

Abstract
Neurofibromatosis 1 (NF1) patients develop multiple neurofibromas, with 8–15% of patients experiencing malignant peripheral nerve sheath tumor (MPNST) during their lifetime. Prediction of transformation, typically from plexiform neurofibroma, is clinically and histologically challenging. In this overview, following a consensus meeting in October 2016, we outline the histopathologic features and molecular mechanisms involved in the malignant transformation of neurofibromas. Nuclear atypia alone is generally insignificant. However, with atypia, loss of neurofibroma architecture, high cellularity, and/or mitotic activity >1/50 but <3/10 high power fields, the findings are worrisome for malignancy. We propose the term “atypical neurofibromatous neoplasms of uncertain biologic potential (ANNUBP)” for lesions displaying at least two of these features. This diagnosis should prompt additional sampling, clinical correlation, and possibly, expert pathology consultation. Currently such tumors are diagnosed inconsistently as atypical neurofibroma or low-grade MPNST. Most MPNSTs arising from neurofibromas are high-grade sarcomas and pose little diagnostic difficulty, although rare non-necrotic tumors with 3–9 mitoses/10 HPFs can be recognized as low-grade variants. While neurofibromas contain numerous S100 protein/SOX10-positive Schwann cells and CD34-positive fibroblasts, both components are reduced or absent in MPNST. Loss of p16/CDKN2A expression, elevated Ki67 labeling, and extensive nuclear p53 positivity are also features of MPNST that can to some degree already occur in ANNUBP. Complete loss of trimethylated histone 3 lysine 27 (H3K27me3) expression is potentially more reliable, being immunohistochemically detectable in about half of MPNSTs. Correlated clinicopathologic, radiologic, and genetic studies should increase our understanding of malignant transformation in neurofibromas, hopefully improving diagnosis and treatment soon.
Keywords: Neurofibroma, atypia, neurofibromatosis 1, malignant peripheral nerve sheath tumor, transformation
Neurofibromatosis type 1 (NF1) is an autosomal dominant condition with a birth incidence of approximately 1:3000. Its genetic basis is a loss of function germline mutation in the NF1 gene at 17q11.2. The NF1 gene encodes neurofibromin, a negative regulator of RAS-signaling related to cell survival and proliferation. 1 Disease penetrance is virtually 100% in adults with NF1, although there is great phenotypic variability in neurofibroma disease burden among patients, even those carrying identical NF1 germline mutations.
The key clinical manifestation of NF1 is occurrence of neurofibromas, and in some patients, development of malignant peripheral nerve sheath tumor (MPNST), generally from a pre-existing neurofibroma, especially the plexiform type. In NF1, neurofibromas develop following somatic loss of the remaining wildtype NF1 allele in Schwann cells, whereas somatic bi-allelic inactivation occurs in sporadic neurofibromas [1]. Additional somatic alterations, such as loss of cell cycle regulators (especially p16/CDKN2A) and polycomb repressor complex components EED and SUZ12, are commonly associated with MPNST [2–4]. The lifetime risk for development of MPNST in NF1 patients has been estimated as 8–16% in two population-based studies [5,6]. Malignant transformation can occur in childhood, but most often occurs in the 3rd to 4th decades and onwards. High-grade MPNST has the highest risk of sarcoma-specific death [7]. The 5-year overall survival is approximately 20% to 50% [8–10], and outcome is especially dismal in those with unresectable or metastatic disease [11]. Most clinicopathologic series of MPNSTs report NF1-associated MPNSTs to be approximately equally common as sporadic cases [8,11–15].
NF1-associated nerve sheath tumors include externally visible circumscribed cutaneous and subcutaneous neurofibromas that vary from few to innumerable and can evolve into infiltrative diffuse masses of varying sizes, the latter termed diffuse neurofibromas, and often containing Meissnerian corpuscles. Cutaneous and diffuse neurofibromas only rarely evolve into MPNST [16].
Neurofibromas developing in peripheral nerves or nerve trunks are termed intraneural neurofibromas. Plexiform neurofibroma is a structurally complex conglomerate of intraneural neurofibroma or a tortuous mass arising in large nerve trunk, wherein multiple fascicles are involved simultaneously. It is a typical manifestation of NF1, potentially involving almost any nerve in the body. Frequent sites include paraspinal nerves, brachial or sacral plexus, sciatic nerve, and nerves in the chest wall and neck. Most MPNSTs in patients with NF1 develop in intraneural and/or plexiform neurofibromas, although the reverse is not true, since only a small subset of these neurofibromas eventually undergo malignant transformation. This therefore presents a clinical dilemma as to how best to monitor these neurofibromas and effectively intervene at a sufficiently early stage to prevent a locally aggressive and/or metastasizing malignancy. Growing, painful, or otherwise troublesome lesions are common indications for surgical excision or diagnostic biopsy.
Volumetric MRI analysis of plexiform neurofibromas has allowed characterization of the natural history of these tumors. Most plexiform neurofibromas grow rapidly during early childhood, whereas significant plexiform neurofibroma growth in adults should raise concern for malignant transformation [17]. MRI analysis also allows for identification of nodular lesions, which are distinctly different from surrounding plexiform components (Fig.1). These nodules typically display more rapid growth compared to conventional plexiform elements in the same patient [18]. In addition, these nodular lesions are typically fluorodeoxy-glucose (FDG)-positron emission tomography (PET) avid compared to plexiform neurofibromas, which have only low-level uptake on FDG-PET. A subset of these lesions was recently described as “atypical neurofibromas”, which are considered precursor lesions for MPNST even though their incidence of malignant transformation is unknown [19]. Painful lesions, or those that demonstrate excessive growth or have concerning magnetic resonance imaging (MRI) characteristics, should undergo diagnostic biopsy or surgical excision. Atypical neurofibromatous tumors and low grade MPNSTs typically require less aggressive surgery compared to high grade MPNST, which requires wide excision with negative surgical margins for potential cure [20]. A standardized pathologic definition of nerve sheath tumors including atypical or indeterminate neural tumors, low grade-, and high-grade MPNST would greatly assist physicians in making informed treatment decisions for these lesions.
Fig. 1.
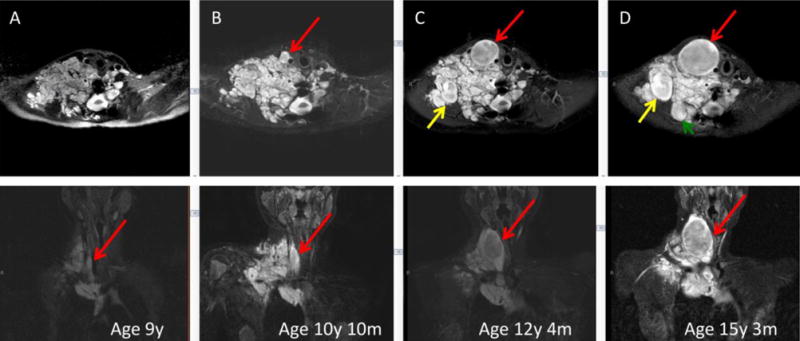
Development and growth of distinct nodules in a paraspinal retroperitoneal plexiform neurofibroma, as shown in successive MR images over a 6-year period. Three separate nodular growths are highlighted with red, yellow, and green arrows. Upper row is axial and lower sagittal projection.
In this consensus overview, based on the outcome of a meeting held at the NIH in October 5, 2016, we discuss the pathology of neurofibroma and its transformation into MPNST, in the specific context of NF1 patients. We also present recommendations for terminology and pathologic diagnosis. However, we realize that current knowledge is incomplete and further research is needed to establish more reliable diagnostic definitions based on clinicopathologic correlations, especially regarding minimal criteria for the diagnosis of malignant transformation, for example, defining mitotic counts. Additional immunohistochemical and molecular genetic data are needed to better understand and define the earliest forms of malignant transformation. While the diagnosis of neurofibroma and high-grade MPNST are generally straightforward, atypical, cellular, and mitotically active tumors pose a problem of classification in the spectrum of nerve sheath tumors potentially evolving into an MPNST and the development of a ‘risk assessment’ scheme would have clinical value.
Based on the published literature and the personal experience of the authors, we propose the following practice-based recommendations for the challenging spectrum of NF1-associated nerve sheath tumors falling between the classic examples of atypical neurofibroma and a high-grade MPNST. We discuss 3 diagnostic categories: 1) neurofibroma with cytologic atypia or hypercellularity, 2) atypical neurofibromatous tumor of uncertain biologic potential (ANNUBP), as proposed here, and 3) MPNST. The key features of these categories are summarized in Table 1.
Table 1.
Proposed Nomenclature for the Spectrum of NF1-associated Nerve Sheath Tumors
| Diagnosis | Proposed Definition |
|---|---|
| Neurofibroma (NF) | Benign Schwann cell neoplasm with thin, often wavy nuclei, wispy cell processes, and a myxoid to collagenous (‘shredded carrots’) matrix. IHC includes extensive but not diffuse S100 and SOX10 positivity and a lattice-like CD34+ fibroblastic network |
| Plexiform | NF diffusely enlarging and replacing a nerve, often involving multiple nerve fascicles, delineated by EMA+ perineurial cells |
| Neurofibroma with atypia (“Ancient neurofibroma”) | NF with atypia alone, most commonly manifesting as scattered bizarre nuclei |
| Cellular | NF with hypercellularity, but retained NF architecture and <1 mf/50 HPF |
| ANNUBP | Schwann cell neoplasm with at least 2 of 4 features: cytologic atypia, loss of neurofibroma architecture, hypercellularity, mitotic index >1/50 HPF and <3/10 HPF |
| MPNST, low-grade | Features of ANNUBP, but with mitotic index of 3–9/10 HPF and no necrosis |
| MPNST, high-grade | MPNST with at least 10 mf/10 HPF or 3–9 mf/10 HPF combined with necrosis |
ANNUBP = atypical neurofibromatous neoplasm of unknown biologic potential; loss of NF architecture = fascicular growth pattern and/or lack of CD34+ fibroblastic network; hypercellularity = ‘blue’ appearance at low magnification and nuclear overlap at high magnification; HPF = high power fields
Neurofibroma with cytologic atypia or hypercellularity
Nuclear atypia is known to be present in some sporadic and NF1-associated neurofibromas, and such tumors have been often designated as “atypical neurofibromas” [21]. There are no reliable estimates of their frequency, likely due to the marked variability in use of this terminology among pathologists. When utilized instead within the context of a neurofibroma showing worrisome features, then atypical neurofibromas have been suggested to be premalignant lesions based on their CDKN2A loss, as is recognized in MPNST, and they occasionally progress to MPNST [16]. However, there is currently no clinical evidence for imminent transition into an MPNST. For this reason, it seems reasonable that such cases can be treated relatively conservatively and even excised with positive margins, as supported by one follow-up study [22].
The presence of focal or even more pronounced nuclear atypia in neurofibroma is not worrisome for malignancy when present without increased mitotic activity in the context of classic neurofibroma architecture: haphazardly arranged S100/SOX10 positive tumor cells variably embedded within Alcian blue positive mucin and collagen aggregates resembling “shredded carrots”, as well as a lattice-like network of CD34-positive fibroblasts. Such nuclear atypia may include nuclear enlargement 2–3-fold or more, hyperchromasia, irregular chromatin distribution, and multinucleated or ‘bizarre’ forms (Fig. 2 A–C). The latter scenario (i.e., scattered bizarre nuclei in the absence of hypercellularity, mitotic activity, or loss of neurofibroma architecture) has sometimes been designated as “degenerative atypia”, similar to that in “ancient schwannoma” and other benign neoplasms; as such, the analogous term of ‘ancient neurofibroma’ could be utilized for such cases. This clearly differs from the more clinically significant use of the term “atypical neurofibroma” to indicate “I see some worrisome features, but not enough to call it malignant”. It is only this latter scenario that matters to the treating physicians of NF1 patients, since “degenerative” atypia alone, is thought to have no clinical significance. Unfortunately, there are no scientific criteria for clearly distinguishing “degenerative atypia” from the “real atypia” of malignancy or premalignancy, though as stated earlier, the former may be favored when scattered nuclear atypia is the only finding of concern. In some cases, apparently increased cellularity reflects extensive infiltration by inflammatory cells, especially histiocytes (Fig. 2D). Cellular schwannian nodules, representing well-demarcated nodular schwannoma-like areas within a neurofibroma, commonly occur in NF1-associated neurofibromas and have no known clinical significance in the absence of atypia and mitotic activity.
Fig. 2.
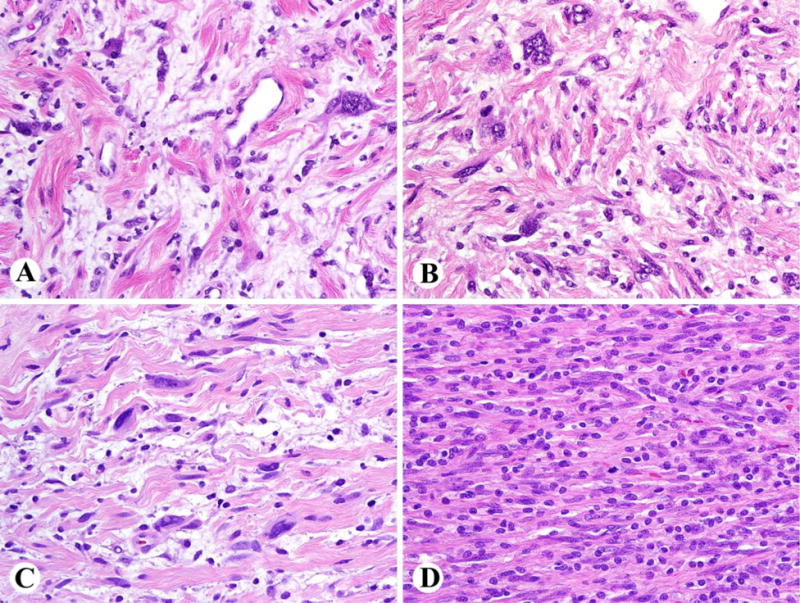
A–C. Examples of plexiform neurofibromas with nuclear atypia (‘ancient neurofibromas’), but preserved neurofibroma architecture. D. Appearance of high cellularity here is caused by extensive lymphohistiocytic infiltration, obscuring neurofibroma architecture.
Cellular neurofibroma is another term utilized to indicate rare neurofibromas wherein hypercellularity is the only worrisome finding (i.e., without mitotic activity, cytologic atypia, or loss of neurofibroma architecture) [12]. Caution is needed to ensure that the higher cellularity is not just due to lymphohistiocytic infiltration (Fig. 2D). As with neurofibromas featuring atypia alone, there are no definitive data on risk for progression to MPNST (Fig 3). However, for cases wherein hypercellularity is accompanied by atypia, loss of neurofibroma architecture, and/or mitotic activity, atypical neurofibromatous neoplasm with uncertain biologic potential (see below) or MPNST are likely better diagnostic considerations.
Fig. 3.
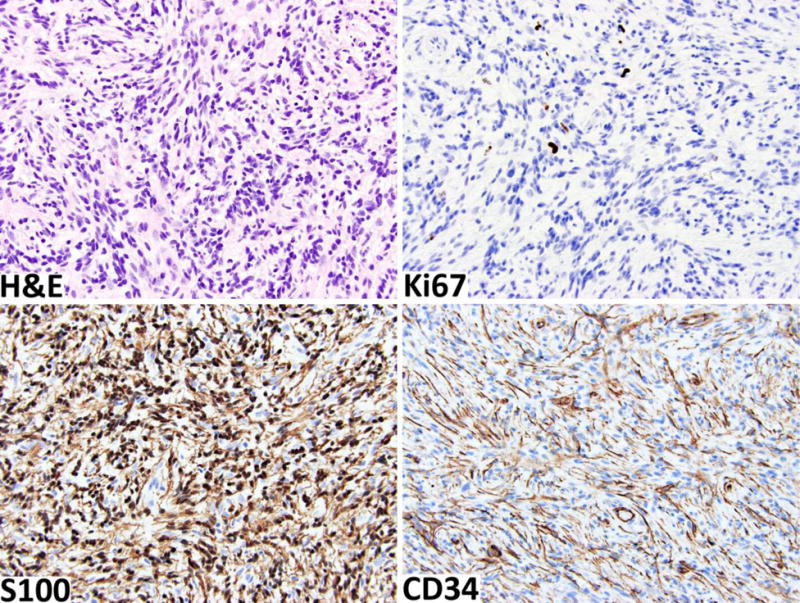
Cellular neurofibroma shows moderate cellularity with no other worrisome features. The Ki67 labeling index is < 3%. The tumor retains strong S100-positivity in the schwannian component and a well-developed CD34-positive fibroblastic network.
Immunohistochemically, low Ki67-labeling indices and low numbers of p53-positive nuclei may also be considered reassuring (Fig 3). S100 protein (cytoplasmic and nuclear) and SOX10 (nuclear) expression highlights the Schwann cell elements, while CD34 identifies a specialized fibroblastic component, the latter often forming a net-like pattern, typical of preserved neurofibroma architecture (Fig. 3).
Another caveat to consider in the diagnosis of NF1-associated nerve sheath tumors is the well-known risk of undersampling in needle biopsies. An FDG-PET CT/MRI-guided approach to obtain multiple cores of radiologically suspicious regions can enhance the accuracy of preoperative diagnosis.
Atypical neurofibromatous neoplasm with uncertain biologic potential (ANNUBP)
Neurofibromatous tumors with nuclear atypia, hypercellularity, variable loss of neurofibroma architecture (e.g., herringbone or storiform fascicular growth and/or loss of CD34 positive network), and/or mitotic activity beyond isolated mitotic figures (>1/50 HPF and <3/10 HPF) should be designated as having uncertain malignant potential when at least two of these features are present (Fig. 4). Although such tumors have been sometimes labeled as low-grade MPNST, they have been associated with mostly a low risk of recurrence and essentially no risk of metastasis [19]. Thus, a malignant designation for these tumors may lead to overly aggressive and sometimes morbid therapy, as well as potential adverse actions from insurance providers. As such, the use of the term low-grade MPNST should be discouraged in this situation, since unpublished data suggest that these lesions are not frankly malignant in the large majority of NF1-related cases. Further research on clinical correlations of such lesions and their histologic, immunohistochemical, and molecular genetic features is sorely needed.
Fig. 4.
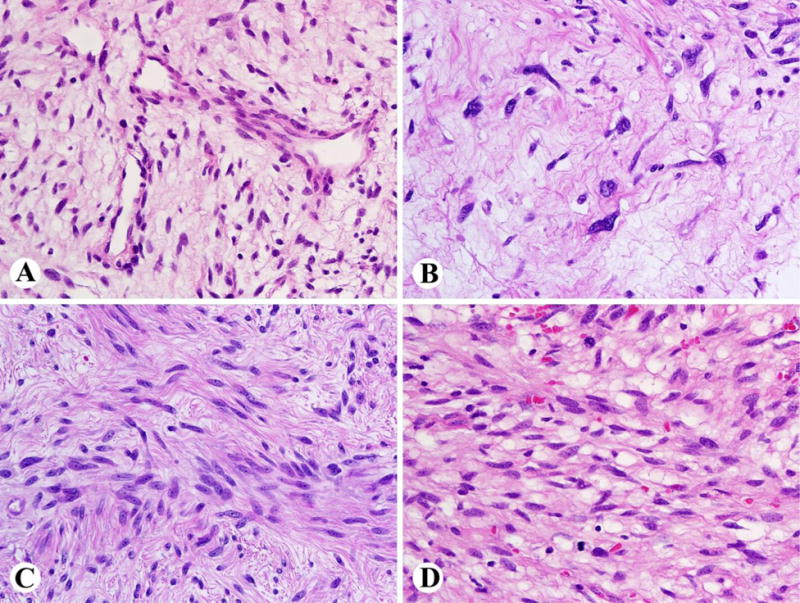
Histological appearances of atypical neurofibromatous tumors of uncertain biologic potential (ANNUBP). A. Tumor with loss of neurofibroma architecture and increased vascularity (not enough for ANNUBP by itself). B. Prominent nuclear atypia without high cellularity or mitoses (not enough for ANNUBP by itself). C. Increased cellularity with a fascicular pattern (loss of neurofibroma architecture), but without mitotic activity. D. Increased cellularity and mitotic activity.
In addition to pooling together a subset of cases previously called atypical neurofibroma or low-grade MPNST, the designation ANNUBP is also applicable to small biopsies, in which atypical features of concern are noted but criteria for MPNST are not met. As such, this designation does not represent a distinct diagnostic entity but rather a clinical situation, which requires additional sampling, clinical correlation, and possibly an expert pathology consultation or clinical follow-up for resolution. Although immunohistochemistry can be helpful to assess neurofibroma architecture and proliferative rate, based on current data, it cannot be used to definitively determine the biological potential.
For the application of ancillary studies, we first emphasize that there is no single immunohistochemical or genetic test to define malignancy status in atypical neurofibromatous tumors. However, the factors that can be somewhat useful, in addition to histologic evaluation, include variable to complete loss of S100 protein/SOX10 expression and loss of the CD34-positive fibroblastic network (Fig. 5 A, B).
Fig. 5.
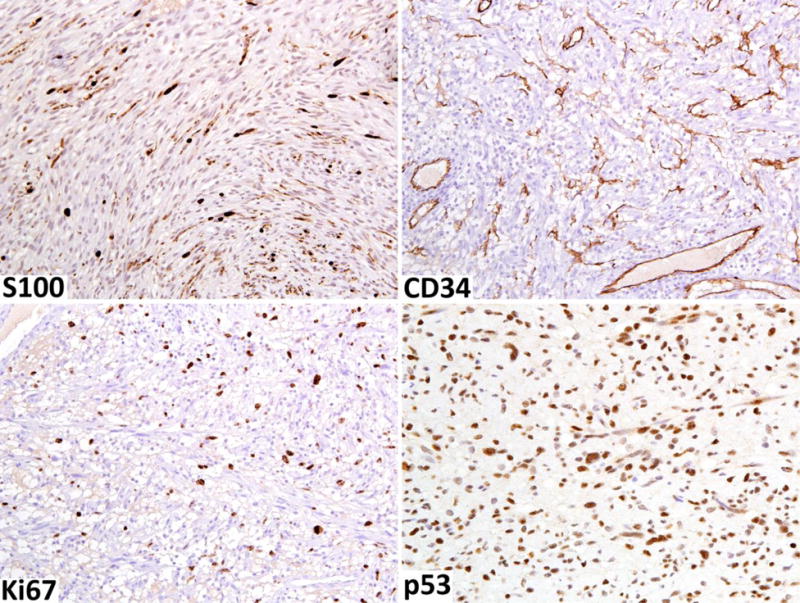
Immunohistochemical findings of neurofibroma transitioning into low-grade MPNST A. S100 protein-positive Schwann cells are greatly reduced. B. CD34-positive fibroblastic network is also reduced. C. Ki67-labeling index is elevated, reaching close to 10%. D. Moderate numbers of nuclei are p53-positive.
The Ki67 (MIB1)-labeling index can also be helpful in assessing nerve sheath tumors in NF1 patients highlighting proliferative hot-spots (Fig. 5C). While ordinary neurofibromas and atypical neurofibromas typically have low labeling indices (<2–5%), higher proliferation rates (>10%) may assist detection of MPNST arising in a neurofibroma.
Loss of the CDKN2A locus at 9p21, encoding the cell cycle regulator p16 among others, characterizes one of the earliest steps in the malignant transformation of neurofibromas [23,24]. Complete immunohistochemical loss of nuclear p16, a common finding in MPNST, can also be detected in atypical and even conventional neurofibromas indicating that it may be an early change in malignant progression, but by itself is not sufficient to prove malignancy. 12
The p53 protein (TP53 gene product) often accumulates in the tumor cell nuclei due to its deregulation or mutation in various malignancies (Fig. 5D). Although expression in high numbers of lesional cells may support a malignant process, there are no convincing data to indicate that early malignant transformation of neurofibroma could be detected by a mildly elevated p53 expression pattern. Furthermore, extensive positivity may be detected in cellular schwannomas representing another diagnostic pitfall [14,25].
Malignant peripheral nerve sheath tumor (MPNST)
Malignant peripheral nerve sheath tumors in NF1 patients usually fulfill the criteria of a high-grade sarcoma with prominent nuclear atypia, showing mitotic indices >10 /10 HPFs and, often, tumor necrosis. However, those rare non-necrotic cases with lower mitotic indices between 3 and 9 mitoses/10 HPFs (1 HPF defined as 0.2 mm2) can be classified as low-grade MPNST. They are most often encountered in the setting of an MPNST arising in a neurofibroma and intermingling with elements of pre-existent neurofibroma.
MPNSTs often show a fascicular or storiform fibrosarcoma-like pattern with enlarged nuclei and variable degrees of nuclear pleomorphism (Fig 6A). Perivascular sparing patterns (i.e., tumor viability limited to perivascular regions), zonal necrosis and glomeruloid vascular proliferations resembling those seen in glioblastoma are common (Fig. 6 B, C). Heterologous rhabdomyosarcoma-like or osteocartilaginous differentiation occur in a minority of cases, and an angiosarcoma-like phenotype is a rare event (Fig. 6D).
Fig. 6.
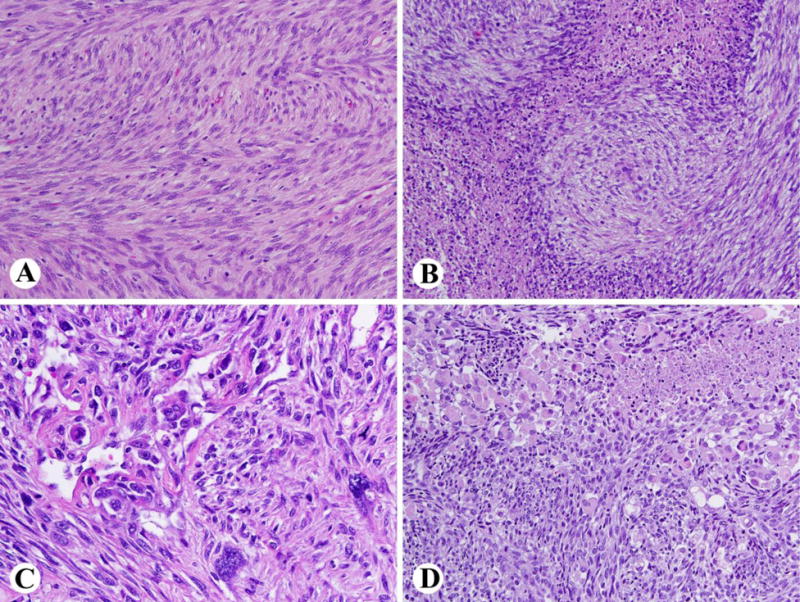
Typical findings in a high-grade MPNST. A. Fibrosarcoma-like highly cellular spindled tumor with mitotic activity. B. Geographic necrosis with a perivascular sparing pattern (i.e., perivascular collections of tumor remain viable). C. Complex vascular proliferation and nuclear pleomorphism. D. Rhabdomyosarcomatous differentiation is seen in a minority of cases.
Immunohistochemically, most MPNSTs are negative for all nerve sheath stains, while others are positive for S100 protein and/or SOX10 in only a small subset of the tumor cells. It is also likely that residual Schwann cells of the pre-existing neurofibroma contribute to the S100 protein/SOX10 positivity of MPNSTs (Fig. 7A). A minority of MPNSTs maintain a schwannian phenotype with extensive S100 protein and SOX10-positivity (Fig. 7B). Other Schwann cell markers, such as GFAP, CD57 (Leu7), and collagen IV similarly suffer from low sensitivities and/or specificities. Loss of the CD34-positive fibroblastic network encountered in neurofibromas can be a helpful clue to the diagnosis of MPNST [13]. However, some MPNST retain a CD34-positive fibroblast-like element. Loss of p16 expression related to losses in the CDKN2A genes is also a typical finding (Fig. 7C). However, because this change can predate morphologic transformation into MPNST, it cannot be used a sole marker of malignancy. The loss of expression of trimethylated histone 3 at lysine residue 27 (H3K27me3), resulting from loss-of-function mutations in EED and SUZ12 genes, is a recently discovered marker of MPNST [2–4]. Such trimethylation is typically ubiquitous in neurofibroma and ANNUBP, but is often lost in MPNST (Fig. 6D). The frequency of H3K27me3-loss has varied between 30–90%, and by some studies, has been more frequent in sporadic and radiation-associated MPNSTs than NF1-associated MPNSTs [26–28]. As with other “loss of expression markers”, positive internal control staining in endothelial, lymphoid or other normal cells is required for validity of staining (Fig. 7D).
Fig. 7.
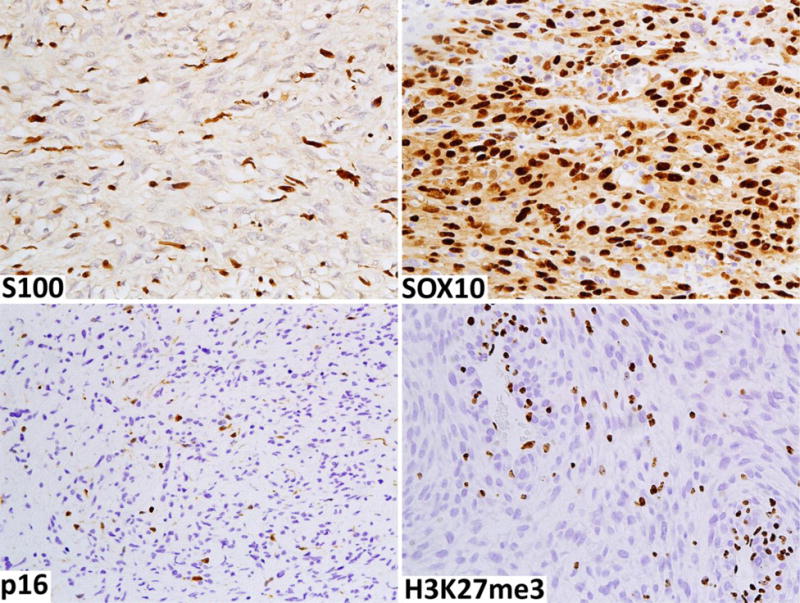
Immunohistochemical findings typical of a high-grade MPNST. A. Only a small number of slender S100-positive Schwann cells are present mostly representing elements of residual neurofibroma. B. This example shows Schwannian differentiation as demonstrated by extensive SOX10 positivity in tumor cells. C. Tumor cells have a loss of p16 expression, with non-neoplastic cells positive. D. There is a loss of expression of trimethylated lysine 27 in histone 3 (H3K27me3) in tumor nuclei. Lymphoid cells and endothelia serve as internal positive controls.
While some studies have suggested that this loss is specific to MPNST among sarcomas and potentially useful in tumor type-diagnosis, others have found frequent losses in other tumors, especially synovial sarcoma, with which there is often a morphologic overlap. A mosaic pattern of expression (loss in some but not all tumor cells) is considerably less specific than complete loss [29]. Therefore, we do not recommend using the mosaic H3K27me3 pattern of loss as evidence of MPNST outside the typical histology and clinical context. Genetic data suggests that loss of H3K27me3 does not occur in cellular schwannoma, and therefore this marker may also be useful in the differential diagnosis between cellular schwannoma and those MPNSTs that have lost this marker [30].
Comments on differential diagnosis of MPNST
Accurate evaluation of biologic potential of nerve sheath tumors requires precise tumor classification as the criteria for atypia and malignancy vary among tumor categories. For example, cellular schwannomas that are mitotically active are not of similar concern as are neurofibromatous tumors with mitotic activity. Malignant neoplasms that can mimic MPNST include desmoplastic and metastatic melanomas, as well as synovial sarcoma, especially when associated with a nerve. As these tumors among others can be S100 protein-positive, such positivity in a sarcomatoid neoplasm is not sufficient evidence for MPNST. Because these problems are less prevalent in the setting of NF1-associated tumors, we do not discuss them in detail but here refer to a review article [31].
Summary
Despite progress in our understanding of MPNST molecular genetics and increased awareness of histologic features associated with clinical malignancy, early detection of malignant transformation in NF1-associated neurofibromas remains difficult and the diagnosis of pathologically worrisome but indeterminate tumor types remains a major challenge. We have therefore recommended a new category designated as “atypical neurofibromatous neoplasm of uncertain biologic potential” for the tumors that show some worrisome features of malignant transformation, but histologically fall short of MPNST. It is our hope that this will eventually be replaced by more precise terminology and diagnostic criteria as greater clinical experience and more accurate biomarkers are characterized. Progress in this field necessitates extensive, coordinated clinical, radiological, histopathologic, and genetic studies now underway in several sarcoma centers. We hope that an improved classification scheme and the use of surrogate biomarkers will further enhance patient care, leading to earlier diagnosis and hopefully prevention of MPNST, thus improving the outlook for NF1 patients.
Highlights.
Clinical evaluation of growing neurofibromas in neurofibromatosis 1 patients is often difficult
We describe the spectrum of pathology in peripheral nerve sheath tumors in neurofibromatosis type 1
While nuclear atypia alone is not decisive, loss of architecture and mitotic activity are worrisome
Intermediate category “atypical neurofibromatous neoplasm” is prosed for borderline lesions
Immunohistochemical studies assist in classification of problematic neurofibromatous tumors
Acknowledgments
This work was supported, in part, by the NCI Center for Cancer Research and Division of Cancer Epidemiology and Genetics, and by the Children’s Tumor Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This work represents the proceeds of the consensus meeting on Pathology of NF1-associated atypical nerve sheath tumors, held in October, 2016, at the NCI/NIH, Bethesda, Maryland.
References
- 1.Gutmann DH, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–7. [PubMed] [Google Scholar]
- 2.De Raedt T, Beert E, Pasmant E, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature. 2014;514:247–251. doi: 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 3.Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227–32. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang M, Wang Y, Jones S, et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46:1170–1172. doi: 10.1038/ng.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans DG, Baser ME, McGaughran J, et al. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311–4. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Uusitalo E, Leppävirta J, Koffert A, et al. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135:904–6. doi: 10.1038/jid.2014.465. [DOI] [PubMed] [Google Scholar]
- 7.Kattan MW, Leung DH, Brennan MF. Postoperative nomogram for 12-year sarcoma- specific death. J Clin Oncol. 2002;20:791–796. doi: 10.1200/JCO.2002.20.3.791. [DOI] [PubMed] [Google Scholar]
- 8.LaFemina J1, Qin LX, Moraco NH, et al. Oncologic outcomes of sporadic, neurofibromatosis-associated, and radiation-induced malignant peripheral nerve sheath tumors. Ann Surg Oncol. 2013;20:66–72. doi: 10.1245/s10434-012-2573-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolberg M, Høland M, Agesen TH, et al. Survival meta-analyses for >1800 malignant peripheral nerve sheath tumor patients with and without neurofibromatosis type 1. Neuro Oncol. 2013;15:135–147. doi: 10.1093/neuonc/nos287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Valentin T, Le Cesne A, Ray-Coquard I, et al. Management and prognosis of malignant peripheral nerve sheath tumors: The experience of the French Sarcoma Group (GSF-GETO) Eur J Cancer. 2016;56:77–84. doi: 10.1016/j.ejca.2015.12.015. [DOI] [PubMed] [Google Scholar]
- 11.Watson KL, Al Sannaa GA, Kivlin CM, et al. Patterns of recurrence and survival in sporadic, neurofibromatosis Type 1-associated, and radiation-associated malignant peripheral nerve sheath tumors. J Neurosurg. 2017;126:319–329. doi: 10.3171/2015.12.JNS152443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Antonescu CR, Scheithauer BW, Woodruff JM. AFIP atlas of tumor pathology. ARP Press; Silver Spring Maryland: Tumors of the peripheral nervous system; p. 2013. (Series 4). [Google Scholar]
- 13.Zhou H, Coffin CM, Perkins SL, et al. Malignant peripheral nerve sheath tumor: a comparison of grade, immunophenotype, and cell cycle/growth activation marker expression in sporadic and neurofibromatosis 1-related lesions. Am J Surg Pathol. 2003;27:1337–1345. doi: 10.1097/00000478-200310000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Pekmezci M, Reuss DE, Hirbe AC, et al. Morphologic and immunohistochemical features of malignant peripheral nerve sheath tumors and cellular schwannomas. Mod Pathol. 2015;28:187–200. doi: 10.1038/modpathol.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Guellec S, Decouvelaere AV, Filleron T, et al. Malignant Peripheral Nerve Sheath Tumor Is a Challenging Diagnosis: A Systematic Pathology Review, Immunohistochemistry, and Molecular Analysis in 160 Patients From the French Sarcoma Group Database. Am J Surg Pathol. 2016;40:896–908. doi: 10.1097/PAS.0000000000000655. [DOI] [PubMed] [Google Scholar]
- 16.Schaefer IM, Fletcher CD. Malignant peripheral nerve sheath tumor (MPNST) arising in diffuse-type neurofibroma: clinicopathologic characterization in a series of 9 cases. Am J Surg Pathol. 2015;39:1234–41. doi: 10.1097/PAS.0000000000000447. [DOI] [PubMed] [Google Scholar]
- 17.Dombi E, Solomon J, Gillespie AJ, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology. 2007;68:643–647. doi: 10.1212/01.wnl.0000250332.89420.e6. [DOI] [PubMed] [Google Scholar]
- 18.Meany H, Dombi E, Reynolds J, et al. 18-fluorodeoxyglucose-positron emission tomography (FDG-PET) evaluation of nodular lesions in patients with Neurofibromatosis type 1 and plexiform neurofibromas (PN) or malignant peripheral nerve sheath tumors (MPNST) Pediatr Blood Cancer. 2013;60:59–64. doi: 10.1002/pbc.24212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beert E, Brems H, Daniëls B, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer. 2011;50:1021–1032. doi: 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 20.Widemann B, Bhaumik S, Akshintala S, et al. Identification of lesions concerning for malignant peripheral nerve sheath tumors in NF1; Paper presented at: 21th Annual Meeting of the Connective Tissue Oncology Society; November 4–7, 2015; Salt Lake City, UT. [Google Scholar]
- 21.Lin BT, Weiss LM, Medeiros LJ. Neurofibroma and cellular neurofibroma with atypia: a report of 14 tumors. Am J Surg Pathol. 1997;21:1443–1449. doi: 10.1097/00000478-199712000-00006. [DOI] [PubMed] [Google Scholar]
- 22.Bernthal NM, Putnam A, Jones KB, et al. The effect of surgical margins on outcomes for low grade MPNSTs and atypical neurofibroma. J Surg Oncol. 2014;110:813–816. doi: 10.1002/jso.23736. [DOI] [PubMed] [Google Scholar]
- 23.Kourea HP, Orlow I, Scheithauer BW, et al. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155:1855–1860. doi: 10.1016/S0002-9440(10)65504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen GP, Stemmer-Rachamimov AO, Ino Y, et al. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155:1879–84. doi: 10.1016/S0002-9440(10)65507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casadei GP, Scheithauer BW, Hirose T, et al. Cellular schwannoma. A clinicopathologic, DNA flow cytometric, and proliferation marker study of 70 patients. Cancer. 1995;75:1109–19. doi: 10.1002/1097-0142(19950301)75:5<1109::aid-cncr2820750510>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 26.Prieto-Granada CN, Wiesner T, Messina JL, et al. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am J Surg Pathol. 2016;40:479–489. doi: 10.1097/PAS.0000000000000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaefer IM, Fletcher CD, Hornick JL. Loss of H3K27 trimethylation distinguishes malignant peripheral nerve sheath tumors from histologic mimics. Mod Pathol. 2016;29:4–13. doi: 10.1038/modpathol.2015.134. [DOI] [PubMed] [Google Scholar]
- 28.Cleven AH, Sannaa GA, Briaire-de Bruijn I, et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol. 2016;29:582–590. doi: 10.1038/modpathol.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asano N, Yoshida A, Ichikawa H, et al. Immunohistochemistry for trimethylated H3K27 in the diagnosis of malignant peripheral nerve sheath tumours. Histopathology. 2017;70:385–393. doi: 10.1111/his.13072. [DOI] [PubMed] [Google Scholar]
- 30.Röhrich M, Koelsche C, Schrimpf D, et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016;131:877–887. doi: 10.1007/s00401-016-1540-6. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez FJ, Folpe AL, Giannini C, Perry A. Pathology of peripheral nerve sheath tumors: diagnostic overview and update on selected diagnostic problems. Acta Neuropathol. 2012;123:295–319. doi: 10.1007/s00401-012-0954-z. [DOI] [PMC free article] [PubMed] [Google Scholar]