

Abstract
The androgen receptor (AR) is a ligand-dependent transcription factor that controls the expression of androgen-responsive genes. A key step in androgen action, which is amplified in castration resistant prostate cancer (CRPC), is AR nuclear translocation. Small molecules capable of inhibiting AR nuclear localization could be developed as novel therapeutics for CRPC. We developed a high throughput screen and identified two structurally-related pyrroloimidazoles that could block AR nuclear localization in CRPC cells. We show that these two small molecules, 3-(4-ethoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (EPPI) and 3-(4-chlorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (CPPI) can inhibit the nuclear localization and transcriptional activity of AR and reduce the proliferation of AR-positive but not AR-negative prostate cancer cell lines. EPPI and CPPI did not inhibit nuclear localization of the glucocorticoid receptor or the estrogen receptor, suggesting they selectively target AR. In LNCaP tumor xenografts, CPPI inhibited the proliferation of relapsed LNCaP tumors. These findings suggest that EPPI and CPPI could serve as lead structures for the development of therapeutic agents for CRPC.
Keywords: prostate cancer, androgen receptor, small molecule inhibitor, castration resistance, AR nucleocytoplasmic trafficking
INTRODUCTION
According to statistics compiled by the American Cancer Society about 180,890 new cases of prostate cancer and 26,120 prostate cancer deaths were tallied in 2016 in the U.S., marking prostate cancer as the most common malignancy diagnosed in males and the second most common cause of male cancer deaths. Androgen deprivation therapy (ADT) remains the standard treatment of metastatic prostate cancer; however, almost all of these patients ultimately relapse with incurable castration resistant prostate cancer (CRPC) (1). Studies have shown that the androgen receptor (AR) is re-activated via multiple mechanisms including AR overexpression, mutation, hypersensitization, and/or intratumoral androgen synthesis in CRPC that could drive tumor growth under ADT (2–8). Overexpression and knockdown studies have demonstrated that AR is a key molecular determinant and an excellent therapeutic target for CRPC (9–13). Abiraterone, an inhibitor of androgen synthesis (14), and the second generation AR antagonist enzalutamide (MDV3100), which competitively inhibits AR binding to androgens and can inhibit AR nuclear translocation and interaction with DNA (12), have been approved by the FDA for CRPC treatment. Abiraterone and enzalutamide can prolong life span of CRPC patients on average for another 4–6 months (14, 15). The majority of prostate cancers relapsed after abiraterone or enzalutamide treatment were PSA positive, indicating AR was activated again in the relapsed tumors cells (16). Thus, finding novel approaches to inhibit AR may lead to new therapies for CRPC patients resistant to abiraterone and/or enzalutamide.
The AR, a member of the steroid receptor superfamily, is a ligand-dependent transcription factor that controls the expression of androgen-responsive genes (reviewed in (17)). A transcription factor requires localization to the nucleus in order to access target genes, and intracellular trafficking is an important mechanism in the regulation of many transcription factors, including AR. Retention of a transcription factor in the cytoplasm can prevent its transcriptional activity. Thus, a key regulatory step in the action of AR is its nuclear translocation. In androgen-sensitive cells, AR is localized to the cytoplasm in the absence of ligand, and translocates into the nucleus and transactivates target genes upon addition of androgens. However, in CRPC cells, AR remains in the nucleus even in the absence of androgens, transactivating androgen-responsive genes and leading to uncontrolled growth (6, 18, 19). Therefore, small molecules that can specifically block the nuclear localization of AR may provide an effective therapy against CRPC tumors, including those relapsed after abiraterone or enzalutamide treatment.
We have reported on a high throughput, high content screen for small molecules capable of inhibiting nuclear AR levels or localization in C4-2 CRPC cells, which led to the identification of two pyrroloimidazoles, 3-(4-ethoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (EPPI) and 3-(4-chlorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (CPPI) (Fig. 1A) (20). In the current study, we evaluated the inhibition of AR nuclear localization and function by these small molecule inhibitors in cultured cells and tested their ability to inhibit prostate cancer cell growth in vitro and in xenograft tumor models.
Figure 1.
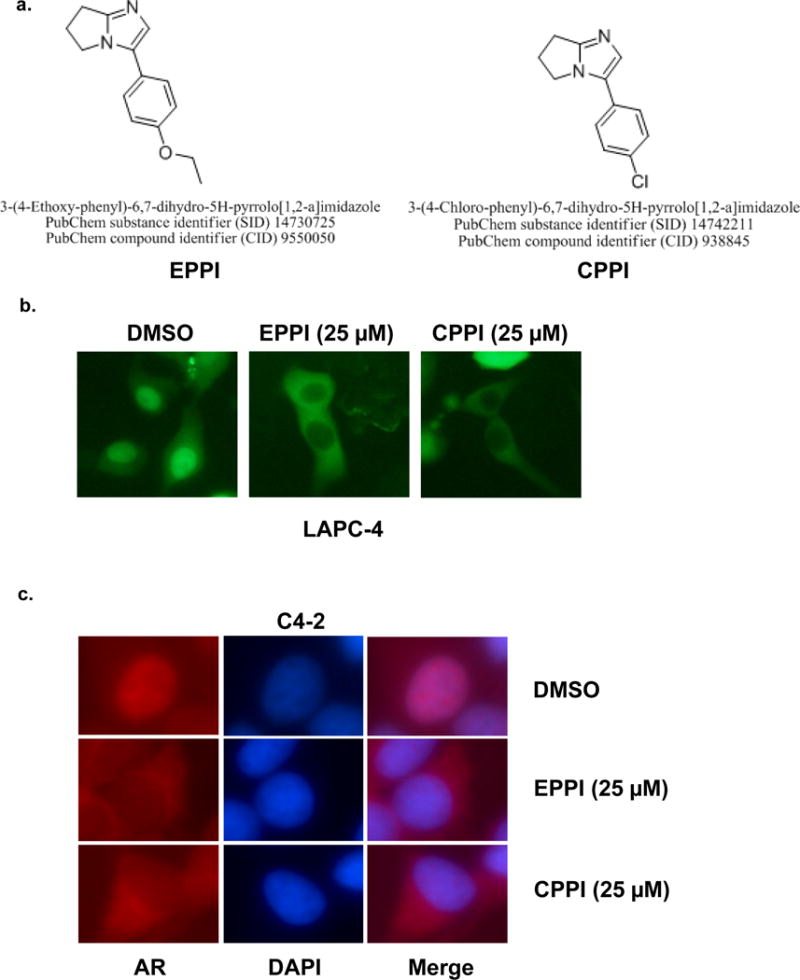
a. Structures of pyrroloimidazole-based small molecules 3-(4-ethoxyphenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (EPPI) and 3-(4-chlorophenyl)-6,7-dihydro-5H-pyrrolo[1,2-a]imidazole (CPPI). The PubChem Substance Identifier (SID) numbers for EPPI and CPPI are 14730725 and 14742211, respectively. b. Inhibition of GFP-AR nuclear localization in LAPC-4 cells by EPPI and CPPI. LAPC-4 cells transfected with GFP-AR (green) were treated with DMSO vehicle or the indicated small molecules at 25 μM overnight in androgen-free conditions. The subcellular localization of GFP-AR was visualized using fluorescent microscopy. c. Inhibition of endogenous AR nuclear localization in C4-2 cells by EPPI and CPPI. Endogenous AR localization was determined with immunofluorescent staining using an anti-AR antibody (red). The nuclei were stained with DAPI (blue). C4-2 cells cultured in androgen-free conditions were treated with 25 μM of EPPI or CPPI overnight prior to fixation and immunofluorescent staining.
MATERIAL AND METHODS
Plasmids
The expression vector pEGFP-C1 (Clontech, Mountain View, CA) was used to generate fusion protein constructs with GFP at the N terminus of AR and the NAR mutant for convenient visualization using fluorescent microscopy as described earlier (21). PSA promoter-driven luciferase reporter vector (pPSA6.1) was kindly provided by Dr. Marianne Sadar and a tk promoter-driven Renilla luciferase reporter (pRL-TK) was purchased from Promega (Madison, WI). Glucocorticoid receptor (GR) expression vector and MMTV-luciferase reporter were kindly provided by Dr. Donald DeFranco.
Small molecules
EPPI and CPPI were purchased from Princeton Biomedical Research, Inc. (Princeton, NJ). MDV3100 was purchased from Selleckchem (Houston, TX).
Cell culture experiments
Human C4-2 prostate cancer cells were obtained from Dr. Leland Chung in 2014 and maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 1% glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA) at 37 °C in the presence of 5% CO2 in a humidified incubator. LNCaP, PC-3, 22Rv1, DU145 and HEK293 cells were obtained from American Type Culture Collection (Manassas, VA). LNCaP PC-3, 22Rv1 and DU145 were maintained in RPMI 1640 medium and HEK293 was maintained in DMEM medium. LAPC4 cells were obtained from Dr. Robert Reiter in 2014. Cell lines LNCaP, 22Rv1, and C4-2 were authenticated in 2016 using DNA fingerprinting by examining microsatellite loci in a multiplex PCR reaction (AmpFlSTR® Identifiler® PCR Amplification Kit, Applied Biosystems, Foster City, CA) by the University of Pittsburgh Cell Culture and Cytogenetics Facility. HEK293 and PC-3 cell lines were obtained from ATCC in 2016. ATCC performed authentication for HEK293 and PC-3 cell lines using short tandem repeat profiling. No authentication was performed for DU145 or LAPC4. RPMI 1640 medium was supplemented with 5–10% FBS stripped 2 times with charcoal, for experiments performed in androgen-free conditions.
AR localization
GFP-AR and GFP-NAR expression vectors was transiently transfected into LAPC4, PC-3, C4-2 and HEK293 cells using Polyjet according to the manufacturer’s protocol (SignaGen Laboratories). Cells were transfected at >60% confluence in phenol red-free OptiMEM. The localization of GFP fusion proteins was imaged 16 h after transfection, or at the indicated times after exposure to small molecules dissolved in DMSO, or DMSO vehicle control, with fluorescence microscopy using either a Nikon TE 2000U, Nikon TS100, or Leica DM-IL microscope as described previously (22). Cytoplasmic localization in transfected cells was defined as GFP fluorescence that was both predominantly in the cytoplasm and more intense relative to nuclei. Nuclei were stained with 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI) (Sigma-Aldrich, St. Louis, MO) or Hoechst 33342 (Sigma-Aldrich). Nuclear localization was defined as GFP fluorescence that was both predominantly in the nuclei and more intense than in the cytoplasm. Even distribution was defined as when GFP fluorescence was evenly distributed between the nucleus and cytoplasm in transfected cells. Quantification of subcellular localization of GFP-tagged fusion proteins was determined by counting 20–200 transfected cells/dish in at least three dishes from each experiment. All experiments were performed in triplicate and repeated at least twice.
C4-2 cells were transfected with GFP-ERα or GFP-GR expression vectors and cultured overnight. The following day, cells were treated with the indicated concentrations of the small molecules EPPI or CPPI dissolved in DMSO, or with the DMSO vehicle control. C4-2 cells transfected with GFP-GR were also treated with 0.5 μM dexamethasone to induce GFP-GR nuclear localization in the presence of small molecules (23). Subcellular localization of GFP-ERα or GFP-GR in transfected C4-2 cells was determined by fluorescent microscopy 24 h after treatment with the small molecules.
Western blot analysis
C4-2 cells cultured in complete medium were treated with 0, 20 or 40 μM EPPI or CPPI for 48 h. Cells were lysed in modified radioimmune precipitation assay (RIPA) buffer [50 mM Tris-Cl (pH7.4), 1mM EDTA, 1% NP-40, 0.1% sodium deoxycholate, 0.1% SDS, 150mM NaCl] with 1% protease inhibitor cocktail (Sigma-Aldrich). Protein concentration was determined by BCA assay (Pierce Chemical Co., Rockford, IL). Western blotting was conducted using primary antibodies against AR (sc-816, Santa Cruz Biotechnology, Santa Cruz, CA), PSA (sc-7638, Santa Cruz Biotechnology) and GAPDH (sc-25778, Santa Cruz Biotechnology), followed with horseradish peroxidase-labeled secondary antibody (sc-2004 and sc-2771, Santa Cruz Biotechnology). Signals were visualized using chemiluminescence (ECL Western Blotting Detection Reagents; GE Healthcare, Piscataway, NJ) and detected by ChemiDoc™ Imaging Systems (Bio-Rad, Hercules, CA).
Luciferase assay
C4-2 cells were plated in 12-well or 24-well plates and transfected with appropriate luciferase plasmid(s) in androgen-free conditions. Cells were treated with 0.1 nM R1881 to induce PSA promoter-driven luciferase activity. After 24–48 h, cells were lysed and luciferase activity was measured using a Promega luciferase assay kit (Promega) and an LmaxII384 luminometer (Molecular Devices, Sunnyvale, CA) following the manufacturer’s recommendations. The firefly luciferase activity was normalized to Renilla luciferase activity. All experiments were performed in triplicate and repeated at least three times.
The MMTV-firefly luciferase reporter, tk-renilla luciferase reporter, and GFP-GR expression vector were co-transfected into C4-2 cells to test the effect of CPPI on GR transactivation. The transiently transfected cells were treated with 0.5 μM dexamethasone in the presence or absence of 10 μM CPPI for 24 h. The cells were then harvested for luciferase detection as described above.
Xenograft tumor studies
Male BALB/c strain of athymic SCID mice were obtained from Charles River Laboratory, Wilmington, MA, USA, and were kept in accordance with the National Institute of Health guidelines under standard animal housing conditions for the Care and Use of Experimental Animals. All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pittsburgh and were conducted in strict accordance with the standards for humane animal care and use as set by the Animal Welfare Act and the National Institutes of Health guidelines for the use of laboratory animals.
Approximately 2×106 LNCaP or PC-3 cells suspended in 250 μL media were gently mixed with 250 μL Matrigel (Becton Dickinson Labware, Bedford, MA) and then inoculated subcutaneously in the flank region of 6~8-week old male athymic SCID mice using a 25-gauge needle to establish xenograft tumors.
PC-3 or LNCaP xenograft tumors were measured three times per week and volume was calculated by the modified ellipsoid formula: length × width2 × 0.52 (24). Trans-scrotal castration was performed under isoflurane anesthesia with proper aseptic and antiseptic technique when tumor volume reached >200 mm3 as reported previously (25). Mice were randomized into two experimental groups at the time of castration. One group of castrated mice received subcutaneous injections of CPPI at 50 mg/kg every alternate day and the other group received vehicle control egg phospholipids (EPL, NSC 704057, National Cancer Institute) in parallel. The mice were euthanized when tumors reached 2,000 mm3 under approval by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
BrdU Cell Proliferation Assays
Cells were seeded at 1,000 cells/well in four-chambered slides and allowed to attach for 24 h. Cells were cultured in phenol red free RPMI medium containing 10% charcoal-stripped FBS (cFBS) for two days. Media was then replaced with fresh cFBS media containing different concentrations of EPPI or CPPI (0.4, 2.0, 10 and 50 μM) for 48 h. BrdU labeling media was added 12 h prior to staining. Cells were then washed with PBS, and BrdU incorporation was assayed according to the manufacturer’s instruction (Invitrogen BrdU staining kit; catalog #: 93-3943). Slides were imaged using a Zeiss Axioplan2 microscope and analyzed using Axiovision Rel. 4.5 imaging software. BrdU-positive cell density was determined according to the presence or absence of nuclear specific staining when compared to the negative controls for 3 independent experiments, with at least 10 fields imaged at 40× magnification for each treatment group.
Immunohistochemistry
Tumor tissue samples were fixed immediately after dissection in 10% formalin overnight, dehydrated in ethanol, cleared in xylene, and embedded in paraffin blocks, as previously described (26). Six micrometer-thick sections were prepared, mounted on positive charged slides (Fisher Biotech, Pittsburgh, PA), and air-dried overnight. After de-waxing and rehydration, slides were subjected to the heat-mediated antigen retrieval procedure using a domestic microwave oven at a maximum power of 750 W. Slides were stained with caspase-3 (Cas 3) (1:400, H-277, sc-7148, Santa Cruz Biotechnology) and Ki-67 (1:50, sc-15402, Santa Cruz Biotechnology) antibodies. Cas 3 and Ki-67-positive cell density was determined by analysis of sections from all five xenografts from each group. Apoptotic and proliferative index were determined from at least 20 fields imaged at 40 × magnification with no overlap, Cas 3 and Ki-67-positive cells were counted to determine the average number of apoptotic and proliferating cells for each section. Sections were imaged using a Zeiss Axioplan2 microscope and analyzed using Axiovision Rel. 4.5 imaging software. Composite images were constructed with Photoshop CS (Adobe Systems, San Jose, CA).
Statistical analysis
GraphPad Prism 5.0 (GraphPad Software, Inc) and MS Excel 2003 (Microsoft) were used for statistical analysis and graphical composition. Data were expressed as the mean ± SEM, and statistical significance was determined by one-way ANOVA or Student’s t-test as appropriate. Tumor volume data were expressed as the mean ± SEM or SD, and statistical significance was determined by Mantel-Cox log-rank or Student’s t-test as appropriate. A p-value of < 0.05 was considered statistically significant.
RESULTS
Small molecules EPPI and CPPI inhibited AR nuclear localization in prostate cancer cells
The structurally related pyrroloimidazoles EPPI and CPPI inhibited GFP-AR nuclear localization in LAPC4 prostate cancer cells (Fig. 1a,b). Both EPPI and CPPI also enhanced cytoplasmic localization of endogenous AR in C4-2 cells (Fig. 1c). The effects of EPPI and CPPI on endogenous AR or GFP-AR subcellular localization were indistinguishable in these experiments, suggesting that these two structurally related compounds are also functionally equivalent.
Although EPPI or CPPI inhibited AR nuclear localization in C4-2 and LAPC4 cells cultured in complete RPMI media, which contains castrate levels of androgens (27), it was not clear if EPPI and CPPI could also inhibit androgen-induced AR nuclear localization. In AR-negative PC-3 and HEK293 cells, transfected GFP-AR was localized to the cytoplasm of cells that were cultured in regular media (Fig. 2), providing an opportunity to test the effect of these compounds on androgen-induced GFP-AR nuclear localization. In the presence of EPPI or CPPI, higher concentrations of R1881 were required to induce GFP-AR nuclear localization in both transfected PC-3 and HEK293 cells (Fig. 2). GFP-AR nuclear localization in the presence 0.2 or 1 nM R1881 in HEK293 cells was significantly inhibited by EPPI or CPPI. Similarly, GFP-AR nuclear localization in the presence 0.04–0.2 nM R1881 in PC-3 cells was significantly inhibited by EPPI or CPPI. At R1881 concentrations of 5 nM or higher, GFP-AR translocated into the nuclei of both PC-3 and HEK293 cells even in the presence of EPPI or CPPI. These observations demonstrated that EPPI and CPPI can inhibit AR nuclear localization at low androgen concentrations, but that higher androgen concentrations could overcome EPPI and CPPI inhibition of AR nuclear translocation.
Figure 2.
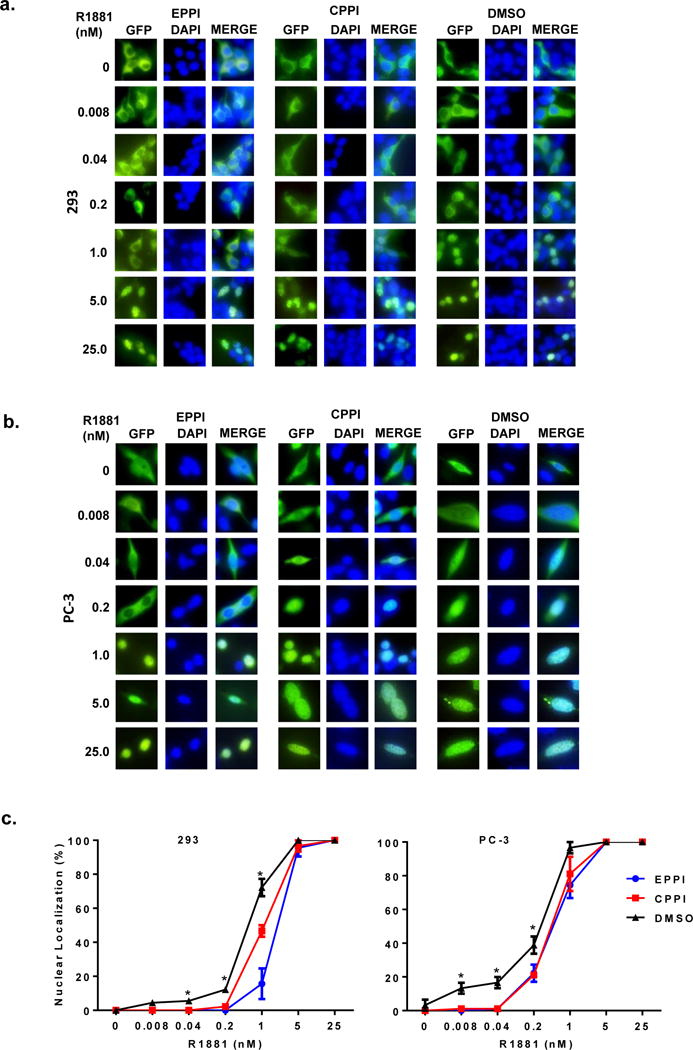
The effect of EPPI and CPPI on R1881-induced GFP-AR nuclear localization. a. HEK293 cells were transiently transfected with GFP-AR (green) in androgen-free conditions. The transfected cells were treated with vehicle DMSO, EPPI or CPPI at 25 μM in the presence of indicated concentrations of R1881 overnight. The subcellular localization of GFP-AR was visualized using fluorescent microscopy. The nuclei were stained with DAPI (blue). b. PC-3 cells were transiently transfected with GFP-AR as in a. c. Quantification of cells exhibiting nuclear localization in transfected HEK293 (left panel) or PC-3 cells (right panel). Data are expressed as mean ±SD (*, p < 0.05).
EPPI and CPPI inhibited androgen induction of PSA expression in C4-2 cells
Since EPPI and CPPI inhibited AR nuclear localization in prostate cancer cells, we tested if these small molecules could inhibit AR induction of its target gene PSA. Western blot analysis demonstrated that EPPI and CPPI inhibited endogenous PSA expression in C4-2 cells (Fig. 3a), indicating that AR transcriptional activity was inhibited. AR expression appeared to be slightly inhibited in response to EPPI or CPPI treatment. To further evaluate the inhibition of AR transactivation by EPPI and CPPI, an androgen responsive PSA promoter-driven luciferase assay was performed. C4-2 cells were transiently transfected with the PSA6.1-luc reporter construct and treated with increasing concentrations of EPPI (Fig. 3b) or CPPI (Fig. 3c) for 48 h in the presence or absence of 0.1 nM R1881. Both small molecules significantly inhibited androgen induced PSA-promoter driven luciferase activity at 10–40 μM concentrations, indicating that they behave similarly to AR antagonists.
Figure 3.
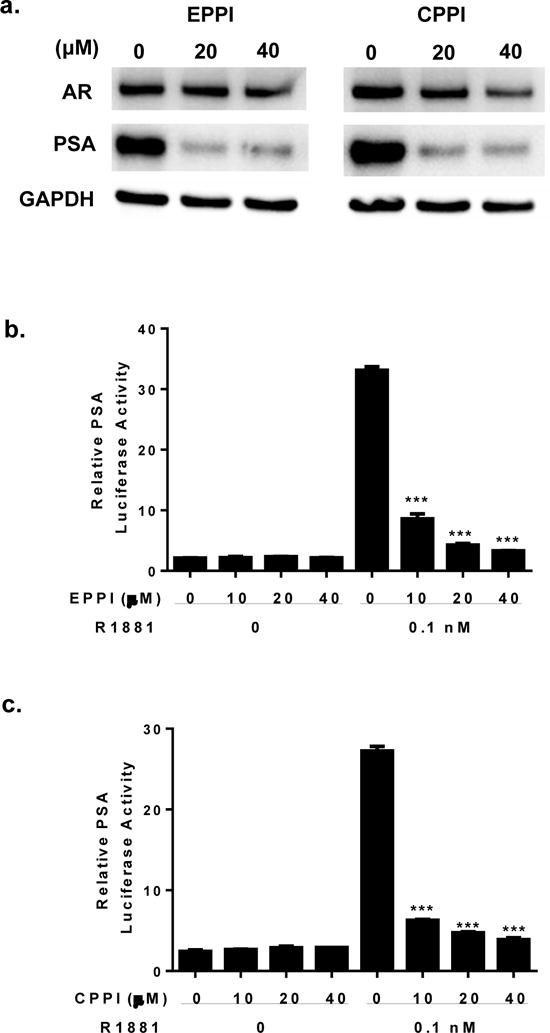
Inhibition of AR transcriptional activation of PSA by EPPI or CPPI in C4-2 cells. a. C4-2 cells cultured in complete media were treated with 25 μM EPPI or CPPI for 48 and immunoblotted with AR and PSA antibodies. GAPDH served as internal loading control. b. The effect of EPPI on PSA promoter-driven luciferase activity in C4-2 cells. The indicated concentrations of EPPI were added to C4-2 cells pre-cultured in androgen-free media and transiently transfected with PSA6.1-luciferase and tk-renilla luciferase reporters in the presence of 0.1 nM R1881 or vehicle ethanol for 48 h. c. The effect of CPPI on PSA promoter-driven luciferase activity in C4-2 cells as described in b. Firefly luciferase activity was normalized to tk promoter-driven renilla luciferase activity. Data are expressed as mean ±SEM (***, p < 0.001).
EPPI and CPPI did not inhibit nuclear localization of GFP-GR and GFP-ERα
To test the specificity of EPPI and CPPI inhibition of AR nuclear localization, we transiently transfected GFP-GR and GFP-ERα into C4-2 cells separately. High doses of EPPI and CPPI (50–100 μM) did not affect the subcellular localization of either ER or GR-GFP (Fig. 4a & 4b). These results indicate that the inhibition of AR nuclear localization by EPPI and CPPI is specific to AR in C4-2 cells. CPPI also did not inhibit the GFP-GR mediated transactivation of a MMTV-luciferase reporter (Fig. 4c), confirming that these small molecules selectively inhibit AR, but not other steroid receptors.
Figure 4.
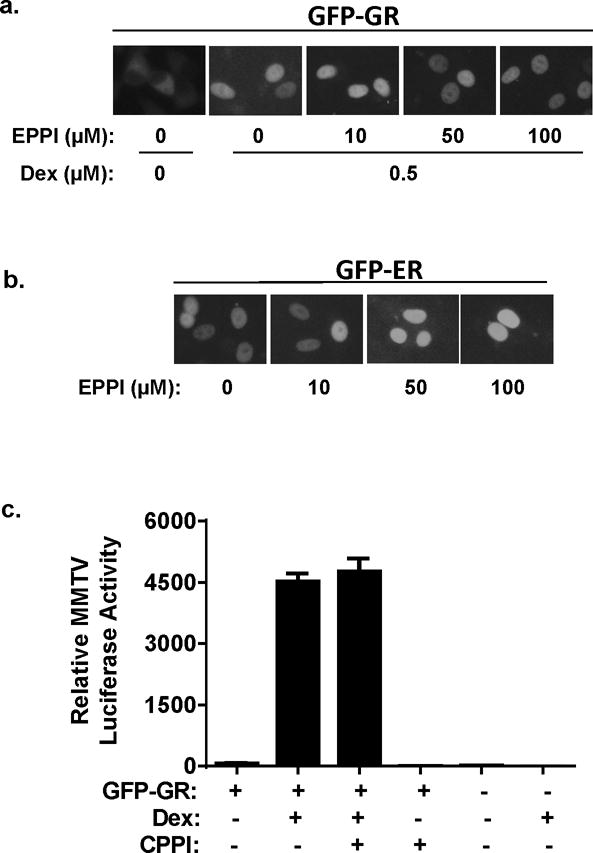
The effect of EPPI and CPPI on subcellular localization of GFP-GR and GFP-ERα in C4-2 cells cultured in androgen-free conditions. a. The effect of EPPI on dexamethasone (Dex)-induced nuclear localization of GFP-GR. C4-2 cells were transiently transfected with GFP-GR expression vector overnight followed by treatment with indicated concentrations of EPPI along with 0.5 μM dexamethasone. Subcellular localization of GFP-GR was visualized by fluorescent microscopy 24 h after the treatment with EPPI. b. The effect of EPPI on transiently transfected GFP-ERα was determined the same as described in a, except no dexamethasone was used. c. Inhibition of GR transactivation of MMTV promoter-driven luciferase activity by CPPI in C4-2 cells. The GR transactivation was induced by 0.5 μM dexamethasone in the presence or absence of 10 μM CPPI. Firefly luciferase activity was normalized to tk promoter-driven renilla luciferase activity. C4-2 cells without GFP-GR transfection were used as controls. Data are expressed as mean ±SEM.
EPPI and CPPI inhibited proliferation of AR-positive prostate cancer cells
Since EPPI and CPPI inhibit AR nuclear localization and transactivation, we performed a BrdU incorporation assays to determine if these small molecules inhibited the proliferation of AR-positive prostate cancer cell lines, consisting of LNCaP, C4-2 and 22RV1. AR-negative PC-3 and DU145 prostate cancer cells were also included as controls. As expected, EPPI and CPPI inhibited the proliferation of LNCaP, C4-2 and 22RV1cells (Fig. 5a & 5b), presumably via targeting AR. EPPI and CPPI also inhibited R1881-stimulated proliferation of LNCaP cells (Fig. 5c). CPPI did not inhibit proliferation of either PC-3 or DU145 cells (Fig. 5b), suggesting that its inhibition of prostate cancer cell growth was mediated through AR. EPPI inhibited DU145, although it did not inhibit PC-3 cells (Fig. 5a), suggesting that EPPI could also inhibit prostate cancer cell growth via AR-independent pathways, due to off-target effects. These data suggest that CPPI might be more specific and therefore less toxic than EPPI.
Figure 5.
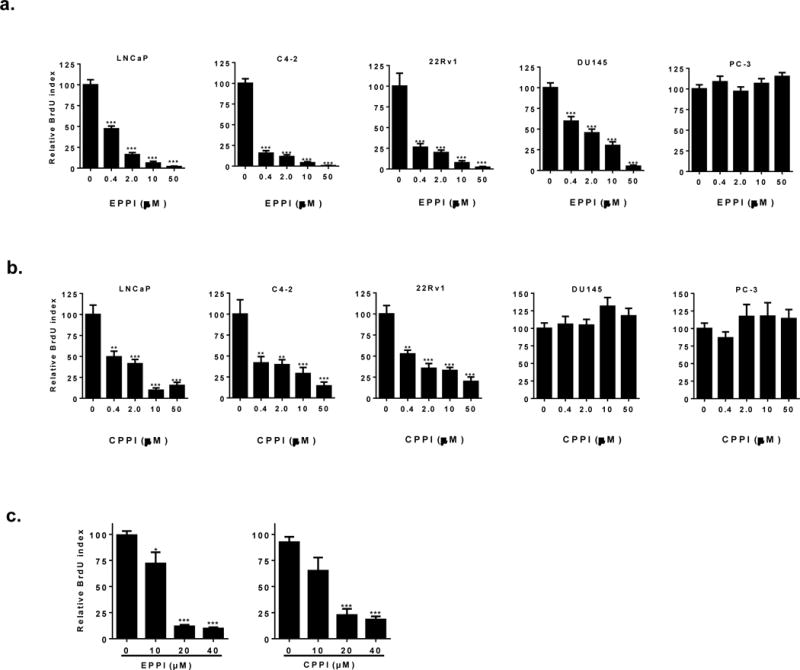
Effects of EPPI and CPPI on cellular proliferation in cultured LNCaP, C4-2, 22Rv1, DU145, and PC-3 cell lines. a. Quantification of BrdU-positive LNCaP, C4-2, 22Rv1, DU145, and PC-3 cells treated with EPPI. Cells were pre-cultured in RPMI media and subsequently treated with DMSO vehicle control, 0.4, 2.0, 10 and 50 μM EPPI for 48 h. BrdU was added 4-12 h before fixing and staining. b. Quantification of BrdU-positive LNCaP, C4-2, 22Rv1, DU145, and PC-3 cells treated with CPPI. Cells were pre-cultured in RPMI media and subsequently treated with DMSO vehicle control, 0.4, 2.0, 10 and 50μM CPPI for 48 h. BrdU was added 4-12 h before fixing and staining. c. EPPI and CPPI inhibition of R1881-stimulated BrdU incorporation. LNCaP cells were pre-cultured in androgen-free conditions for 48 h, and subsequently treated with DMSO vehicle control, 0.1 nM R1881, and DMSO vehicle control, 10, 20 and 40 μM EPPI and CPPI, respectively for 48 h. Data are expressed as mean ±SEM (*, p < 0.05; **, p < 0.01; ***, p < 0.0001).
EPPI and CPPI did not inhibit nuclear localization of GFP-NAR
We investigated the effect of CPPI and EPPI on full-length GFP-AR and GFP-NAR, which consists of the NTD and DBDH, but lacks the LBD (28). C4-2 cells with transiently transfected GFP-AR and GFP-NAR treated with 25 μM EPPI or CPPI overnight demonstrated the inhibition of nuclear localization of GFP-AR, but not GFP-NAR (Fig. 6), suggesting that EPPI and CPPI influence of AR subcellular localization requires LBD. However, this finding cannot rule out the possibility that CPPI/EPPI may target NTD and subsequently influence LBD function because N-terminus and C-terminus of AR can interact (29). Overnight MDV3100 treatment (25 μM) had no effect on GFP-AR or GFP-NAR nuclear localization in C4-2 cells (Fig. 6).
Figure 6.
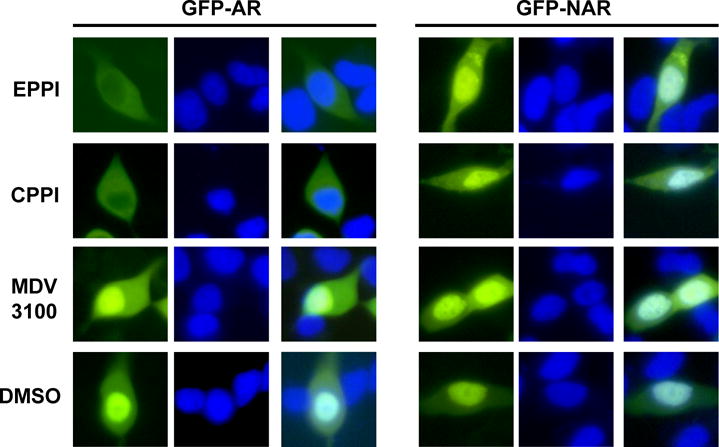
Effect of EPPI, CPPI and MDV3100 on subcellular localization of GFP-AR and GFP-NAR in C4-2 cells cultured in complete media. C4-2 cells were transiently transfected with GFP-AR or GFP-NAR overnight followed by treatment with EPPI, CPPI or MDV3100 at 25 μM, or DMSO vehicle control for 24 h. The subcellular localization of GFP-AR and GFP-NAR was visualized using fluorescent microscopy. The nuclei were stained with Hoechst (blue).
CPPI inhibited growth of relapsed LNCaP xenografts
Since CPPI only inhibited AR-positive, but not AR-negative, prostate cancer cells in culture (Fig. 5a), we examined its ability to inhibit AR-positive prostate xenograft tumor growth. LNCaP xenograft tumors were established in testis-intact nude mice and mice were castrated to induce tumor relapse. Relapsed LNCaP tumors were treated with CPPI via subcutaneous injection every other day at 50 mg/kg body weight or vehicle (EPL) treatment only. CPPI significantly inhibited the growth of relapsed LNCaP xenograft tumors (Fig. 7a, P<0.0001).
Figure 7.
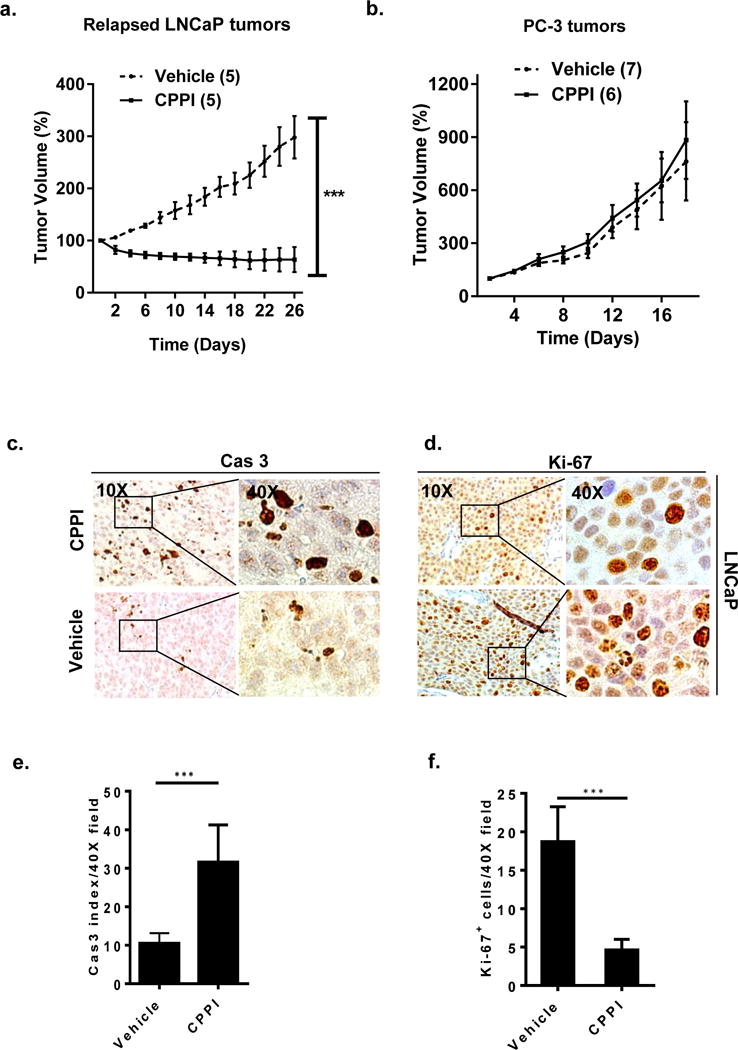
a. CPPI inhibition of relapsed LNCaP xenograft tumor growth. Response of LNCaP tumors relapsed after castration to CPPI treatment or vehicle control. b. Effect of CPPI on PC-3 xenograft tumor growth. Response of PC-3 tumors relapsed after castration. Number of animals in each group is indicated in parentheses. c. Representative images (10–40X) of relative Cas-3+ immunostaining in transverse sections of relapsed LNCaP xenograft tumors treated with CPPI or vehicle control. d. Ki-67 immunostaining in transverse sections of LNCaP xenograft tumors treated with CPPI or vehicle control. e. Quantification of Cas-3 positive cells in LNCaP tumors. f. Quantification of Ki-67 positive cells in LNCaP. Data are representative of 5 different readings from each tumor. Values are presented as mean ± SEM (*, p < 0.05, ***, p < 0.0001).
CPPI did not alter PC-3 xenograft growth compared to controls (Fig. 7b). These findings demonstrate that CPPI inhibited the proliferation of AR-positive, but not AR-negative prostate cancer cells in vivo. The inhibition of relapsed LNCaP tumors by CPPI was consistent with the Cas-3 and Ki-67 staining analysis of tumors (Fig. 7c–f). Tumors in CPPI treated mice had a 3-fold increase in Cas-3 apoptotic index (P < 0.0001) and a 4-fold decrease in Ki-67 index compared to tumors in the vehicle control group.
DISCUSSION
We show that EPPI and CPPI inhibit AR nuclear localization and transactivation of its target gene PSA in prostate cancer cells. The inhibition of AR appeared to be specific because the small molecules failed to inhibit the nuclear localization of two other steroid receptors, GR and ERα, and failed to inhibit GR-mediated transactivation. Although both small molecules inhibited the proliferation of AR-positive prostate cancer cells, CPPI did not inhibit the growth of AR-negative prostate cancer cells, suggesting a high specificity of AR-targeting. CPPI also inhibited the relapsed growth of AR-positive LNCaP xenograft tumors but not AR-negative PC-3 xenograft tumors in SCID mice. These data suggest that pyrroloimidazole-based small molecules could be developed to target AR signaling for the treatment of CRPC.
Our findings provide evidence that preventing AR nuclear localization is a viable approach to inhibit AR-positive prostate cancer cell growth. Although some AR antagonists such as enzalutamide could reduce AR protein in the nucleus, the nuclear AR level was still higher than the cytoplasmic AR in prostate cancer cells treated with enzalutamide (12). In our study, CPPI and EPPI, but not enzalutamide, inhibited GFP-AR nuclear localization in C4-2 cells cultured in complete media (Fig. 6). Nuclear AR was significantly less than cytoplasmic AR in prostate cancer cells treated with EPPI or CPPI (see Fig. 1 and Fig. 6). CPPI and EPPI inhibition of AR nuclear localization in prostate cancer cells was effective under androgen-deprived conditions but not when high doses of androgens were present (see Fig. 2), indicating that high levels of androgens could overcome the inhibition of AR nuclear localization. This limitation should not be a concern for developing these small molecules as potential therapy for CRPC because CRPC patients are on ADT, and these small molecules could be used in combination with traditional ADT.
EPPI, but not CPPI, exhibited growth inhibitory effect on AR-negative DU145 cells. Although the scaffold of both molecules is identical, EPPI has an ethoxy group whereas CPPI has a chloride substituent. The EtO-Ph-C=C substructure in EPPI is flagged in the Registry of Toxic Effects of Chemical Substances (RTECS) and therefore a candidate for unspecific toxicity pathways. Thus, AR-independent inhibition of DU145 by EPPI appears to be a function of the ethoxy arene, but not to the heterocyclic scaffold shared by EPPI and CPPI. Therefore, this scaffold provides an excellent starting point for the synthesis of more effective analogs to inhibit AR.
In a study reported by Demchenko et al., structural analogs of EPPI and CPPI have antifungal effects at a concentration of 500 μg/mL or higher (30). It is likely that the antifungal effects are mediated through targeting proteins other than AR since fungus does not have AR. It is possible that some analogs of these small molecules will inhibit AR without antifungal activities, considering that CPPI and EPPI can inhibit AR transcriptional activity at a low 10 μM concentration, equivalent to 2.19 μg/mL and 2.65 μg/mL for CPPI and EPPI, respectively. Importantly, EPPI was well tolerated in animals. Thus, the structural feature responsible for the antifungal effects in this scaffold may not influence mammalian cells.
EPPI and CPPI are structurally very different from some of the well-known AR antagonists, including flutamide, bicalutamide, enzalutamide and EPI-001(19) (12, 31) and thus may represent a new class of small molecules capable of inhibiting AR function. The mechanisms of action for these small molecules are likely very different from LBD-targeting AR antagonists because their pyrroloimidazole core is structurally different from LBD-targeting AR antagonists. Thus, these small molecules may be synergistic with LBD-targeting AR antagonists at inhibiting AR signaling in prostate cancer cells.
CPPI and EPPI inhibition of AR appeared to be specific, because these small molecules did not inhibit the nuclear localization of GFP-GR and GFP-ERα and transactivation of GR. However, the mechanisms of CPPI and EPPI inhibition of AR nuclear localization and function have not yet been resolved. This class of small molecules may interact with AR directly or indirectly through a co-factor. Future work will focus on whether CPPI or EPPI could directly bind to AR. Differential scanning fluorimetry assays could be used to measure potential interactions of CPPI/EPPI with AR (32). LBD-targeting AR antagonists such as bicalutamide are known to modulate AR recruitment of co-factors (33, 34). NTD targeting small molecule EPI-001 was also reported to inhibit AR interaction with co-factor CBP (31). Thus, if CPPI/EPPI could directly bind to AR, they may also interfere with AR recruitment of its co-factors. Another possibility is that CPPI/EPPI could target AR co-factors capable of modulating AR subcellular localization. However, AR co-factor modulation of endogenous AR nuclear localization is not well studied. For example, Prp8 is an AR co-factor and its knockdown increased GFP-AR nuclear localization in transfected PC-3 cells (35). However, Prp8 knockdown did not affect endogenous AR nuclear localization in LNCaP cells. Instead, Prp8 knockdown significantly enhanced the ubiquitination of endogenous AR. Therefore, it is unlikely that Prp8 is a molecular target of CPPI or EPPI. If CPPI or EPPI targets an AR co-factor, instead of directly targets AR, one approach may be to develop analogs of CPPI/EPPI that can be conjugated to agarose beads for affinity purification of protein(s) that can bind to CPPI/EPPI. Future studies such as these will be required to determine how these agents inhibit AR and to develop analogs with higher potency, minimal toxicity, and favorable pharmacokinetic profiles for the treatment of patients with CRPC.
Acknowledgments
We are grateful to Marie Acquafondata, Megan Lambert, Katie Leschak, Krystal Roskov, Kathleen J. O’Malley and Aiyuan Zhang for technical support. We would like to thank Dr. Leland W. K. Chung for providing C4-2 cells, Dr. Robert Reiter for LAPC4 cells, Dr. Marianne Sadar for PSA6.1 luciferase reporter, and Dr. Donald B. DeFranco for GFP-GR, GFP-ERα, and MMTV-luciferase reporter vectors. This work was funded in part by the Department of Urology, University of Pittsburgh and NIH grants 9R01 CA186780, 1P50 CA180995, and 1R50 CA211242. This project used the UPCI Animal Facility, which was supported in part by award P30 CA047904.
Author’s financial support:
Z. Wang, National Cancer Institute, 9R01 CA186780
L.E. Pascal, National Cancer Institute, 1R50 CA211242
Z. Wang, National Cancer Institute, 1P50 CA180995
J.T. Newsome, National Cancer Institute, P30 CA047904
References
- 1.Rini BI, Small EJ. Hormone-refractory Prostate Cancer. Current treatment options in oncology. 2002;3:437–446. doi: 10.1007/s11864-002-0008-1. [DOI] [PubMed] [Google Scholar]
- 2.Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinanen R, Palmberg C, Palotie A, Tammela T, Isola J, Kallioniemi OP. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nature Genetics. 1995;9:401–406. doi: 10.1038/ng0495-401. [DOI] [PubMed] [Google Scholar]
- 3.Ford OH, 3rd, Gregory CW, Kim D, Smitherman AB, Mohler JL. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J Urol. 2003;170:1817–1821. doi: 10.1097/01.ju.0000091873.09677.f4. [DOI] [PubMed] [Google Scholar]
- 4.Brown RS, Edwards J, Dogan A, Payne H, Harland SJ, Bartlett JM, Masters JR. Amplification of the androgen receptor gene in bone metastases from hormone-refractory prostate cancer. J Pathol. 2002;198:237–244. doi: 10.1002/path.1206. [DOI] [PubMed] [Google Scholar]
- 5.Veldscholte J, Berrevoets C, Ris-Stalpers C, Kuiper G, Jenster G, Trapman J, Brinkmann A, Mulder E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. [Review] Journal of Steroid Biochemistry & Molecular Biology. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 6.Gregory CW, Johnson RT, Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 7.Mohler JL. Castration-recurrent prostate cancer is not androgen-independent. Adv Exp Med Biol. 2008;617:223–234. doi: 10.1007/978-0-387-69080-3_21. [DOI] [PubMed] [Google Scholar]
- 8.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 9.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 10.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 11.Salem M, Garcia JA. Abiraterone acetate, a novel adrenal inhibitor in metastatic castration-resistant prostate cancer. Curr Oncol Rep. 2011;13:92–96. doi: 10.1007/s11912-011-0153-4. [DOI] [PubMed] [Google Scholar]
- 12.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SC, Dehm SM. Constitutive activity of the androgen receptor. Advances in pharmacology. 2014;70:327–366. doi: 10.1016/B978-0-12-417197-8.00011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB, Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Flechon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K, de Wit R, Mulders P, Chi KN, Shore ND, Armstrong AJ, Flaig TW, Flechon A, Mainwaring P, Fleming M, Hainsworth JD, Hirmand M, Selby B, Seely L, de Bono JS, Investigators A Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 16.Boudadi K, Antonarakis ES. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clinical Medicine Insights Oncology. 2016;10:1–9. doi: 10.4137/CMO.Ss34534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Z, Wong C, Sar M, Wilson E. The androgen receptor: an overview. [Review] Recent Progress in Hormone Research. 1994;49:249–274. doi: 10.1016/b978-0-12-571149-4.50017-9. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Johnson M, Le KH, Sato M, Ilagan R, Iyer M, Gambhir SS, Wu L, Carey M. Interrogating androgen receptor function in recurrent prostate cancer. Cancer Res. 2003;63:4552–4560. [PubMed] [Google Scholar]
- 19.Tian X, He Y, Zhou J. Progress in antiandrogen design targeting hormone binding pocket to circumvent mutation based resistance. Frontiers in pharmacology. 2015;6:57. doi: 10.3389/fphar.2015.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston PA, Nguyen MM, Dar JA, Ai J, Wang Y, Masoodi KZ, Shun T, Shinde S, Camarco DP, Hua Y, Huryn DM, Wilson GM, Lazo JS, Nelson JB, Wipf P, Wang Z. Development and Implementation of a High-Throughput High-Content Screening Assay to Identify Inhibitors of Androgen Receptor Nuclear Localization in Castration-Resistant Prostate Cancer Cells. Assay Drug Dev Technol. 2016;14:226–239. doi: 10.1089/adt.2016.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dar JA, Masoodi KZ, Eisermann K, Isharwal S, Ai J, Pascal LE, Nelson JB, Wang Z. The N-terminal domain of the androgen receptor drives its nuclear localization in castration-resistant prostate cancer cells. The Journal of steroid biochemistry and molecular biology. 2014;143:473–480. doi: 10.1016/j.jsbmb.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dar JA, Eisermann K, Masoodi KZ, Ai J, Wang D, Severance T, Sampath-Kumar SD, Wang Z. N-terminal domain of the androgen receptor contains a region that can promote cytoplasmic localization. The Journal of steroid biochemistry and molecular biology. 2014;139:16–24. doi: 10.1016/j.jsbmb.2013.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dekelbab BH, Witchel SF, DeFranco DB. TNF-alpha and glucocorticoid receptor interaction in L6 muscle cells: a cooperative downregulation of myosin heavy chain. Steroids. 2007;72:705–712. doi: 10.1016/j.steroids.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Euhus DM, Hudd C, LaRegina MC, Johnson FE. Tumor measurement in the nude mouse. Journal of surgical oncology. 1986;31:229–234. doi: 10.1002/jso.2930310402. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Gupta S, Hua V, Ramos-Garcia R, Shevrin D, Jovanovic BD, Nelson JB, Wang Z. Prolongation of off-cycle interval by finasteride is not associated with survival improvement in intermittent androgen deprivation therapy in LNCaP tumor model. Prostate. 2010;70:147–154. doi: 10.1002/pros.21046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Masoodi KZ, Ramos Garcia R, Pascal LE, Wang Y, Ma HM, O’Malley K, Eisermann K, Shevrin DH, Nguyen HM, Vessella RL, Nelson JB, Parikh RA, Wang Z. 5alpha-reductase inhibition suppresses testosterone-induced initial regrowth of regressed xenograft prostate tumors in animal models. Endocrinology. 2013;154:2296–2307. doi: 10.1210/en.2012-2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sedelaar JP, Isaacs JT. Tissue culture media supplemented with 10% fetal calf serum contains a castrate level of testosterone. Prostate. 2009;69:1724–1729. doi: 10.1002/pros.21028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong Y, Wang D, Dar JA, Singh P, Graham L, Liu W, Ai J, Xin Z, Guo Y, Wang Z. Nuclear export signal of androgen receptor (NESAR) regulation of androgen receptor level in human prostate cell lines via ubiquitination and proteasome-dependent degradation. Endocrinology. 2012;153:5716–5725. doi: 10.1210/en.2012-1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Askew EB, Minges JT, Hnat AT, Wilson EM. Structural features discriminate androgen receptor N/C terminal and coactivator interactions. Mol Cell Endocrinol. 2012;348:403–410. doi: 10.1016/j.mce.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demchenko AM, Sinchenko VG, Prodanchuk NG, Kovtunenko VA, Patratii VK, Tyltin AK, Babichev FS. Synthesis and antifungal activity of 3-aryl-6,7-dihydro-5h-pyrrolo[1,2-a]imidazoles. Pharmaceutical Chemistry Journal. 1987;21:789–791. [Google Scholar]
- 31.Andersen RJ, Mawji NR, Wang J, Wang G, Haile S, Myung JK, Watt K, Tam T, Yang YC, Banuelos CA, Williams DE, McEwan IJ, Wang Y, Sadar MD. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 32.Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- 33.Kang Z, Janne OA, Palvimo JJ. Coregulator recruitment and histone modifications in transcriptional regulation by the androgen receptor. Mol Endocrinol. 2004;18:2633–2648. doi: 10.1210/me.2004-0245. [DOI] [PubMed] [Google Scholar]
- 34.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–610. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Nguyen MM, Masoodi KZ, Singh P, Jing Y, O’Malley K, Dar JA, Dhir R, Wang Z. Splicing Factor Prp8 Interacts With NES(AR) and Regulates Androgen Receptor in Prostate Cancer Cells. Molecular endocrinology. 2015;29:1731–1742. doi: 10.1210/me.2015-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]