 2009;81:1130. doi: 10.1021/ac802262w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bereman MS, Young DD, Deiters A, Muddiman DC. J Proteome Res. 2009;8:3764. doi: 10.1021/pr9002323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang ZQ, Varghese RS, Bekesova S, Loffredo CA, Hamid MA, Kyselova Z, Mechref Y, Novotny MV, Goldman R, Ressom HW. J Proteome Res. 2010;9:104. doi: 10.1021/pr900397n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hua S, An HJ, Ozcan S, Ro GS, Soares S, DeVere-White R, Lebrilla CB. Analyst. 2011;136:3663. doi: 10.1039/c1an15093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hua S, Lebrilla C, An HJ. Bioanalysis. 2011;3:2573. doi: 10.4155/bio.11.263. [DOI] [PubMed] [Google Scholar]
- 21.Bones J, Mittermayr S, O’Donoghue N, Guttman A, Rudd PM. Anal Chem. 2010;82:10208. doi: 10.1021/ac102860w. [DOI] [PubMed] [Google Scholar]
- 22.Isailovic D, Plasencia MD, Gaye MM, Stokes ST, Kurulugama RT, Pungpapong V, Zhang M, Kyselova Z, Goldman R, Mechref Y, Novotny MV, Clemmer DE. J Proteome Res. 2011;11:576. [Google Scholar]
- 23.Alley WR, Vasseur JA, Goetz JA, Svoboda M, Mann BF, Matei DE, Menning N, Hussein A, Mechref Y, Novotny MV. J Proteome Res. 2012;11:2282. doi: 10.1021/pr201070k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adamczyk B, Tharmalingam T, Rudd PM. BBA – Gen Subjects. 2012;1820:1347. doi: 10.1016/j.bbagen.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Zhao J, Simeone DM, Heidt D, Anderson MA, Lubman DM. J Proteome Res. 2006;5:1792. doi: 10.1021/pr060034r. [DOI] [PubMed] [Google Scholar]
- 26.Reggi M, Capon C, Gharib B, Wieruszeski JM, Michel R, Fournet B. Eur J Biochem. 1995;230:503. doi: 10.1111/j.1432-1033.1995.tb20589.x. [DOI] [PubMed] [Google Scholar]
- 27.Fernandez-Rodriguez J, Dwir O, Alon R, Hansson GC. Glycoconjugate J. 2001;18:925. doi: 10.1023/a:1022208727512. [DOI] [PubMed] [Google Scholar]
- 28.Schulz BL, Sloane AJ, Robinson LJ, Sebastian LT, Glanville AR, Song Y, Verkman AS, Harry JL, Packer NH, Karlsson NG. Biochem J. 2005;387:911. doi: 10.1042/BJ20041641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prien JM, Huysentruyt LC, Ashline DJ, Lapadula AJ, Seyfried TN, Reinhold VN. Glycobiology. 2008;18:353. doi: 10.1093/glycob/cwn010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prien JM, Prater BD, Cockrill SL. Glycobiology. 2010;20:629. doi: 10.1093/glycob/cwq012. [DOI] [PubMed] [Google Scholar]
- 31.Isailovic D, Kurulugama RT, Plasencia MD, Stokes ST, Kyselova Z, Gold-man R, Mechref Y, Novotny MV, Clemmer DE. J Proteome Res. 2008;7:1109. doi: 10.1021/pr700702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wuhrer M, de Boer AR, Deelder AM. Mass Spectrom Rev. 2009;28:192. doi: 10.1002/mas.20195. [DOI] [PubMed] [Google Scholar]
- 33.Pabst M, Grass J, Toegel S, Liebminger E, Strasser R, Altmann F. Glycobiology. 2012;22:389. doi: 10.1093/glycob/cwr138. [DOI] [PubMed] [Google Scholar]
- 34.Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Anal Chem. 2007;79:5051. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 35.Wuhrer M, Koeleman CAM, Deelder AM, Hokke CH. Anal Chem. 2003;76:833. doi: 10.1021/ac034936c. [DOI] [PubMed] [Google Scholar]
- 36.Wu S, Tao N, German JB, Grimm R, Lebrilla CB. J Proteome Res. 2010;9:4138. doi: 10.1021/pr100362f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu S, Grimm R, German JB, Lebrilla CB. J Proteome Res. 2010;10:856. doi: 10.1021/pr101006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aldredge D, An HJ, Tang N, Waddell K, Lebrilla CB. J Proteome Res. 2012;11:1958. doi: 10.1021/pr2011439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hua S, Nwosu C, Strum J, Seipert R, An H, Zivkovic A, German J, Lebrilla C. Analytical and Bioanalytical Chemistry. 2012;403:1291. doi: 10.1007/s00216-011-5109-x. [DOI] [PubMed] [Google Scholar]
- 40.Hua S, An HJ. BMB Rep. 2012;45:323. doi: 10.5483/bmbrep.2012.45.6.132. [DOI] [PubMed] [Google Scholar]
- 41.Pabst M, Wu SQ, Grass J, Kolb A, Chiari C, Viernstein H, Unger FM, Altmann F, Toegel S. Carbohydr Res. 2010;345:1389. doi: 10.1016/j.carres.2010.02.017. [DOI] [PubMed] [Google Scholar]
- 42.Hashii N, Kawasaki N, Itoh S, Hyuga M, Kawanishi T, Hayakawa T. Proteomics. 2005;5:4665. doi: 10.1002/pmic.200401330. [DOI] [PubMed] [Google Scholar]
- 43.Chu CS, Niñonuevo MR, Clowers BH, Perkins PD, An HJ, Yin H, Killeen K, Miyamoto S, Grimm R, Lebrilla CB. Proteomics. 2009;9:1939. doi: 10.1002/pmic.200800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ninonuevo M, An H, Yin H, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Electrophoresis. 2005;26:3641. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 45.Fortier MH, Bonneil E, Goodley P, Thibault P. Anal Chem. 2005;77:1631. doi: 10.1021/ac048506d. [DOI] [PubMed] [Google Scholar]
- 46.Yin H, Killeen K, Brennen R, Sobek D, Werlich M, van de Goor T. Anal Chem. 2004;77:527. doi: 10.1021/ac049068d. [DOI] [PubMed] [Google Scholar]
- 47.Niñonuevo M, An H, Yin H, Killeen K, Grimm R, Ward R, German B, Lebrilla C. Electrophoresis. 2005;26:3641. doi: 10.1002/elps.200500246. [DOI] [PubMed] [Google Scholar]
- 48.Yin H, Killeen K. J Sep Sci. 2007;30:1427. doi: 10.1002/jssc.200600454. [DOI] [PubMed] [Google Scholar]
- 49.Kronewitter SR, de Leoz MLA, Peacock KS, McBride KR, An HJ, Miyamoto S, Leiserowitz GS, Lebrilla CB. J Proteome Res. 2010;9:4952. doi: 10.1021/pr100202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kronewitter SR, An HJ, de Leoz MLA, Lebrilla CB, Miyamoto S, Leiserowitz GS. Proteomics. 2009;9:2986. doi: 10.1002/pmic.200800760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pabst M, Altmann F. Anal Chem. 2008;80:7534. doi: 10.1021/ac801024r. [DOI] [PubMed] [Google Scholar]
- 52.Abd Hamid UM, Royle L, Saldova R, Radcliffe CM, Harvey DJ, Storr SJ, Pardo M, Antrobus R, Chapman CJ, Zitzmann N, Robertson JF, Dwek RA, Rudd PM. Glycobiology. 2008;18:1105. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 53.Alley WR, Madera M, Mechref Y, Novotny MV. Anal Chem. 2010;82:5095. doi: 10.1021/ac100131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu JP, Knežević A, Wang YX, Rudan I, Campbell H, Zou ZK, Lan J, Lai QX, Wu JJ, He Y, Song MS, Zhang L, Lauc G, Wang W. J Proteome Res. 2011;10:4959. doi: 10.1021/pr2004067. [DOI] [PubMed] [Google Scholar]
- 55.Vasseur JA, Goetz JA, Alley WR, Novotny MV. Glycobiology. 2012;22:1684. doi: 10.1093/glycob/cws108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markowski CA, Markowski EP. Am Stat. 1990;44:322. [Google Scholar]
- 57.Hakomori SI, Zhang Y. Chem Biol. 1997;4:97. doi: 10.1016/s1074-5521(97)90253-2. [DOI] [PubMed] [Google Scholar]
- 58.Kim Y, Varki A. Glycoconjugate J. 1997;14:569. doi: 10.1023/a:1018580324971. [DOI] [PubMed] [Google Scholar]
- 59.Dall’Olio F, Chiricolo M, Mariani E, Facchini A. Eur J Biochem. 2001;268:5876. doi: 10.1046/j.0014-2956.2001.02536.x. [DOI] [PubMed] [Google Scholar]
- 60.Miyoshi E, Moriwaki K, Nakagawa T. J Biochem. 2008;143:725. doi: 10.1093/jb/mvn011. [DOI] [PubMed] [Google Scholar]
- 61.Kuzmanov U, Jiang N, Smith CR, Soosaipillai A, Diamandis EP. Mol Cell Proteomics. 2009;8:791. doi: 10.1074/mcp.M800516-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dennis J, Laferte S, Waghorne C, Breitman M, Kerbel R. Science. 1987;236:582. doi: 10.1126/science.2953071. [DOI] [PubMed] [Google Scholar]
- 63.Abbott KL, Nairn AV, Hall EM, Horton MB, McDonald JF, Moremen KW, Dinulescu DM, Pierce M. Proteomics. 2008;8:3210. doi: 10.1002/pmic.200800157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Abbott KL, Lim JM, Wells L, Benigno BB, McDonald JF, Pierce M. Proteomics. 2010;10:470. doi: 10.1002/pmic.200900537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wu J, Xie X, Liu Y, He J, Benitez R, Buckanovich RJ, Lubman DM. J Proteome Res. 2012;11:4541. doi: 10.1021/pr300330z. [DOI] [PubMed] [Google Scholar]
- 66.Simon I, Liu Y, Krall KL, Urban N, Wolfert RL, Kim NW, McIntosh MW. Gynecol Oncol. 2007;106:112. doi: 10.1016/j.ygyno.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 67.Cramer DW, Bast RC, Berg CD, Diamandis EP, Godwin AK, Hartge P, Lokshin AE, Lu KH, McIntosh MW, Mor G, Patriotis C, Pinsky PF, Thornquist MD, Scholler N, Skates SJ, Sluss PM, Srivastava S, Ward DC, Zhang Z, Zhu CS, Urban N. Cancer Prev Res. 2011;4:365. doi: 10.1158/1940-6207.CAPR-10-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhu CS, Pinsky PF, Cramer DW, Ransohoff DF, Hartge P, Pfeiffer RM, Urban N, Mor G, Bast RC, Moore LE, Lokshin AE, McIntosh MW, Skates SJ, Vitonis A, Zhang Z, Ward DC, Symanowski JT, Lomakin A, Fung ET, Sluss PM, Scholler N, Lu KH, Marrangoni AM, Patriotis C, Srivastava S, Buys SS, Berg CD. Cancer Prev Res. 2011;4:375. doi: 10.1158/1940-6207.CAPR-10-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zweig MH, Campbell G. Clin Chem. 1993;39:561. [PubMed] [Google Scholar]
- 70.Anderson GL, McIntosh M, Wu L, Barnett M, Goodman G, Thorpe JD, Bergan L, Thornquist MD, Scholler N, Kim N, O’Briant K, Drescher C, Urban N. J Natl Cancer Inst. 2010;102:26. doi: 10.1093/jnci/djp438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mamtani M, Thakre T, Kalkonde M, Amin M, Kalkonde Y, Amin A, Kulkarni H. BMC Bioinformatics. 2006;7:442. doi: 10.1186/1471-2105-7-442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Laxman B, Morris DS, Yu J, Siddiqui J, Cao J, Mehra R, Lonigro RJ, Tsodikov A, Wei JT, Tomlins SA, Chinnaiyan AM. Cancer Res. 2008;68:645. doi: 10.1158/0008-5472.CAN-07-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gomar JJ, Bobes-Bascaran MT, Conejero-Goldberg C, Davis P, Goldberg TE. Arch Gen Psychiat. 2011;68:961. doi: 10.1001/archgenpsychiatry.2011.96. [DOI] [PubMed] [Google Scholar]