

Abstract
Although some patients with acute leukemia have good prognoses, the prognosis of adult and pediatric patients who relapse or cannot tolerate standard chemotherapy is poor. Inhibition of WEE1 with AZD1775 has been shown to sensitize cancer cells to genotoxic chemotherapies including cytarabine in AML and T-ALL. Inhibition of WEE1 impairs homologous recombination by indirectly inhibiting BRCA2. Thus, we sought to determine if AZD1775 could sensitize cells to the PARP1/2 inhibitor olaparib. We found that combined treatment with AZD1775 and olaparib was synergistic in AML and ALL cells, and this combination impaired proliferative capacity upon drug withdrawal. AZD1775 impaired homologous recombination in olaparib-treated cells resulting in enhanced DNA damage accumulation and apoptosis induction. This combination enhanced disease control and increased survival in a murine AML model. Furthermore, we demonstrated that combined treatment with AZD1775 and olaparib reduces proliferation and colony formation and increases apoptosis in AML patient samples. In aggregate, these studies raise the possibility of rational combinations of targeted agents for leukemia in patients for whom conventional chemotherapeutics may not be effective or well tolerated.
Keywords: WEE1, PARP, leukemia, olaparib, AZD1775
Introduction
Acute leukemia is the most common form of pediatric cancer and a leading cause of cancer related deaths in children. While survival rates in children have improved, in part due to dose escalation and optimization of chemotherapy regimens, up to 50% of patients with AML and 20% of patients with ALL relapse, and the prognosis for these patients is poor (1,2). Outcomes for adult patients with acute leukemia are generally worse, particularly in AML where incidence increases with advanced age (3). Due to its high toxicity, the use of chemotherapy is limited in patients over age 60, leaving few treatment options for many AML cases (4). Further dose escalation in pediatric patients or addition of non-targeted genotoxic agents is likely to increase toxicity while imparting minimal improvements in survival to these patients, necessitating the advent of novel therapeutic strategies (5).
One therapeutic strategy that has shown promise in BRCA1/2-mutated solid tumors is inhibition of poly (ADP-ribose) polymerase (PARP) (6,7). The PARP family includes 18 members, 3 of which are enzymes known to function in various DNA-repair processes including the recognition of single strand breaks (SSBs) in base excision repair (BER)(8). Impaired BER in PARP-inhibited cells results in accumulation of single-strand breaks that are converted to double-strand breaks upon collision with replication forks during S phase of the cell cycle (7). PARP-inhibited cells rely on homologous recombination (HR) for DNA damage repair and survival, and cells possessing defects in the HR pathway are particularly susceptible to PARP inhibitors (4,9–11).
Despite its promise in some solid tumors, the clinical application of PARP inhibitors in acute leukemia has been limited. This limited success is partly because mutations in DNA damage response (DDR)-associated genes are not common in acute leukemia, which is often driven by transcription factor mutations or gene fusions (4,12,13). However, pharmacologic impairment of the HR pathway could potentially sensitize acute leukemia cells to PARP inhibition. One approach to impairing the HR pathway is through inhibition of WEE1. WEE1 is a tyrosine kinase that regulates the cell cycle during the G2/M- and S-phases via inhibitory phosphorylation of CDK1 and CDK2, respectively. In response to DNA damage, CHK1 phosphorylates and activates WEE1 to promote cell cycle arrest and DNA damage repair (14,15). A small molecule inhibitor of WEE1, AZD1775, has been shown to abrogate cell cycle arrest and enhance DNA damage induction and apoptosis in cells exposed to genotoxic chemotherapies such as cytarabine, gemcitabine, and cisplatin (16–19). In addition to its effects on the cell cycle, inhibition of WEE1 impairs HR-mediated repair through forced activation of CDK1 and subsequent inhibitory phosphorylation of BRCA2 (20). Therefore, we hypothesized that inhibition of WEE1 via AZD1775 would sensitize acute leukemia cells to the PARP inhibitor olaparib. Our data confirm that inhibition of WEE1 impairs HR in olaparib-treated cells. Combined inhibition of WEE1 and PARP1/2 results in a synergistic reduction in cellular proliferation in AML and ALL cell lines, likely through impaired HR and subsequent DNA damage accumulation and induction of apoptosis. This combination extended survival of mice with an MLL-rearranged murine AML. Finally, addition of AZD1775 to olaparib treated AML patient samples enhanced apoptosis and reduced colony formation in methylcellulose. In aggregate, these studies indicate that combined treatment with olaparib and AZD1775 could be an effective treatment of acute leukemia.
Methods
Cell Culture
Jurkat, Molm13, MV4;11, REH, and OCI-AML3 cell lines were generous gifts from the laboratories of Drs. Douglas Graham and James DeGregori. 32D cells were purchased from ATCC. Cell lines were DNA fingerprinted by multiplex PCR using the Profiler Plus or Identifier Kits (ABI) as previously described (21), and periodically tested for Mycoplasma by PCR. Cells were cultured in RPMI with 10% FBS and penicillin/streptomycin at 37°C in humidified air supplemented with 5% CO2 and maintained in culture for no longer than 2 months. WEHI3 conditioned media was added to the culture media at 10% as a source of IL-3 for 32D cells. Isogenic cell lines were generated by transduction of target cells with Murine Stem Cell Virus expressing genes of interest followed by an internal ribosome entry site and green fluorescence protein (MSCV-iresGFP), as previously described (21). Primary AML samples were collected after informed consent with approval of the Colorado Multiple Institutional Review Board. Ficoll-separated mononuclear cells were cultured in IMDM supplemented with 20% BIT Serum Substitute, LDL, B-ME, penicillin/streptomycin, L-glutamine, IL-3, Flt-3 ligand, and SCF. To assess colony-forming ability, primary samples were plated in Methocult H4434 Classic (Stemcell Technologies, Vancouver, BC, Canada). Colonies were counted after 10–14 days of continuous drug exposure.
Chemotherapies, Antibodies, and Reagents
Olaparib and AZD1775 were provided by AstraZeneca (Wilmington, DE). The chemical structure of AZD1775 has previously been described (22). Antibodies against phosphorylated CDK1 (Y15), Total CDK1, γH2AX, tubulin, and caspase 3 were purchased from Cell Signaling Technology (Danvers, MA).
Comet Assays
Formation of DNA double strand breaks was assessed using CometAssay kits (Trevigen, Gaithersburg, MD). Cells were collected after 72 hours of drug treatment and embedded in agarose on slides. Slides were subjected to electrophoresis in TAE buffer followed by staining with SYBR Green (Trevigen) and visualization by fluorescent microscopy at 4x magnification. CometScore software (TriTek, Summerduck, VA) was used to calculate the olive moment, the product of the mean tail migration distance and the fraction of total DNA in the tail, of a minimum of 50 cells per condition (23).
Immunofluorescence
Cells were treated with AZD1775 and/or olaparib for 48 hours in poly-D-lysine coated chamber slides (Sigma-Aldrich, St Louis, MO). Cells were then fixed with 4% paraformaldehyde for 15 minutes at room temperature and permeabilized with 0.2% Triton X in PBS for 10 minutes. Cells were incubated in blocking solution (5% milk in 0.05% Triton X-PBS) for 30 minutes followed by an overnight incubation in anti-Rad51antibody (Cell Signaling Technology) at a dilution of 1:500. After several washes, Alexa Fluor 488 conjugated anti-rabbit antibody (1:500) was applied for 1 hour. ProLong Gold Antifade Mountant with DAPI (Life Technologies, Waltham, MA) was used for mounting. Images were acquired using an inverted epifluorescence microscope at 100X magnification.
Flow Cytometry
Cell viability was determined with the Guava EasyCytePlus (Millipore, Billerica, MA) by measuring cell counts with propdium iodide exclusion. Apoptosis was assessed using Guava Nexin reagent according to the manufacturer’s protocol (Millipore). Cell cycle analysis was performed using Guava Cell Cycle Reagent according the manufacturer’s protocol (Millipore).
Beta-Galactosidase Staining
Cells were treated with AZD1775 and/or olaparib for 48 hours. After drug treatment, cells were placed in drug-free media and cultured in poly-D-lysine coated chamber slides (Sigma-Aldrich) for an additional 96 hours. Beta-galactosidase staining was performed using the senescence β-galactosidase staining kit according to the manufacturer’s protocol (Cell Signaling Technology).
Animal Experiments
Female 6-week old C57BL/6J mice were purchased from The Jackson Laboratory and housed in sterile micro-isolators in the Center for Comparative Medicine at the University of Colorado Anschutz Medical Campus (Aurora, CO). Five mice per treatment group were used for each of the two experiments for a total of ten mice per treatment group. Five hundred thousand luciferase tagged AML cells were injected by tail vein injection into un-irradiated recipients to induce leukemia (24). Beginning 3 days later, mice were treated 5 days/week with olaparib 50 mg/kg and/or AZD1775 80 mg/kg once per day by oral gavage. Luciferase activity was measured 5 minutes after injection of luciferin using a Xenogen IVIS2000 imaging system. Animal studies were approved by the Institutional Animal Care and Use Committee of the University of Colorado Denver.
Statistical Analysis
Data analysis and graphing was performed using Graphpad Prism 5 (GraphPad Software, La Jolla, CA). Unless otherwise indicated, graphs represent the mean from a minimum of three biological replicate experiments, and error bars portray the standard error of the mean. One-way ANOVA was used to compare 3 or more samples with a single variable. Two-way ANOVA was used to compare 3 or more samples with 2 variables. The Bonferroni correction was applied to determine significance between any two conditions. Combination Index (CI) values were calculated using the equation of Chou and Talay with CalcuSyn (Biosoft, Cambridge, United Kingdom) (25). CI values were classified as CI >1.1, antagonism; CI = 0.9–1.1, additive; CI = 0.85–0.9, slight synergism; CI = 0.7–0.85, moderate synergism; CI = 0.3–0.7, synergism; CI = 0.1–0.3, strong synergism; and CI < 0.1, very strong synergism (26). The Mantel-Cox (log-rank) test was used to determine significant differences in survival.
Results
AZD1775 Impairs Homologous Recombination
Cells treated with PARP inhibitors require an intact HR pathway to prevent DNA damage accumulation. Inhibition of WEE1 impairs HR by promoting increased CDK1 activity and subsequent inhibitory phosphorylation of BRCA2 (20). Thus, we sought to determine if inhibition of WEE1 could reduce HR in acute leukemia cells treated with the PARP1/2 inhibitor olaparib. To assess this, we selected five cell lines representing 3 subtypes of acute leukemia. MV4;11 and Molm13 are AML cells lines that possess MLL rearrangements and FLT3 internal tandem duplications, while OCI-AML3 cells harbor mutations in NPM1 and DNMT3A. Jurkat is a T-cell ALL line with a mutated TP53 gene, and REH is a B-cell ALL cell line that harbors an ETV6-RUNX1 fusion.
First, we examined the anti-proliferative effect of olaparib and AZD1775 as single agents in short-term liquid culture. Both olaparib and AZD1775 had similar single agent activity in MV4;11, Molm13, Jurkat, and REH cells (Supplemental Figure 1). In light of the inhibitory effect of olaparib on these cell lines in this short-term assay, we next addressed whether they were competent in homologous recombination. Query of the Cancer Cell Line Encyclopedia revealed heterozygous mutations in homologous recombination pathway genes, BRCA (Molm13 and REH) and NBN (MV4;11), although the extent to which these mutations impair homologous recombination is unknown. Thus, we sought to determine whether these cell lines had intact HR in a functional assay. PARP1 deficiency or inhibition enhances the accumulation of Rad51 foci in cells in which the HR machinery is functional (27). In response to olaparib treatment, each cell line displayed a significant increase in Rad51 foci formation indicating these cell lines are capable of activating the HR pathway (Supplemental Figure 2). After demonstrating an increase in Rad51 foci in response to olaparib treatment, we sought to determine whether addition of AZD1775 could impair activation of the HR pathway. Indeed, when MV4;11 cells were treated with AZD1775 and olaparib, we observed reduced Rad51 foci compared to cells treated with olaparib alone (Figure 1a,b). Thus, inhibition of WEE1 impairs HR in olaparib-treated leukemia cells, consistent with previous findings in breast cancer cells (20).
Figure 1.
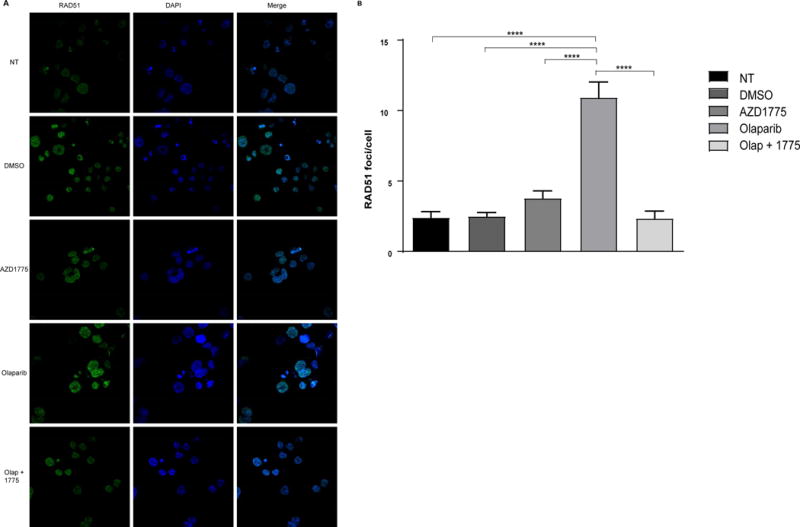
AZD1775 inhibits homologous recombination in olaparib-treated cells. A. MV;411 cells were treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 48 hr after which cells were fixed and stained with anti-Rad51 (primary antibody), Alexa Fluor 488 conjugated secondary antibody, and DAPI. Representative images are shown. B. Rad51 foci per cell was calculated from a minimum of 10 immunofluorescent images. Results are displayed as mean ± SEM from a minimum of 100 cells per condition. ****, P < 0.0001.
AZD1775 Synergizes with Olaparib in Acute Leukemia Cell Lines and Reduces Proliferative Capacity Upon Drug Withdrawal
As AZD1775 impaired HR in cells treated with olaparib, we sought to determine whether cells treated with AZD1775 and olaparib demonstrate reduced viability compared to cells treated with olaparib alone. At doses achievable in the plasma of patients (28), AZD1775 potentiated the reduction in proliferation induced by olaparib in four out of five cell lines (Figure 2a and Supplemental Figure 3). In order to better model a clinical setting in which cancer cells are exposed to varying drug concentrations over time, we examined a wider range of drug combinations in MV4;11 cells. Using Chou and Talay median effects analysis, we determined that 18 out of the 20 combinations analyzed were synergistic (CI < 1.0), with higher concentrations of olaparib and AZD1775 resulting in greater levels of synergy (Table 1). Treatment with 6 dose combinations of olaparib and AZD1775 yielded CI values that were additive to moderately synergistic in Molm13 and REH cells (CI: 0.931–0.627 and CI: 0.983–0.654, respectively) and additive to slightly synergistic in Jurkat cells (CI: 1.025–0.805; not shown). CI values from OCI-AML3 cells were antagonistic to additive (CI: 1.817–0.994).
Figure 2.
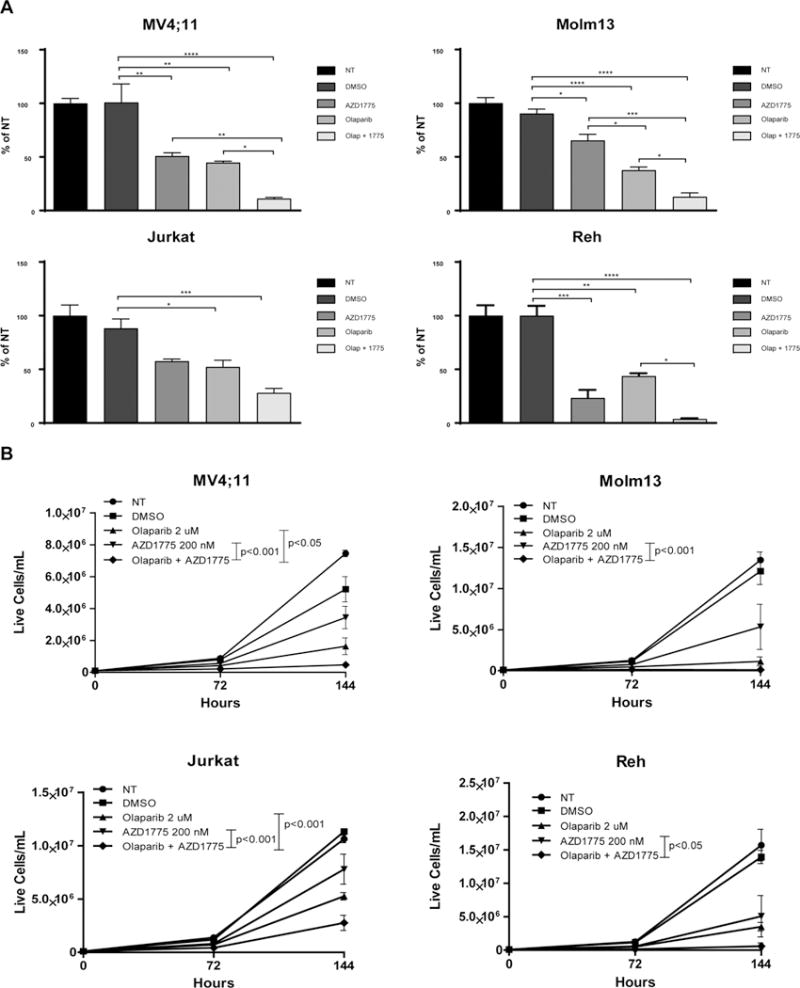
AZD1775 enhances the anti-proliferative effect of olaparib on leukemia cells. A. Relative numbers of viable cells treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 72 hr. Viable cell counts are normalized to cells receiving no treatment (NT). Results are shown as mean ± SEM from three independent experiments. *, P < 0.05. **, P < 0.01. ***, P < 0.001, ****, P < 0.0001. B. Live cell number of NT AML and ALL cell lines or treated with the indicated concentrations of DMSO (vehicle control), olaparib, and/or AZD1775 for 72 hr, removed from drug and cultured in fresh media for another 72 hr. Results are displayed as mean ± SEM from three independent experiments. Displayed P values were calculated by two-way ANOVA.
Table 1.
Combination Index Values for MV4;11 Cells Treated with Olaparib and AZD1775 for 72 Hours
| AZD1775 (nM) | |||||
|---|---|---|---|---|---|
| Olaparib (uM) | 50 | 100 | 200 | 400 | 800 |
| 0.25 | 0.787 | 0.963 | 0.914 | 0.641 | 0.804 |
| 0.5 | 0.867 | 0.881 | 0.994 | 0.891 | 0.597 |
| 1.0 | 0.976 | 1.138 | 0.814 | 0.546 | 0.678 |
| 2.0 | 1.028 | 0.943 | 0.547 | 0.420 | 0.588 |
Previous reports by others have suggested that cells lacking functional P53 are reliant on WEE1 to maintain genomic integrity when exposed to DNA damaging agents (29). Since OCI-AML3 cells (with functional P53) had the least response to the combination of AZD1775 and olaparib, we used these cells to test whether impaired P53 function sensitizes these cells to combination therapy. We inhibited P53 in OCI-AML3 cells, by stably expressing the dominant negative dimerization domain of P53 (DDp53) from MSCV-ires-GFP as we have done before (21). AZD1775 did not sensitize OCI-AML3-ddP53 cells to olapararib (Supplemental Figure 3), indicating that the sensitivity to AZD1775 and olaparib is independent of P53 functionality and consistent with our data from cell lines with (Jurkat) and without (MV4;11, Molm13, REH) TP53 mutation. As sensitive AML cell lines (Molm13, MV4;11) harbor FLT3-ITD, while the insensitive OCI-AML3 cell lines do not, and FLT3-ITD is associated with impaired DNA damage response (30,31), we expressed FLT3-ITD, in the 32D murine myeloid progenitor cell line, but did not observe enhanced reduction in proliferation in cells expressing these oncogenes (Supplemental Figure 4), suggesting that FLT3-ITD is not a marker predictive of sensitivity to AZD1775 and olaparib.
As cancer cells often recover proliferative capacity after treatment with chemotherapeutics, we sought to determine if AZD1775 could prevent proliferation in olaparib-treated cells once drug was removed. To accomplish this, cells were treated with olaparib and/or AZD1775 for 72 hours then re-plated without drug and cultured for an additional 72 hours. While olaparib treatment resulted in decreased proliferative capacity, addition of AZD1775 prevented the recovery of cells compared to single agent treatment (Figure 2b), suggesting that this combination induces prolonged proliferative defects or DNA-damage induced senescence. Collectively, these results reveal the potential therapeutic utility of olaparib and AZD1775 in a variety of, but not all, acute leukemia subtypes, independent of P53 functionality.
AZD1775 Enhances DNA Damage, Apoptosis, and Senescence Induced by Olaparib
As cells with impaired PARP activity require HR to resolve single strand breaks that occur spontaneously, we hypothesized that impaired HR induced by WEE1 inhibition would result in accumulation of DNA damage in cells treated with olaparib and AZD1775 (7). To test this hypothesis, we used the alkaline comet assay to visualize double strand breaks (DSBs) in cells treated with olaparib and/or AZD1775. AZD1775 enhanced DNA damage in olaparib-treated cells as evidenced by the prevalence of distinct comet tails (Figure 3a). Indeed, the addition of AZD1775 enhanced the average olive moment by approximately 10-fold compared to olaparib alone (Figure 3b). This increase in DNA damage was confirmed by increased γH2AX in cells with both PARP and WEE1 inhibition (Figure 3c).
Figure 3.
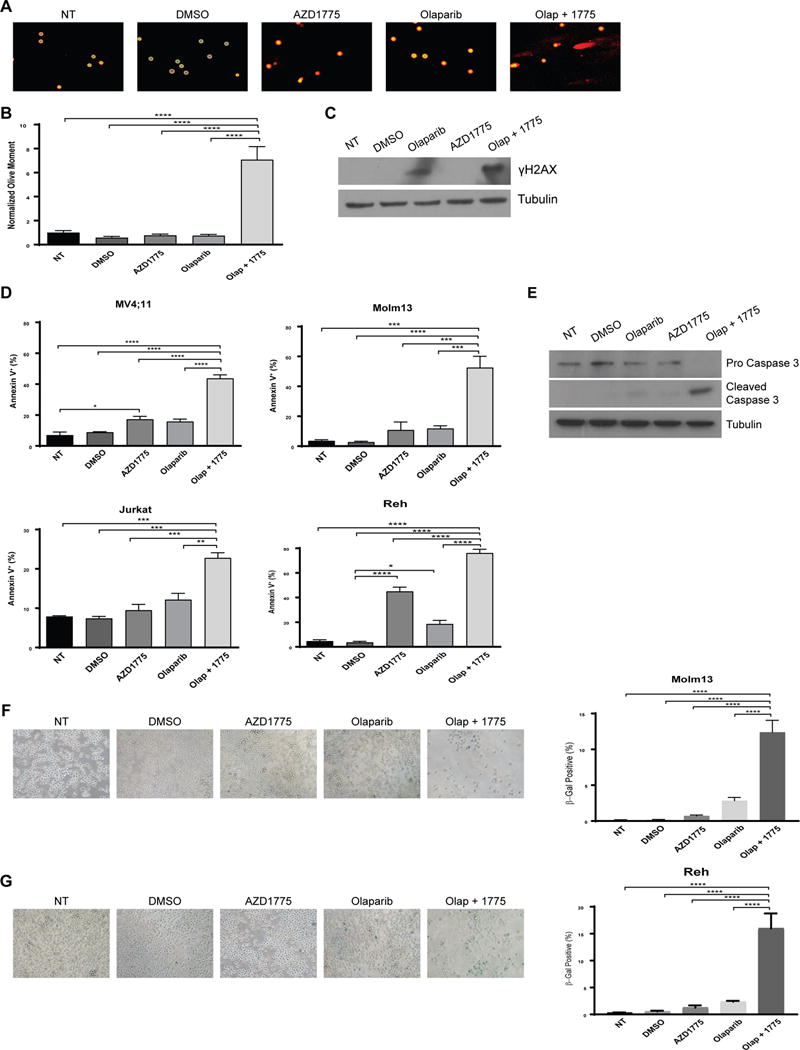
Combined AZD1775 and olaparib treatment enhances DNA damage accumulation, apoptosis induction, and senescence. A. MV4;11 cells were treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 72 hr and analyzed via Comet assay. Images are representative from one of three independent experiments. B. Olive moment values normalized to MV4;11 cells receiving no treatment (NT). Results are shown as mean ± SEM from one of three independent experiments. A minimum of 50 cells per experiment were analyzed. ****, P < 0.0001. C. MV4;11 cells were treated with DMSO (vehicle control), olaparib (2 uM) and/or AZD1775 (200 nM) for 48 hr after which protein lysates were subjected to western blotting using antibodies specific to γH2AX and tubulin. D. Indicated cell lines were treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 72 hr and stained with Annexin V-PE. Results are displayed as mean ± SEM of three independent experiments. *, P < 0.05. **, P < 0.01. ***, P < 0.001. ****, P < 0.0001. E. MV4;11 cells were treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 48 hr after which protein lysates were subjected to western blotting using antibodies specific to caspase 3 and tubulin. F and G. Molm13 and REH cells were treated with DMSO (vehicle control), olaparib (1 uM), and/or AZD1775 (100 nM) for 48 hr, re-plated in drug-free media for an additional 96 hr, then fixed and stained for β-galactosidase. Images are representative from one of two independent experiments. Percent of β-gal positive cells are quantified to the right. A minimum of 200 cells per condition was analyzed. ****, P < 0.0001.
To determine whether addition of AZD1775 to olaparib induced more apoptosis compared to olaparib alone, we measured apoptosis by Annexin V staining after 72 hours of treatment. The addition of AZD1775 to olaparib lead to significantly more apoptosis than with either agent alone (Figure 3d). We also observed an increase in cleaved caspase 3 accompanied by reduced pro caspase 3 in MV4;11 cells treated with olaparib and AZD1775, further suggesting increased apoptosis in cells treated with both PARP and WEE1 inhibitors (Figure 3e).
As cells treated with olaparib and AZD1775 fail to recover proliferative capacity upon drug removal (Figure 2b), we sought to determine whether the drug combination induced senescence in surviving cells. To test this, Molm13 and REH cells were treated with AZD1775 and/or olaparib for 48 hours. After drug treatment, cells were re-plated in drug-free media for an additional 96 hours then fixed and stained for beta-galactosidase. In Molm13, we observed a 6-fold increase in beta-galactosidase positive cells in cells treated with olaparib and AZD1775 compared to cells treated with olaparib alone (Figure 3f). REH cells treated with the drug combination displayed an 8-fold increase in beta-galactosidase positive cells compared to cells treated with only olaparib (Figure 3g).
Our lab and others have previously demonstrated that AZD1775 enhances the efficacy of some genotoxic agents by abrogating the cell cycle arrest that occurs in cells treated with chemotherapy alone (16,32). Therefore, we examined whether changes in the cell cycle occur in cells treated with olaparib and AZD1775. Jurkat cells treated with olaparib display a G2/M phase arrest, and this is abrogated with addition of AZD1775 (Supplemental Figure 5a,c). Additionally, Molm13 cells treated with the drug combination have a decreased percentage of cells in the G2/M phase, although olaparib alone does not induce a G2/M phase arrest in this cell line (Supplemental Figure 5a). However, we did not observe similar changes in other cell lines tested indicating that altered cell cycle progression is not a unifying mechanism responsible for the combinatorial activity of olaparib and AZD1775 (Supplemental Figure 5a,b). Together, these results indicate that combined inhibition of WEE1 and PARP1/2 enhances accumulation of DNA DSBs, apoptosis, and senescence, independent of cell cycle effects of the drugs.
Combined AZD1775 and Olaparib Treatment Enhances Survival of Mice with Murine AML
We then asked whether AZD1775 and olaparib treatment could slow leukemia progression in vivo. For these studies, we selected a murine MLL-ENL, FLT3-ITD+ AML cell line that responds to combined treatment with olaparib and AZD1775 in vitro (Supplemental Figure 6). The murine AML cells were injected via tail vein into mice as previously described (21). Single agent treatment with either olaparib or AZD1775 failed to enhance survival compared to vehicle controls (olaparib or AZD1775 median survival: 17 days; vehicle median survival: 14 days) (Figure 4c). However, the combination treatment slowed leukemia progression as measured by luciferase expression and extended median survival to 31 days (Figure 4a–c). Mouse weights and complete blood counts were assessed 7 days after treatment initiation to evaluate toxicity. Mice receiving combination treatment had reduced hemoglobin levels compared to mice treated with vehicle control or olaparib; however, there was no change in weight, red blood cell counts, white blood cell counts, or platelet levels with either single agent or combination treatment (Supplemental Figure 7). Two of the ten mice treated with AZD1775 and olaparib were euthanized due to ill appearance, although no evidence of leukemia was observed at the time of sacrifice.
Figure 4.
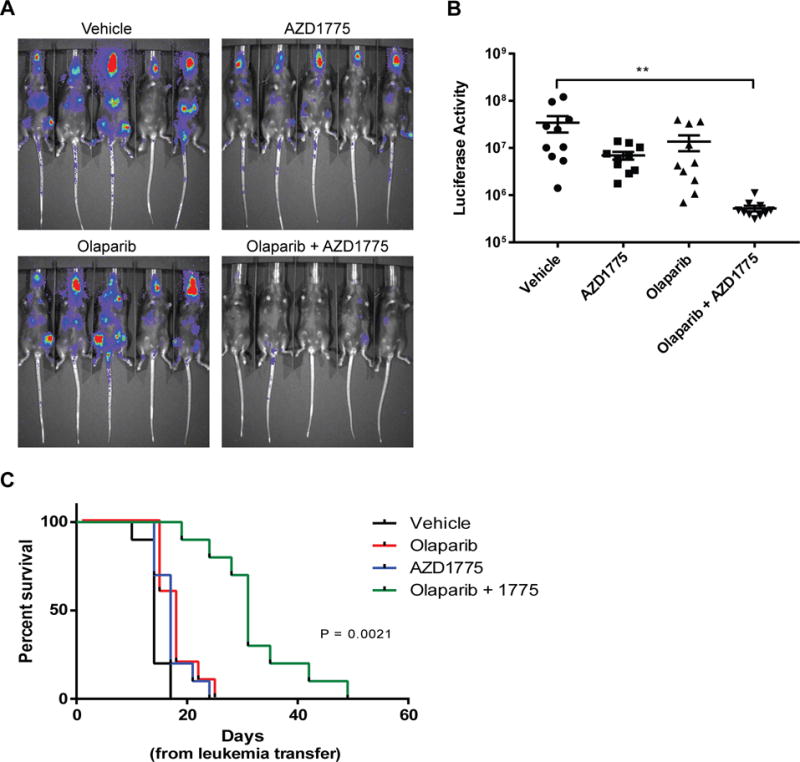
Combined treatment with AZD1775 and olaparib improves survival in a murine AML model. A. Murine AML cells were injected intravenously into C57BL/6J mice, and treatment was initiated 3 days after leukemia transfer. Mice were treated 5 days per week until leukemia burden as measured by luciferase intensity reached the threshold for sacrifice. Displayed images are representative IVIS imaging acquired 10 days after leukemia transfer. B. Quantified luciferase activity 10 days after leukemia transfer. Results are displayed as mean ± SEM from two independent experiments. **, P < 0.01 (vehicle vs. olaparib + AZD1775). C. Kaplan-Meier curve of mice with murine AML treated as indicated. Results are shown from 2 independent experiments.
AZD1775 Enhances Apoptosis Induction and Reduces Colony Formation in AML Patient Samples
We next examined the effects of AZD1775 and olaparib on cell viability, apoptosis induction, and colony formation on AML patient samples. Samples with baseline apoptosis greater than 40% 72 hours after plating or that did not form colonies under no treatment conditions were excluded from analysis (Supplemental Figure 8). Patient sample AML 10-5-10 harbors a FLT3-ITD mutation (33), but genetic information for the other patient samples was not available. Exposure to the drug combination resulted in a reduction in viable cell numbers, an increase in apoptotic cells, and a decline in colony formation (Figure 5a, b, c). Further supporting reduced colony forming ability, the colonies that did form in cells exposed continuously to AZD1775 and olaparib were smaller than those in cells exposed to olaparib (Figure 5d). Similar to our observations in cell lines, combined treatment with olaparib and AZD1775 enhanced DNA damage as evidenced by elevated γH2AX (Figure 5e). Thus, combined olaparib and AZD1775 treatment could be an effective therapeutic approach for acute leukemia.
Figure 5.
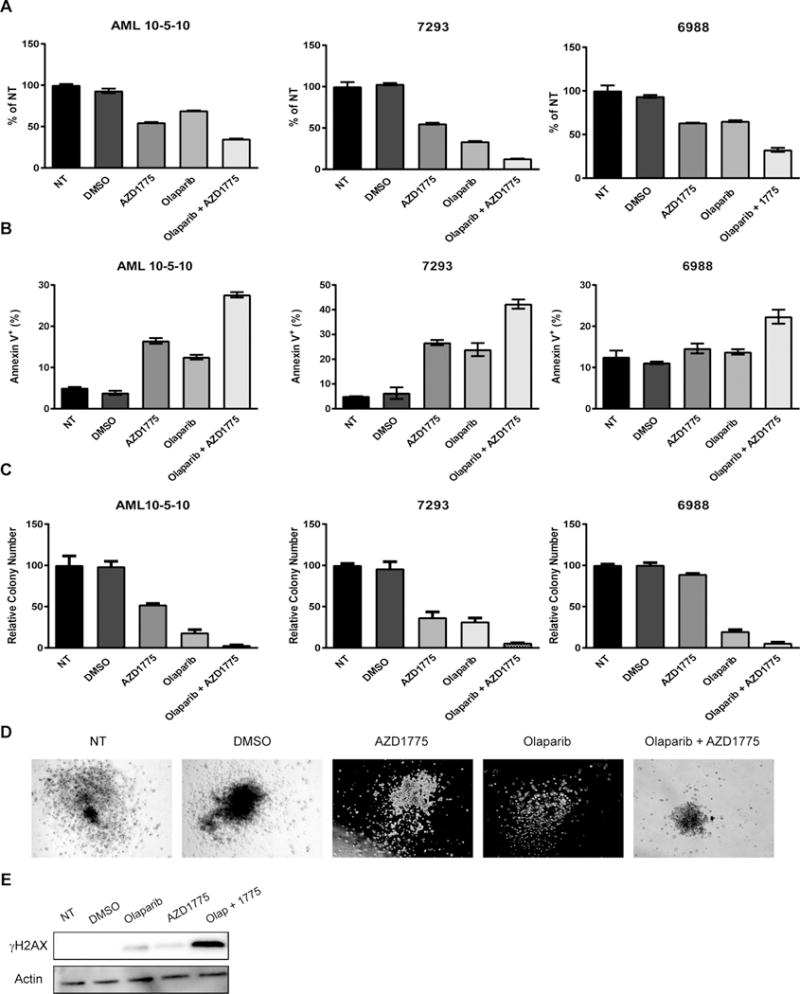
AZD1775 and olaparib reduce proliferation and colony formation and induce apoptosis in AML patient samples. A and B. AML patient samples were treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 72 hr and analyzed for cell viability (A) and apoptosis induction (B). Results are displayed as mean ± SEM of two technical replicates. C. Quantification of colonies formed in methylcellulose after continuous exposure to DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM) for 10–14 days. Results are normalized to cells receiving no treatment (NT) and are displayed as mean ± SEM of three technical replicates. D. Representative images of AML sample 7293 colonies formed after 10 days of continuous exposure to DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (200 nM). E. Patient sample AML 10-5-10 was treated with DMSO (vehicle control), olaparib (2 uM), and/or AZD1775 (nM) for 48 hr after which protein lysates were subjected to western blotting using antibodies specific to γH2AX and actin.
Discussion
While improvements to chemotherapy regimens have increased survival of patients with acute leukemia, outcomes remain poor for patients that relapse and for elderly patients that cannot tolerate standard treatment (34,35). Thus, novel treatment options are necessary to improve outcomes in these patient populations. In this study, we have demonstrated that the rational combination of AZD1775 and olaparib inhibits proliferation and enhances apoptosis in AML and ALL cell lines and AML patient samples. This combination also significantly prolonged survival of mice with murine AML.
While olaparib has proven to be an effective treatment in BRCA1/2-mutant malignancies, the expansion of this drug to cancers lacking defects in the HR pathway has been limited (6,7,36,37). Here we demonstrate that inhibition of WEE1, a known regulator of HR, sensitizes BRCA1/2 wild type and mutant cell lines to olaparib. The doses of AZD1775 that we used in vitro are comparable to the IC50 of this compound in cell based assays in which phosphorylation of CDK is inhibited, and lower than achieved in vivo in an early phase clinical trial (38). Similarly, the concentrations of olaparib used in vitro can be achieved in vivo (39), although they are higher than is necessary to inhibit poly-(ADP)-ribosylation in vitro (40). We have demonstrated that inhibition of WEE1 impairs HR in olaparib-treated cells. The precise mechanisms of cell death due to PARP inhibition in the context of impaired homologous recombination remain unresolved, with at least 4 models proposed (8). These models share the common endpoint of unresolved and accumulated DNA damage and apoptosis, which our data support. While the impairment of HR may contribute to the observed sensitization, WEE1 has multiple effects on the cell cycle and DNA damage repair pathways through its regulation of CDK1 and CDK2 that could also contribute to the sensitization to olaparib. Indeed, WEE1 inhibition results in stalled replication forks that require processing to avoid collapse, and PARP is required for recruitment of DDR proteins to stalled forks for processing and repair, suggesting that failure to repair stalled replication forks could also contribute to the observed combinatorial activity of AZD1775 and olaparib (41,42).
Early work describing WEE1 inhibition as an effective chemosensitizing agent suggested combinations with WEE1 inhibitors were only effective in cells lacking functional P53 (19,43–45). However, previous work from our lab has demonstrated that inhibition of WEE1 sensitizes AML and lung cancer cells to antimetabolite chemotherapeutics independent of P53 functionality (21). Likewise, this work demonstrates that AZD1775 synergizes with olaparib in both TP53 wild-type (MV4;11, Molm13, REH) and mutant (Jurkat) cell lines. Furthermore, in an isogenic model, inhibition of endogenous P53 failed to enhance the combinatorial effect of AZD1775 and olaparib suggesting that sensitivity to this combination is independent of P53 functionality in this context. Although this combination appears to be effective regardless of TP53 status, it is still likely to display selectivity for cancer cells as a number of malignancies, including AML, have increased expression of WEE1 compared to noncancerous controls (16,46–48). As noted, AZD1775 did not significantly enhance the combinatorial effect of olaparib in all cell lines; thus, this combination is likely to be effective in some, but not all, acute leukemia cells. Further work is required to identify biomarkers to predict sensitivity to this combination.
In addition to our in vitro data, we have demonstrated that combined AZD1775 and olaparib treatment enhanced the survival of mice injected with a murine AML cell line. Notably, this cell line possesses a MLL-ENL fusion which has been shown to mediate resistance to PARP inhibitors via HOXA9-mediated upregulation of DDR genes (4,24). Thus, this combination could provide a novel treatment option for MLL-rearranged leukemias, which are frequently resistant to standard chemotherapies. It should be noted that two of the ten mice treated with the combination died with no evidence of leukemia. While mouse weights and complete blood counts suggest minimal drug toxicity after 7 days of treatment, future studies are necessary to assess cumulative toxicity with prolonged treatment. Although optimizations to dosing and the timing of treatments may be necessary, this combination is not expected to be particularly toxic, as both olaparib and AZD1775 have been used in multiple clinical trials with reasonable toxicity profiles (6,38). Indeed, early results from a phase 1b clinical trial combining olaparib with AZD1775 for the treatment of refractory solid tumors suggest no dose-limiting toxicities (49).
In summary, this report describes a novel synergistic combination, AZD1775 and olaparib, for the treatment of acute leukemia. We have demonstrated that pharmacologic inhibition of WEE1 has potential to broaden the application of PARP inhibitors beyond the current use in BRCA1/2-mutant cancers. Taken together, these data provide support for clinical trials testing AZD1775 and olaparib for acute leukemia.
Supplementary Material
Acknowledgments
Financial support: This work was supported by grants from the National Institutes of Health to C.C. Porter (CA172385), the University of Colorado Cancer Center (CA046934) to C.C. Porter, and the CU Medical Scientist Training Program (GM008497) to T.B. Garcia.
Abbreviation list
- ALL
acute lymphoblastic leukemia
- AML
acute myeloid leukemia
- ANOVA
analysis of variance
- BER
break excision repair
- CDK
cyclin-dependent kinase
- CI
combination index
- DDR
DNA damage response
- DNA
deoxyribonucleic acid
- DSBs
double-strand breaks
- HR
homologous recombination
- IVIS
in vivo imaging system
- MLL
mixed lineage leukemia
- PARP
poly (ADP-ribose) polymerase
- PBS
phosphate-buffered saline
- PCR
polymerase chain reaction
- SSBs
single-strand breaks
- TAE
tris base, acetic acid, and EDTA
References
- 1.Pui C-H, Carroll WL, Meshinchi S, Arceci RJ. Biology, Risk Stratification, and Therapy of Pediatric Acute Leukemias: An Update. Journal of Clinical Oncology. 2011;29:551–65. doi: 10.1200/JCO.2010.30.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandez HF, Sun Z, Yao X, Litzow MR, Luger SM, Paietta EM, et al. Anthracycline dose intensification in acute myeloid leukemia. N Engl J Med. 2009;361:1249–59. doi: 10.1056/NEJMoa0904544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lowenberg B, Rowe JM. Introduction to the review series on advances in acute myeloid leukemia (AML) Blood. 2016;127 doi: 10.1182/blood-2015-10-662684. [DOI] [PubMed] [Google Scholar]
- 4.Esposito MT, Zhao L, Fung TK, Rane JK, Wilson A, Martin N, et al. Synthetic lethal targeting of oncogenic transcription factors in acute leukemia by PARP inhibitors. Nat Med. 2015;21:1481–90. doi: 10.1038/nm.3993. [DOI] [PubMed] [Google Scholar]
- 5.Burnett AK, Hills RK, Milligan DW, Goldstone AH, Prentice AG, McMullin MF, et al. Attempts to optimize induction and consolidation treatment in acute myeloid leukemia: results of the MRC AML12 trial. J Clin Oncol. 2010;28:586–95. doi: 10.1200/JCO.2009.22.9088. [DOI] [PubMed] [Google Scholar]
- 6.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015;21:4257–61. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 7.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 8.De Lorenzo SB, Patel AG, Hurley RM, Kaufmann SH. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Frontiers in Oncology. 2013;3:228. doi: 10.3389/fonc.2013.00228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mo W, Liu Q, Lin CC, Dai H, Peng Y, Liang Y, et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22:1699–712. doi: 10.1158/1078-0432.CCR-15-1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ashworth A. A Synthetic Lethal Therapeutic Approach: Poly(ADP) Ribose Polymerase Inhibitors for the Treatment of Cancers Deficient in DNA Double-Strand Break Repair. J Clin Oncol. 2008;26:3785–90. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 11.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R, et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Eng J Med. 2015;373:1697–708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang L, Hamard P, Nimer S. PARP inhibitors: a treatment option for AML? Nat Med. 2015;21:1393–4. doi: 10.1038/nm.4007. [DOI] [PubMed] [Google Scholar]
- 13.Esposito MT, So CWE. DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma. 2014;123:545–61. doi: 10.1007/s00412-014-0482-9. [DOI] [PubMed] [Google Scholar]
- 14.Indovina P, Giordano A. Targeting the checkpoint kinase WEE1. Cancer Biol Ther. 2010;9:523–5. doi: 10.4161/cbt.9.7.11276. [DOI] [PubMed] [Google Scholar]
- 15.Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, et al. Preclinical Evaluation of the WEE1 Inhibitor MK-1775 as a Single Agent Anticancer Therapy. Mol Cancer Ther. 2013;3:1442–52. doi: 10.1158/1535-7163.MCT-13-0025. [DOI] [PubMed] [Google Scholar]
- 16.Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D, et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26:1266–76. doi: 10.1038/leu.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ford JB, Baturin D, Burleson TM, Van Linden AA, Kim YM, Porter CC. AZD1775 sensitizes T cell acute lymphoblatic leukemia cells to cytarabine by promoting apoptosis over DNA repair. Oncotarget. 2015;6:28001–10. doi: 10.18632/oncotarget.4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris PS, Venkataraman S, Alimova I, Birks DK, Balakrishnan I, Cristiano B, et al. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Mol Cancer. 2014;13 doi: 10.1186/1476-4598-13-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rajeshkumar V, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in 53-deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17 doi: 10.1158/1078-0432.CCR-10-2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krajewska M, Heijink AM, Bisselink YJWM, Seinstra RI, Sillje HHW, de Vries EGE, et al. Forced activation of Cdk1 via wee1 inhibition impairs homologous recombination. Oncogene. 2013;32:3001–8. doi: 10.1038/onc.2012.296. [DOI] [PubMed] [Google Scholar]
- 21.Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Mol Cancer Ther. 2013;12:2675–84. doi: 10.1158/1535-7163.MCT-13-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol Cancer Ther. 2009;8:2992–3000. doi: 10.1158/1535-7163.MCT-09-0463. [DOI] [PubMed] [Google Scholar]
- 23.Duez P, Dehon G, Kumps A, Dubois J. Statistics of the Comet assay: a key to discriminate between genotoxic effects. Mutagenesis. 2002;18:159–66. doi: 10.1093/mutage/18.2.159. [DOI] [PubMed] [Google Scholar]
- 24.Pardee TS, Zuber J, Lowe SW. Flt3-ITD alters chemotherapy response in vitro and in vivo in a p53-dependent manner. Exp Hematol. 2011;39 doi: 10.1016/j.exphem.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 26.Bhatla T, Wang J, Morrison DJ, Raetz EA, Burke MJ, Brown P, et al. Epigenetic reprogramming reverses the relapse-specific gene expression signature and restores chemosensitivity in childhood B-lymphoblastic leukemia. Blood. 2012;119:5201–10. doi: 10.1182/blood-2012-01-401687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Research. 2003;31:4959–64. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leijen S, van Geel RM, Pavlick AC, Tibes R, Rosen L, Razak AR, et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol. 2016 doi: 10.1200/JCO.2016.67.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marusyk A, Wheeler LJ, Mathews CK, DeGregori J. p53 Mediates Senescence-Like Arrest Induced by Chronic Replicational Stress. Molecular and Cellular Biology. 2007;27:5336–51. doi: 10.1128/MCB.01316-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: implications for genomic instability and therapy. Blood. 2010;116:5298–305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sallmyr A, Fan J, Datta K, Kim KT, Grosu D, Shapiro P, et al. Internal tandem duplication of FLT3 (FLT3/ITD) induces increased ROS production, DNA damage, and misrepair: implications for poor prognosis in AML. Blood. 2008;111:3173–82. doi: 10.1182/blood-2007-05-092510. [DOI] [PubMed] [Google Scholar]
- 32.Indovina P, Marcelli E, Di Marzo D, Casini N, Forte IM, Giorgi F, et al. Abrogating G(2)/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer Biol Ther. 2014;15:380–8. doi: 10.4161/cbt.27623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee-Sherick AB, Zhang W, Menachof KK, Hill AA, Rinella S, Kirkpatrick G, et al. Efficacy of a Mer and Flt3 tyrosine kinase small molecule inhibitor, UNC1666, in acute myeloid leukemia. Oncotarget. 2015;6:6722–36. doi: 10.18632/oncotarget.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll W, Raetz E. Clinical and Laboratory Biology of Childhood Acute Lymphoblastic Leukemia The Journal of Pediatrics. 2012;160:10–8. doi: 10.1016/j.jpeds.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Krug U, Buchner T, Berdel WE, Muller-Tidow C. The Treatment of Elderly Patients With Acute Myeloid Leukemia. Dtsch Arztebl Int. 2011;108:863–70. doi: 10.3238/arztebl.2011.0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fong PC, Yap TA, Boss DS, Carden CP, Mergui-Roelvink M, Gourley C, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28:2515–9. doi: 10.1200/JCO.2009.26.9589. [DOI] [PubMed] [Google Scholar]
- 37.Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N Engl J Med. 2012;366:1382–92. doi: 10.1056/NEJMoa1105535. [DOI] [PubMed] [Google Scholar]
- 38.Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, et al. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. J Clin Oncol. 2015;33:3409–15. doi: 10.1200/JCO.2014.60.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Del Conte G, Sessa C, von Moos R, Vigano L, Digena T, Locatelli A, Gallerani E, Fasolo A, Tessari A, Cathomas R, Gianni L. Phase I study of olaparib in combination with liposomal doxorubicin in patients with advanced solid tumors. Br J Cancer. 2014;111:651–9. doi: 10.1038/bjc.2014.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51:6581–91. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 41.Bryant HE, Petermann E, Schultz N, Jemth A, Loseva O, Issaeva N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. The EMBO Journal. 2009;28:2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dominquez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VAJ, Medema RH, et al. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J Cell Biol. 2011;194:567–79. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hirai H, Arai T, Okada M, Nishibata T, Kobayashi M, Sakai N, et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther. 2010;9:514–22. doi: 10.4161/cbt.9.7.11115. [DOI] [PubMed] [Google Scholar]
- 44.Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, et al. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001;61:8211–17. [PubMed] [Google Scholar]
- 45.Mizuarai S, Yamanaka K, Itandani H, Arai T, Nishibata T, Hirai H, et al. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol Cancer. 2009;8 doi: 10.1186/1476-4598-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.M T, Shiratori Y, Rengifo W, Igarashi K, Yamagata M, Kurokohchi K, et al. Cyclins and Cyclin-Dependent Kinases: Comparitive Study of Hepatocellular Carcinoma Versus Cirrhosis. Hepatology. 2003;37:534–43. doi: 10.1053/jhep.2003.50112. [DOI] [PubMed] [Google Scholar]
- 47.I E, Lord CJ, Grigoriadis A, McDonald S, Fenwick K, MacKay A, et al. Intergrated Functional, Gene Expression and Genomic Analysis for the Identification of Cancer Targets. PLoS One. 2009;4 doi: 10.1371/journal.pone.0005120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.GI M, Holm R, Emilsen E, Rosnes AKR, Slipicevic A, Florenes VA. High Expression of Wee1 Is Associated with Poor Disease-Free Survival in Malignant Melanoma: Potential for Targeted Therapy. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamilton EP, Wang JS, Falchook G, Jones SF, Cook C, Mugundu G, et al. A phase Ib study of AZD1775 and olaparib combination in patients with refractory solid tumors. J Clin Oncol 2016 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.