

Abstract
Objective
Congenital heart disease (CHD) with increased pulmonary blood flow (PBF) results in progressive pulmonary vascular endothelial dysfunction and associated increased perioperative morbidity. Utilizing our ovine model of CHD with increased PBF, we have previously demonstrated progressive endothelial dysfunction associated with disruption in carnitine homeostasis, mitochondrial dysfunction, decreased nitric oxide (NO) signaling, and enhanced reactive oxygen species (ROS) generation. However, potential alterations in these parameters in patients with CHD have not been investigated. The objective of this study was to test the hypothesis that children with increased PBF will have evidence of altered carnitine homeostasis, mitochondrial dysfunction, decreased NO levels, and increased ROS generation.
Design
A prospective single center cohort study
Setting
A tertiary care CICU/PICU
Patients
Arterial blood samples from 18 patients with CHD associated with increased PBF (ventricular septal defect, VSD), 20 with CHD without increased PBF (tetralogy of Fallot, TOF), and 10 without heart disease (controls) were obtained.
Interventions
Plasma levels of total carnitine (TC), free carnitine (FC), acylcarnitine (AC); and lactate-to-pyruvate ratios, an indicator of mitochondrial function, were determined and compared. In addition, levels of superoxide and hydrogen peroxide were determined and compared in patients with VSD and controls. Statistical analysis was performed using an unpaired t-test and ANOVA.
Measurements and Main Results
Baseline AC levels (25.7 ± 13 vs. 12.7 ± 8.3, P<0.05), the AC:FC ratio (0.8 ± 0.1 vs. 0.3 ± 0.05, P<0.05), and the lactate/pyruvate ratio were higher in VSD (27.5 ± 3.8 vs. 11.1 ± 4.1, P<0.05) than TOF; there were no differences between TOF and control. Superoxide and H2O2 levels were also higher in VSD compared to controls, and NOx levels were lower in VSD patients compared to TOF and controls (P<0.05).
Conclusions
These data suggest that increased PBF from VSD results in altered carnitine and mitochondrial homeostasis, decreased NO signaling, and increased ROS production. These data are consistent with our animal data demonstrating that altered carnitine homeostasis results in mitochondrial dysfunction, increased ROS production, and decreased bioavailable NO. Since disruption of carnitine metabolism may contribute to endothelial dysfunction, carnitine supplementation may attenuate endothelial dysfunction associated with increased PBF and warrants further investigation.
Keywords: carnitine, congenital heart disease, nitric oxide, pulmonary hypertension, superoxide, pulmonary blood flow
INTRODUCTION
Pulmonary vascular dysfunction is perhaps the most important complication for children with common congenital heart defects (CHD) that result in increased pulmonary blood flow (PBF) and pressure, such as large ventricular septal defects (VSD) and atrioventricular septal defects.(1, 2) Beginning immediately after birth, the pulmonary vasculature in these infants is subjected to pathologic mechanical forces, which result in early functional abnormalities of the vascular endothelium.(3, 4) Normally, repairing these defects in infancy or early childhood prevents the development of irreversible pulmonary vascular disease. However, perioperative pulmonary vascular dysfunction, particularly following cardiopulmonary bypass, continues to contribute to the morbidity and mortality of these patients.(5, 6) Integrated in vitro and in vivo data from our laboratory and others demonstrate that exposure to these abnormal mechanical forces results in early and progressive abnormalities in pulmonary vascular endothelial structure and function including progressive decreases in bioavailable nitric oxide (NO) and increases in oxidative stress.(3, 7–12) Utilizing our clinically relevant lamb model of CHD, we have generated compelling data demonstrating that increased PBF impairs carnitine homeostasis, a vital role in cellular energy production. Our in vivo and in vitro animal data further suggest that this altered carnitine homeostasis leads to mitochondrial dysfunction, increased reactive oxygen species (ROS) production, and decreased bioavailable NO.(7–11, 13) However, potential alterations in these parameters in patients with CHD have not been investigated.
Carnitine is present in the body as free carnitine (FC) or as acylcarnitines (AC, esterified form). Adequate carnitine levels and optimal activities of carnitine-dependent enzymes are needed to allow the carnitine system to work. The main function of L-carnitine is the transport of long-chain fatty acids from the cytosol to the mitochondrial matrix for β-oxidation and ATP production. L-carnitine, however, also plays a key regulatory role in intermediary metabolism by modulating the cellular acyl-CoA/CoA ratio. CoA is an obligate cofactor for many enzymes involved in intermediary metabolism. It remains compartmentalized in limited pools within the cell, mainly in the mitochondria, and is normally kept in equilibrium with carnitine. The reversible transfer of acyl groups from CoA to carnitine ensures the vital maintenance of free CoA pools within the mitochondria and prevents the accumulation of poorly metabolized short-chain acyl-CoA compounds which are exported out of the mitochondria as carnitine esters. Therefore, the carnitine system is crucial for normal mitochondrial function, as the accumulation of acyl groups and the unavailability of free CoA result in a metabolic roadblock within the mitochondria with subsequent impaired oxidative metabolism, increased mitochondrial ROS generation, and decreased energy production.(14–16)
Thus, we hypothesized that children with increased PBF will have evidence of altered carnitine homeostasis, mitochondrial dysfunction, decreased NO signaling, and increased ROS generation. To this end, we determined and compared plasma levels of carnitine, lactate/pyruvate ratios (an indirect determinant of mitochondrial function), bioavailable NO (NOx) levels (a marker of NO generation), and ROS, in children with CHD associated with increased PBF (VSD), children with CHD without increased PBF (tetralogy of Fallot, TOF), and children without heart disease (controls).
METHODS
We conducted a prospective cohort study in the Pediatric Intensive Care Units (PICU) at the UCSF Benioff Children’s Hospital, San Francisco. Parents of all eligible patients were approached for informed consent and study enrollment. Eligible subjects included all infants (<1 year of age) with a large VSD or TOF undergoing elective surgical repair. ECHO criteria used to define that the VSD was large was: (1) a non-restrictive flow pattern across the VSD; (2) a VSD size larger than the aortic annulus; and (3) evidence of left atrial and left ventricular enlargement. Ten patients admitted in the PICU without heart disease served as controls. The University of California, San Francisco Hospital review board approved the study.
Blood samples were obtained from an arterial catheter < 24 hours preoperatively. The samples were placed immediately on ice in chilled ethylenediamine tetraacetic acid-treated tubes. Plasma was frozen at −80°C within 15 minutes of collection until analysis.
Plasma Acylcarnitine and free carnitine levels
Plasma Acylcarnitine and free carnitine levels were determined using High Performance Liquid Chromatography.(13) NOx levels were determined by chemiluminescence.(11, 13)
Lactate and Pyruvate levels
Lactate and Pyruvate levels were determined by spectrophometric measurements as previously described.(13)
Plasma Superoxide levels
Plasma Superoxide levels were estimated by electron paramagnetic resonance (EPR) assay using the spin-trap compound 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine·HCl (CMH) in the presence and absence of polyethylene glycol-superoxide dismutase (PEG-SOD), as we have previously described. (22)
Plasma Hydrogen peroxide levels
Plasma Hydrogen peroxide levels were determined utilizing a modified H2DCFDA oxidation method (17,18). Five microlitres of patient’s plasma was incubated with 25 μM H2DCFDA (Calbiochem) for 15 min in the dark. The samples were measured with excitation at 485 nm and emission at 530 nm in Fluoroskan Ascent FL (Thermo Electron, Waltham, MA). The values were normalized with protein concentration in their respected plasma samples.
Statistical analyses
Statistical analyses were performed using an unpaired t-test and one-way ANOVA for differences between more than two groups. Analyses were performed using SAS 9.4 (SAS Institute, Cary NC). A p value <0.05 was considered significant.
RESULTS
The median age of the patients with VSD [4 – 3,7 (months, IQR)] and the patients with TOF [3.5 - 3,7 (months, IQR)] were similar. Control patients were older (16.0 – 1, 7.2 (months, IQR)](P<0.05). Eleven of the 48 patients (45.8%) were male. Diagnoses within the control group admitted to the PICU included pertussis, seizures, other neurologic disorders, pyloric stenosis, and jejunal atresia.
Baseline AC levels (25.7±13 vs. 12.7 ± 8.3 μM, P<0.05) and the AC:FC ratio (0.8 ± 0.1 vs. 0.3 ± 0.05, P<0.05) were higher in patients with VSD than TOF and controls, suggestive of abnormal carnitine homeostasis in children with VSD (Figure 1, A–B). The lactate/pyruvate ratio was higher in patients with VSD (27.5 ±3.8 vs. 11.1±4.1, P<0.05) than in patients with TOF and controls, suggestive of mitochondrial dysfunction in patients with VSD (Figure 1C). There were no differences in the lactate/pyruvate ratio between patients with TOF and control. NOx levels were decreased in children with VSD compared to children with TOF and controls, suggestive of decreased bioavailable NO in children with VSD (Figure 2A), and superoxide and H2O2 levels were increased in patients with VSD compared to controls, suggestive of increased ROS production in VSD patients (Figure 2, B–C)(P<0.05).
Figure 1.
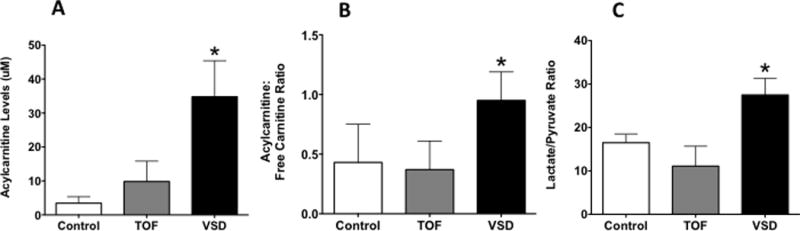
Plasma acetylcarnitine levels (A), the acetylcarnitine:free carnitine ratio (B), and the lactate/pyruvate ratio (C) was higher in VSD than TOF and controls. There were no differences between TOF and control. *P<0.05 vs. control and TOF (ANOVA). Values are mean±SD. N=18 for VSD; 20 for TOF, and 10 for control.
Figure 2.
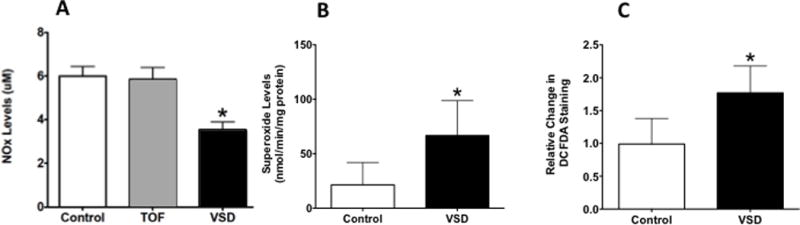
Plasma NOx levels were decreased in VSD patients compared to TOF and controls (A), and superoxide (B) and H2O2 (C) levels were increased in VSD compared to controls. *P<0.05 vs. control (ANOVA). Values are mean±SD. N=18 for VSD; 20 for TOF, and 10 for control.
DISCUSSION
Endothelial dysfunction is well documented in pulmonary vascular diseases. For example, Giard et al demonstrated decreased NO and increased endothelin-1 gene expression in lungs from patients with advanced pulmonary vascular disease. In addition, human data suggests a role for increased ROS production and oxidative stress in advanced disease.(17–20) Since most forms of pulmonary vascular disease present clinically in advanced forms, little is known about the initiating events that lead to its development. Importantly, in CHD, the initiating events, abnormal mechanical forces, and natural history are better understood. This provides a framework in order to elucidate early perturbations in vascular function. In fact, Rabinovitch et al demonstrated structural abnormalities of the vascular endothelium in young children undergoing surgical repair of CHD with pulmonary hypertension, and Celemajer et al identified functional abnormalities of the endothelium in children with CHD and increased pulmonary blood flow prior to the establishment of pulmonary hypertension.(2, 3)
Utilizing our clinically relevant large animal model of CHD with increased pulmonary blood flow, we have previously demonstrated a mechanism for flow/pressure-induced endothelial dysfunction mediated via the down-regulation of PPARγ that results in altered carnitine homeostasis, mitochondrial dysfunction, and a ROS-mediated decrease in bioavailable NO.(21, 22) The current study is the first investigation seeking to examine these potential aberrations in humans with CHD. Infants with VSD represented those with increased PBF; infants with TOF represented those with normal or decreased PBF; and infants and children admitted to the ICU without CHD served as controls. Supporting the animal data, we found that infants with increased PBF had altered carnitine homeostasis, altered lactate/pyruvate ratios, increased plasma superoxide and hydrogen peroxide levels, and decreased NO metabolites (NOx) compared to children with TOF or control children. As only children with VSD, and not children with TOF, exhibited these aberrations, our data are highly suggestive that altered biomechanical forces associated with increased PBF are responsible for the disruption of carnitine homeostasis.
A few limitations are noteworthy. Most importantly all samples were obtained from blood, negating our ability to localize any differences to the pulmonary vasculature and potentially diluting any potential differences. Despite this limitation, all blood samples were consistent with the animal data. Next, our control group was older than both CHD groups. However, there were no differences between the controls and younger patients with TOF suggesting that age was not a confounding factor. Lastly, none of these patients underwent direct measurements of PBF via cardiac catheterization; the status of PBF was assumed by the lesion and echocardiographic findings.
In summary, this study demonstrates early alterations in carnitine homeostasis that are associated with altered mitochondrial function, increased ROS, and decreased bioavailable NO. This is consistent with our previous animal investigations that utilized a clinically relevant ovine model of increased PBF.(14, 22, 23) In fact, we also demonstrated in this model that carnitine supplementation normalized the noted aberrations and endothelial dysfunction. Since disruption of carnitine metabolism may contribute to endothelial dysfunction, carnitine supplementation may attenuate endothelial dysfunction associated with increased PBF, and warrants further investigation.
Acknowledgments
Sources of support: This research was supported in part by grants from the National Institutes of Health (HL61284 to JRF) (Hl60190 and HL067841 to SMB).
Footnotes
Copyright form disclosure: Drs. Black, Keller, Maltepe, Datar, and Fineman received support for article research from the National Institutes of Health (NIH). Dr. Black’s institution received funding from the NIH. Dr. Keller’s institution received funding from the National Heart, Lung, and Blood Institute. The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Hoffman JI, Rudolph AM, Heymann MA. Pulmonary vascular disease with congenital heart lesions: pathologic features and causes. Circulation. 1981;64:873–877. doi: 10.1161/01.cir.64.5.873. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovitch M, Haworth SG, Castaneda AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation. 1978;58:1107–1122. doi: 10.1161/01.cir.58.6.1107. [DOI] [PubMed] [Google Scholar]
- 3.Celermajer DS, Cullen S, Deanfield JE. Impairment of endothelium-dependent pulmonary artery relaxation in children with congenital heart disease and abnormal pulmonary hemodynamics. Circulation. 1993;87:440–446. doi: 10.1161/01.cir.87.2.440. [DOI] [PubMed] [Google Scholar]
- 4.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109:159–165. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 5.Schulze-Neick I, Li J, Penny DJ, et al. Pulmonary vascular resistance after cardiopulmonary bypass in infants: effect on postoperative recovery. The Journal of Thoracic and Cardiovascular Surgery. 2001;121:1033–1039. doi: 10.1067/mtc.2001.113747. [DOI] [PubMed] [Google Scholar]
- 6.Burrows FA, Klinck JR, Rabinovitch M, et al. Pulmonary hypertension in children: perioperative management. Can Anaesth Soc J. 1986;33:606–628. doi: 10.1007/BF03014268. [DOI] [PubMed] [Google Scholar]
- 7.Oishi PE, Wiseman DA, Sharma S, et al. Progressive dysfunction of nitric oxide synthase in a lamb model of chronically increased pulmonary blood flow: a role for oxidative stress. AJP: Lung Cellular and Molecular Physiology. 2008;295:L756–L766. doi: 10.1152/ajplung.00146.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakshminrusimha S, Wiseman D, Black SM, et al. The role of nitric oxide synthase-derived reactive oxygen species in the altered relaxation of pulmonary arteries from lambs with increased pulmonary blood flow. Am J Physiol Heart Circ Physiol. 2007;293:H1491–7. doi: 10.1152/ajpheart.00185.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinhorn RH, Russell JA, Lakshminrusimha S, et al. Altered endothelium-dependent relaxations in lambs with high pulmonary blood flow and pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2001;280:H311–7. doi: 10.1152/ajpheart.2001.280.1.H311. [DOI] [PubMed] [Google Scholar]
- 10.Black S, Fineman J, Johengen M, et al. Increased pulmonary blood flow alters the molecular regulation of vascular reactivity in the lamb. CHEST. 1998;114:39S. doi: 10.1378/chest.114.1_supplement.39s. [DOI] [PubMed] [Google Scholar]
- 11.Grobe AC. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. AJP: Lung Cellular and Molecular Physiology. 2006;290:L1069–L1077. doi: 10.1152/ajplung.00408.2005. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovitch M, Bothwell T, Hayakawa BN, et al. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy Lab Invest. 1986;55:632–653. [PubMed] [Google Scholar]
- 13.Sharma S, Sud N, Wiseman DA, et al. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2008;294:L46–56. doi: 10.1152/ajplung.00247.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zammit VA, Ramsay RR, Bonomini M, et al. Carnitine, mitochondrial function and therapy. Adv Drug Deliv Rev. 2009;61:1353–1362. doi: 10.1016/j.addr.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 15.Ramsay RR, Zammit VA. Carnitine acyltransferases and their influence on CoA pools in health and disease. Mol Aspects Med. 2004;25:475–493. doi: 10.1016/j.mam.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Pande SV, Blanchaer MC. Reversible inhibition of mitochondrial adenosine diphosphate phosphorylation by long chain acyl coenzyme A esters. J Biol Chem. 1971;246:402–411. [PubMed] [Google Scholar]
- 17.Aggarwal S, Gross CM, Sharma S, et al. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol. 2013;3:1011–1034. doi: 10.1002/cphy.c120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wong C-M, Bansal G, Pavlickova L, et al. Reactive oxygen species and antioxidants in pulmonary hypertension. Antioxid Redox Signal. 2013;18:1789–1796. doi: 10.1089/ars.2012.4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333:214–221. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 20.Giaid A, Yanagisawa M, Langleben D, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328:1732–1739. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 21.Sharma S, Barton J, Rafikov R, et al. Chronic inhibition of PPAR-γ signaling induces endothelial dysfunction in the juvenile lamb. Pulm Pharmacol Ther. 2013;26:271–280. doi: 10.1016/j.pupt.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma S, Sun X, Rafikov R, et al. PPAR-γ regulates carnitine homeostasis and mitochondrial function in a lamb model of increased pulmonary blood flow. PLoS ONE. 2012;7:e41555. doi: 10.1371/journal.pone.0041555. [DOI] [PMC free article] [PubMed] [Google Scholar]