

Abstract
Molecularly targeted drugs have yielded significant therapeutic advances in oncogene-driven non-small cell lung cancer (NSCLC), but a majority of patients eventually develop acquired resistance. Recently, the relation between proinflammatory cytokine interleukin-6 (IL6) and resistance to targeted drugs has been reported. We investigated the functional contribution of IL6 and the other members of IL6 family proinflammatory cytokine pathway to resistance to targeted drugs in NSCLC cells. In addition, we examined the production of these cytokines by cancer cells and cancer-associated fibroblasts (CAFs). We also analyzed the prognostic significance of these molecule expressions in clinical NSCLC samples. In NSCLC cells with acquired resistance to targeted drugs, we observed activation of the IL6-cytokine pathway and STAT3 along with epithelial-to-mesenchymal transition (EMT) features. In particular, IL6 family cytokine oncostatin-M (OSM) induced a switch to the EMT phenotype and protected cells from targeted drug-induced apoptosis in OSM receptors (OSMRs)/JAK1/STAT3-dependent manner. The cross-talk between NSCLC cells and CAFs also preferentially activated the OSM/STAT3 pathway via a paracrine mechanism and decreased sensitivity to targeted drugs. The selective JAK1 inhibitor filgotinib effectively suppressed STAT3 activation and OSMR expression, and co-targeting inhibition of the oncogenic pathway and JAK1 reversed resistance to targeted drugs. In the analysis of clinical samples, OSMR gene expression appeared to be associated with worse prognosis in patients with surgically resected lung adenocarcinoma. Our data suggest that the OSMRs/JAK1/STAT3 axis contributes to resistance to targeted drugs in oncogene-driven NSCLC cells, implying that this pathway could be a therapeutic target.
Keywords: non-small cell lung cancer, molecularly targeted therapy, drug resistance, proinflammatory cytokines, epithelial-to-mesenchymal transition
Introduction
The emergence of molecularly targeted therapies against driver mutations and gene fusions has resulted in significant therapeutic advances for a subset of non-small cell lung cancer (NSCLC) patients (1). However, even when targeted therapy is effective, patients eventually develop resistance; thus more effective therapies that overcome this acquired resistance are being sought (2). A variety of mechanisms are believed to contribute to intrinsic and acquired resistance to anticancer drugs, especially molecularly targeted drugs. In oncogene-driven cancers, nongenetic mechanisms have been widely reported in addition to the genetic mechanisms such as gatekeeper mutations. For example, growth factors secreted from the tumor microenvironment promote proliferative or anti-apoptotic signaling and protect cancer cells from targeted drugs (3). Furthermore, cancer cells produce several factors, including heregulin, amphiregulin, and hepatocyte growth factor, that contribute to intrinsic and acquired resistance to EGFR inhibitors in NSCLC (4–6).
Several molecularly targeted drugs, including EGFR tyrosine kinase inhibitors (TKIs) and MET inhibitors, promote activation of STAT3 signaling (7, 8). Lee et al. reported the relation between proinflammatory cytokine interleukin-6 (IL6) and resistance to targeted drugs (9). They showed that inhibition of MEK functioning downstream of various receptor tyrosine kinases, including EGFR, MET, ALK, and HER2, triggers the feedback activation of STAT3 through IL6 secretion, significantly contributing to resistance to pathway-targeted drugs. The family of IL6 cytokines comprises IL6, IL11, oncostatin-M (OSM), leukemia inhibitory factor (LIF), ciliary neurotrophic factor, cardiotrophin-1, and cardiotrophin-like cytokine. These cytokines activate target genes associated with cell differentiation, survival, apoptosis, and proliferation (10). Among this family, IL6, OSM, and LIF are the most widely expressed in different organs and are associated with cancer progression (11). These proinflammatory cytokines have individual receptors (e.g., IL6R, OSMR, and LIFR), which generally work as heterodimers with IL6ST (gp130) (12, 13). These cytokines are reported to be robust stimulators of STAT3 and to be strong promoters of epithelial-to-mesenchymal transition (EMT), cancer metastasis, and cancer stem cells (CSCs) in several types of cancer (14).
In this study, we describe for the first time that activation of the other members of IL6 family proinflammatory cytokine pathway, in particular the OSM-OSMR duo, can contribute to resistance to molecularly targeted drugs in oncogene-driven NSCLC cells. In addition, an inhibitor of Janus kinase 1(JAK1), a key mediator of IL6 cytokine pathway activation in NSCLC cells, effectively reversed resistance to targeted drugs in these cells. Our data suggest that the OSMRs/JAK1/STAT3 axis contributes to resistance to targeted drugs in oncogene-driven NSCLC cells, implying that this pathway could be a therapeutic target.
Materials and Methods
Cell lines and reagents
We used 6 human oncogene-driven NSCLC cell lines, all provided by Dr. Adi F. Gazdar (The University of Texas Southwestern Medical Center, Dallas, TX) and co-author (JDM). For a control, we utilized the nonmalignant human bronchial epithelial cell line HBEC3KT (15). The identities of all cell lines were confirmed by short tandem repeat (STR) DNA fingerprinting using the Promega 16 High Sensitivity STR Kit. We used normal lung fibroblasts (NLFs) obtained from a normal lung specimen and cancer-associated fibroblasts (CAFs) obtained from a lung cancer specimen in co-cultures to mimic the tumor microenvironment. These fibroblasts were used in passages 5 through 7.
Targeted inhibitors selumetinib (AZD6244) (16), erlotinib, crizotinib, filgotinib (GLP0634) (17), momelotinib (CYT387) (18), and stattic (19) were purchased from Selleckchem. Recombinant human (rh) IL6, OSM, and LIF proteins were purchased from EMD Millipore.
Cell proliferation
Cell proliferation was quantified by a modified MTS assay with CellTiter 96 AQueous One Solution Reagent (Promega) as previously reported (20). For experiments testing the effect of proinflammatory cytokines, JAK1, or STAT3 on cell proliferation, the crystal violet staining or MTT dye reduction method (Sigma) was used. The percentage growth is shown relative to untreated controls. Each experiment was performed at least in triplicate, and three times independently.
mRNA and miRNA expression analysis by qRT-PCR
PCR amplification was conducted on an ABI Real-Time PCR 7900 HT (Applied Biosystems) and gene expression was calculated by the comparative CT method. Three replicates per sample were assayed for each gene. To quantify the relative changes in gene expression, the 2(−ΔΔCT) method was used and reactions were normalized to endogenous control gene GAPDH expression level for mRNA expression analysis and to miR-374 expression level for miRNA expression analysis.
Western blotting analysis
The primary antibodies were as follows: anti-STAT3, p-STAT3 (Tyr705), IL6ST (Gp130), E-cadherin, Vimentin (R28), ZEB1, Snail, Axl, AKT, p-AKT (Ser473), p44/p42 mitogen-activated protein kinase (ERK), p-p44/p42 MAPK (p-ERK1/2), JAK1, p-JAK1, JAK2, p-JAK2, JAK3, Tyk2 (a JAK family menber), PARP, and c-PARP (Cell Signaling Technology) and IL6R, OSMR, and LIFR (Santa Cruz Biotechnology). Monoclonal anti-actin antibody, used as an equal loading control, was purchased from EMD Millipore. Secondary antibodies were goat anti-rabbit and goat anti-mouse horseradish peroxidase-conjugated immunoglobulin G (Santa Cruz Biotechnology). To detect specific signals, the membranes were examined by Western Lightning Plus-ECL (Perkin-Elmer).
Phosphokinase array
A Human Phospho-Kinase Array Kit (R&D Systems) was used to measure the relative level of phosphorylation of 46 distinct intracellular kinases according to the manufacturer’s instructions.
Intracellular kinase activation after cytokine treatment
For RNA interference assays, cells were transfected with Silencer Select siRNA against OSMR, LIFR, JAK1, JAK2, JAK3, Tyk2, or scrambled negative control siRNA. The cells were placed in 60-mm dishes and incubated for 72 hours and then treated with rhIL6, rhOSM, or rhLIF (1 ng/mL) for 1 hour. Cell lysates were analyzed by Western blotting for levels of the indicated proteins.
Enzyme-linked immunosorbent assay
HBEC3K, NLF, and CAF cells were seeded at 2×105 cells per 60-mm cell culture dish and incubated for 72 hours. For measurement of IL6 concentration, cell culture media were collected and subjected to an IL6 ELISA kit (R&D Systems) according to the manufacturer’s instructions. For measurement of OSM, cells were subjected to lysis in cell lysis buffer (Sigma) analyzed with an OSM ELISA kit (R&D Systems).
Co-culture of NSCLC cells with fibroblasts
Tumor cells were treated or not treated with targeted drugs and placed in the lower chambers of transwell polycarbonate membrane cell culture inserts (2×104 cells per chamber). NLFs or CAFs (2×105) were placed in the upper chambers; the lower and upper chambers were separated by 0.4-μm pore size filters (Corning).
Clinical datasets and patient sample characteristics
A total of 855 patients from four clinical datasets, including The Cancer Genome Atlas (TCGA, n=515), Profiling of Resistance patterns and Oncogenic Signaling Pathways in Evaluation of Cancers of the Thorax (PROSPECT, n=180), and the Biomarker integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE-1 [n=57] and BATTLE-2 [n=103]), were analyzed as independent datasets. Clinical and demographic information for 515 mostly early-stage, surgically resected lung adenocarcinomas included in TCGA dataset was obtained from the TCGA portal (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). The PROSPECT dataset includes tumor tissue collected from patients who underwent surgical resection of lung adenocarcinoma with curative intent between 1996 and 2008 at MD Anderson Cancer Center. BATTLE-1 was a randomized phase II, single-center, biomarker-based, biopsy-manded clinical trial of targeted therapy in patients with advanced chemorefractory NSCLC (NCT00409968) (21). BATTLE-2 was a randomized phase II, multi-center, biomarker-integrated, biopsy-mandated trial of targeted therapy in patients with advanced chemorefractory NSCLC (NCT01248247) (22). Clinical characteristics of these samples are described in Table S1.
Statistical analyses
Statistical analyses were conducted by using GraphPad Prism 6 (GraphPad Software) or the R system for statistical computing. Differences between groups were calculated by the t-test. Univariate Cox proportional hazards regression analysis was utilized to assess the association of OSMR on overall survival (OS), recurrent-free survival (RFS), and progression-free survival (PFS). Based on univariate analysis, variables with P-value less than 0.2 were initially included in a multivariable model in PROSPECT cohort. Backward elimination of multivariable Cox regression model was conducted to eliminate variables that were insignificant. Survival curves were plotted by using the Kaplan-Meier method and analyzed by using the log-rank test. Spearman test was used to evaluate a correlation between each cytokine and receptor expression level. A P-value <0.05 was considered significant.
Results
IL6 proinflammatory pathway is activated in NSCLC cells with acquired resistance to molecularly-targeted drugs
From 3 of the oncogene-driven NSCLC cell lines, we established resistant cell lines by long-term exposure to a specific targeted drug according to a previously reported method (20). Cell line H23-SR was established by exposing H23 KRAS-mutant (-mut) cells to MEK inhibitor selumetinib; HCC4006-ER by exposing HCC4006 EGFR-mut cells to EGFR-TKI erlotinib; and H3122-CR by exposing H3122 EML4-ALK fusion cells to ALK inhibitor crizotinib. After more than 4 months of drug exposure, each of these cell lines showed resistance to the pertinent drug (Fig. 1A). IC-50 values against each targeted drug were as follows: H23 and resistant H23-SR, 31.2 μM and > 100 μM; HCC4006 and resistant HCC4006-ER, 0.034 μM and > 10 μM; and H3122 and resistant H3122-CR, 0.47 μM and 9.9 μM, respectively. After acquisition of resistance to each drug, all 3 cell lines showed transcriptional upregulation of IL6 family cytokine receptor genes IL6R, IL6ST, OSMR, and LIFR (Fig. 1B). Western blotting showed upregulation of basal IL6 family cytokine receptors, except for IL6ST in H3122 cells, and their downstream protein phospho-STAT3 (Y705) (p-STAT3) in each acquired-resistance cell line compared to the parental cell line (Fig. 1C). While the reason was unclear, we could not detect the IL6ST protein in both H3122 and H3122-CR cells. As already reported, selumetinib and erlotinib treatment did not suppress p-STAT3 expression in the parental cells, while crizotinib did suppress p-STAT3 expression in H3122 cell (9, 23). Interestingly, 72 hours after targeted drug treatment, all acquired resistance cells showed further overexpression of p-STAT3 (Fig. 1C).
Figure 1. NSCLC cell lines with acquired resistant to targeted drugs show proinflammatory cytokine pathway activation with EMT features.
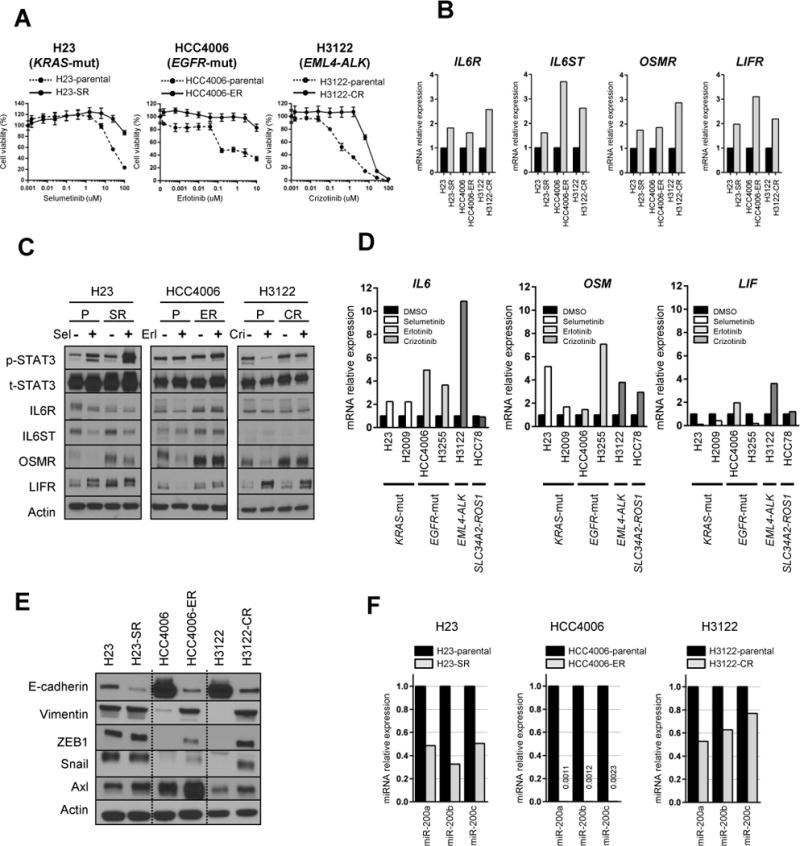
(A) Three oncogene-driven NSCLC cell lines developed acquired resistance after continuous exposure to the indicated molecularly targeted drug. Cell sensitivity to each drug was measured by MTS assay.
(B) IL6 family proinflammatory cytokine receptor gene expression levels in parental and acquired-resistance cells were analyzed by qRT-PCR.
(C) Parental (P) and acquired-resistance (SR, ER, CR) cells were treated with their respective targeted drug for 72 hours and expression levels of proinflammatory cytokine receptors and their downstream p-STAT3 were determined by Western blotting. Sel, selumetinib; Erl; erlotinib; Cri, crizotinib.
(D) IL6 family proinflammatory cytokine gene expression changes in 6 parental NSCLC cell lines after treatment for 24 hours with a targeted drug were analyzed by qRT-PCR.
(E) Expression of EMT-related proteins was measured in parental and acquired-resistance cells by Western blotting.
(F) Expressions of the miR-200 family in parental and acquired-resistance cells were analyzed by qRT-PCR.
We next analyzed expression levels of the IL6 family cytokines after treatment with a targeted drug in the 6 parental NSCLC cell lines. We treated KRAS-mut cell lines H23 and H2009 with selumetinib, EGFR-mut cell lines HCC4006 and H3255 with erlotinib, and EML4-ALK fusion cell line H3122 and SLC34A2-ROS1 fusion cell line HCC78 with crizotinib for 24 hours, and analyzed IL6 family cytokine gene expression levels by qRT-PCR. Crizotinib is a MET and ALK kinase inhibitor that is also reported to have activity against ROS1 (24, 25). IL6 and OSM gene expression levels were upregulated by each drug treatment, while LIF modest increased expression varied (Fig. 1D), suggesting that exposure to these targeted drugs caused an activation of the proinflammatory cytokine pathway preponderantly mediated by IL6 and OSM in these cells.
Interestingly, all 3 acquired resistance cell lines showed changes suggestive of EMT, including downregulation of E-cadherin and upregulation of vimentin, ZEB1, Snail, and Axl, on Western blotting (Fig. 1E). These acquired-resistance cells also showed morphological changes to spindle shapes (Fig. S1). Because we previously reported that long-term EGFR-TKI exposure caused EMT with downregulation of the miR-200 family in EGFR-mut NSCLC cells, we analyzed the expression status of the miR-200 family by qRT-PCR (20) and showed that miR-200a/b/c were downregulated in all acquired-resistance cells with EMT features (Fig. 1F).
OSM is a potent activator of oncogenic pathway molecules
To confirm the functional effect of IL6 family cytokines on their downstream pathway in NSCLC cells, we stimulated NSCLC cells with each of 3 IL6 family cytokines (rhIL6, rhOSM, or rhLIF) and analyzed phospho-kinase activation. Whole-cell extracts from H23 cells treated or not treated with rhIL6, rhOSM, or rhLIF for 48 hours were incubated with a human phospho-kinase array to compare kinase’s phosphorylation induced by each of the IL6 family cytokines (Fig. 2A). Phosphorylation of several kinases, including AKT (S473), ERK1/2, STAT3 (Y705 and S727), p53, and c-JUN (S7), was observed after cytokine treatment. Among the 3 cytokines, rhOSM was the most potent activator of these oncogenic molecules. Further, the 6 parental NSCLC cell lines were cultured with each cytokine for various time intervals under serum-starved condition and analyzed by Western blotting (Fig. 2B, Fig. S2). The rhOSM treatment induced marked STAT3 (Y705) phosphorylation in all cell lines. AKT (S473) and ERK1/2 (T202/Y204) were phosphorylated by rhOSM in all 6 cell lines even under serum-starved conditions.
Figure 2. IL6 family proinflammatory cytokines stimulate activation of downstream pathway molecules in KRAS-mut H23 NSCLC cells, promoting EMT and cell survival.
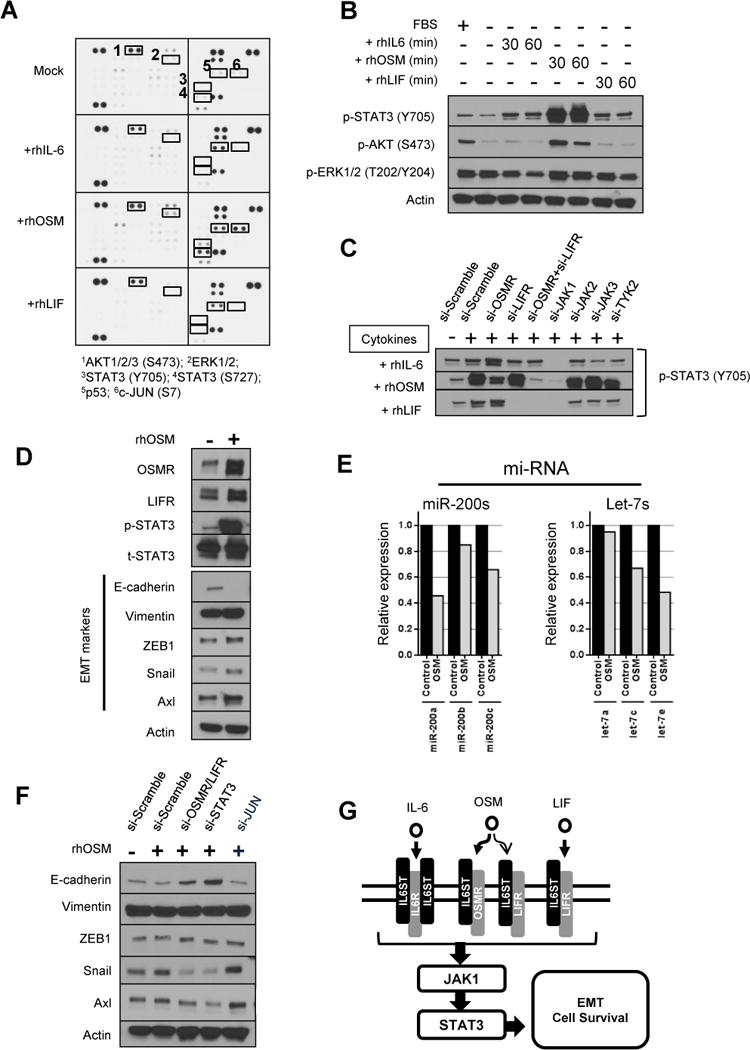
(A) Activated intracellular kinases were analyzed by using the Human Phospho-Kinase Array Kit. Whole-cell extracts from H23 cells, treated or not treated (Mock) with an IL6 family cytokine (rhIL6, rhOSM, or rhLIF) for 48 hours were incubated with the arrays, and phosphorylation status of each kinase was determined.
(B) H23 cells were cultured in one of the IL6 family cytokines (1 ng/mL) for the indicated time periods under serum-starved conditions, and cell lysates were analyzed for the indicated proteins by Western blotting.
(C) H23 cells were transfected with siRNAs specific for IL6 family cytokine receptors, JAK family members, or scrambled negative control for 72 hours. After treatment with one of the cytokines (1 ng/mL) for 1 hour, the cells were analyzed for p-STAT (Y705) protein expression by Western blotting.
(D) H23 cells were incubated with or without rhOSM (10 ng/mL) for 4 weeks and whole-cell proteins were analyzed by Western blotting.
(E) H23 cells treated or not treated (Control) with rhOSM (10 ng/mL) for 1 week were analyzed for miR-200 family and let-7 family expression levels by qRT-PCR.
(F) H23 cells were transfected with siRNAs specific for OSMR/LIFR, STAT3, JUN, or scrambled negative control for 72 hours. After treatment with rhOSM (1 ng/mL) for 1 week, cell lysates were analyzed by Western blotting.
(G) Suggested model of IL6 family proinflammatory cytokines/receptors activation of the downstream pathway causing EMT and cell survival.
JAK1 mediates STAT3 phosphorylation by IL6 family cytokines
To determine which molecule preferentially mediates STAT3 phosphorylation after treatment with these proinflammatory cytokines, we knocked down OSMR, LIFR, and JAK family members with siRNA in 23 cells and treated the knockdown cells with each cytokine (Fig. 2C). Sufficient knockdown efficacy by each siRNA was confirmed by Western blotting (Fig. S3). In rhIL6-stimulated cells, STAT3 phosphorylation was blocked by JAK1 knockdown. In rhOSM-stimulated cells, STAT3 phosphorylation was blocked by co-knockdown of OSMR and LIFR or by knockdown of JAK1. In rhLIF-stimulated cells, STAT3 phosphorylation was blocked by knockdown of LIFR or JAK1. Taken together, knockdown of JAK1 blocked the STAT3 phosphorylation stimulated by each of the 3 cytokines, while knockdown of the other JAK members (JAK2, JAK3, and TYK2) could not block STAT3 phosphorylation. In addition, STAT3 phosphorylation by rhOSM was mediated by both OSMR and LIFR, as previously reported (13). These results suggest that JAK1 is the critical kinase that phosphorylates STAT3 via stimulation by IL6 family cytokines in NSCLC cells. Interestingly, when we knocked down JAK1, expression of OSMR was also downregulated (Fig. S3), suggesting the possibility of feedback activation of OSMR by JAK1 kinase.
OSM-induced EMT is mediated by STAT3 activation
OSM has been reported to enhance migration of several kinds of cancer cells (26–32). We also confirmed that rhOSM treatment resulted in EMT-associated changes in H23 cells, as evinced by repression of the epithelial marker E-cadherin and induction of the mesenchymal markers vimentin, ZEB1, Snail, and Axl, on Western blotting (Fig. 2D). rhOSM treatment caused morphological change to a spindle shape (Fig. S4). In addition, rhOSM treatment caused EMT-related epigenetic changes, with suppression of the miR-200 family and let-7 family members as previously reported (Fig. 2E) (28). On the other hand, rhOSM did not cause major effect on cell viability (Fig. S5). These EMT-related changes induced by rhOSM treatment were prevented by siRNA-mediated co-downregulation of OSMR and LIFR or downregulation of STAT3 (Fig. 2F, S6). In contrast, downregulation of JUN, previously reported to be an EMT-promoting factor in cancer cells (33), could not protect cells from rhOSM-induced EMT-related changes. These results suggest that rhOSM-induced EMT and cell survival are mediated by OSMR/LIFR-STAT3 activation (Fig. 2G).
OSM pathway activation protects cells from targeted drug-induced apoptosis in an OSMRs/JAK1/STAT3-dependent manner
To analyze the effect of OSM/STAT3 pathway activation on sensitivity to molecularly targeted drugs in oncogene-driven NSCLCs, we treated the 6 NSCLC cell lines with molecularly targeted drugs with or without 2 weeks of pretreatment with rhOSM in cell culture medium. Two cell lines harboring KRAS-mut (H23 and H2009) were treated with selumetinib, 2 cell lines harboring EGFR-mut (H3255 and HCC4006) were treated with erlotinib, and 2 cell lines harboring EML4-ALK fusion H3122 and SLC34A2-ROS1 fusion HCC78 were treated with crizotinib. In all cells, rhOSM treatment protected from targeted drug-induced cell death (Fig. 3A).
Figure 3. OSM pathway activation protects cells from targeted drug-induced apoptosis and is dependent on an OSMRs/JAK1/STAT3 pathway.
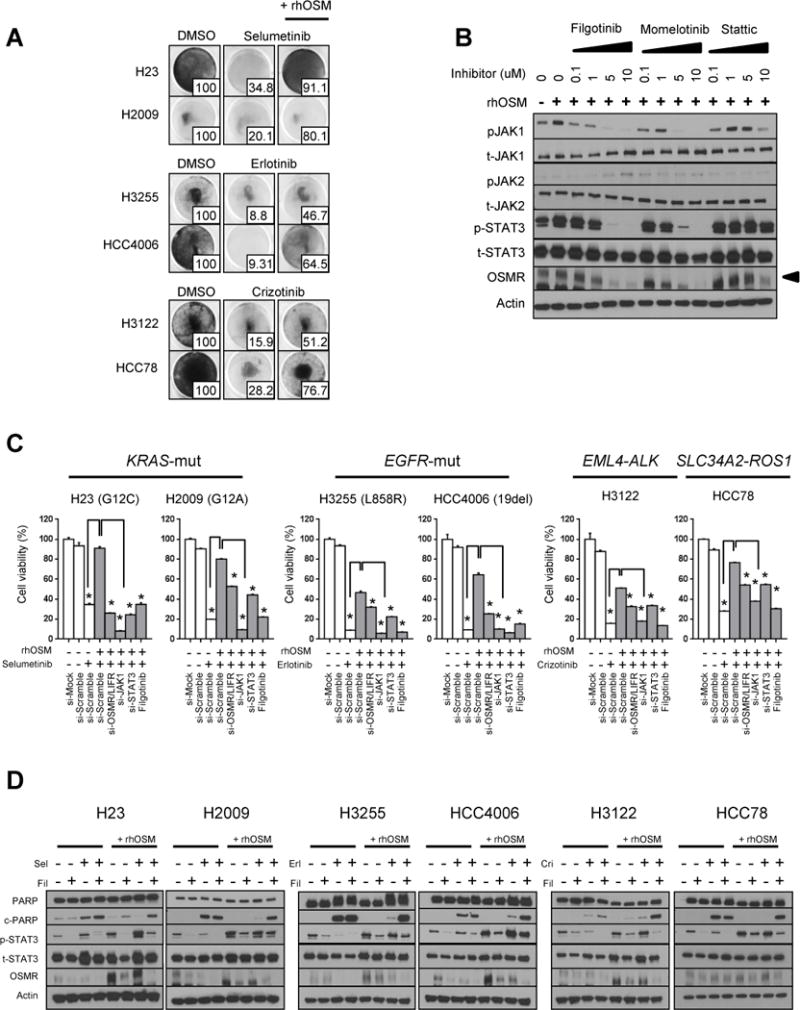
(A) Six NSCLC cell lines were treated with DMSO or a targeted drug: H23 and H2009 (KRAS-mut) with selumetinib (10 μM), H3255 and HCC4006 (EGFR-mut) with erlotinib (100 nM), H3122 (EML4-ALK fusion) and HCC78 (SLC34A2-ROS1 fusion) with crizotinib (500 nM), with or without pretreatment with rhOSM (10 ng/mL) for at least 2 weeks. Cells were stained with crystal violet 7 to 10 days after treatment. Quantification measured by MTT assay (relative to DMSO) is shown at bottom right of each plate.
(B) H2009 cells cultured with or without rhOSM (10 ng/mL) were treated with serial dilutions of selective JAK1 inhibitor filgotinib, JAK1/2 and TBK1 inhibitor momelotinib, or STAT3 inhibitor stattic for 48 hours. Whole-cell proteins were harvested and analyzed by Western blotting. OSMR signals were detected at 180 kD (arrow head).
(C) Six NSCLC cell lines treated or not treated with rhOSM (10 ng/mL) for at least 2 weeks were transfected with siRNA specific for OSMR/LIFR, JAK1, STAT3, or scrambled negative control for 48 hours. The cells then were treated with one of the targeted drugs, as follows, with or without filgotinib: H23 and H2009, selumetinib (10 μM); H3255 and HCC4006, erlotinib (100 nM); H3122 and HCC78, crizotinib (500 nM). After 7 to 10 days of treatment, cell viabilities compared to control cells (without siRNA and drugs) were measured by MTT assay. *P <0.05.
(D) Six NSCLC cell lines were treated with the indicated targeted drugs with or without rhOSM for 72 hours, and lysates obtained from cancer cells were examined by Western blotting with the indicated antibodies. Sel, selumetinib; Fil, filgotinib; Erl, erlotinib; Cri, crizotinib.
We then hypothesized that inhibition of the OSM receptors (OSMRs)/JAK1/STAT3 pathway with each oncogenic pathway-targeted treatment could prevent drug resistance caused by OSM pathway activation. To explore JAK/STAT3 inhibitor activities, we treated H2009 cells with novel selective JAK1 inhibitor filgotinib, JAK1/2 and TBK1/IKKε inhibitor momelotinib, or STAT3 inhibitor stattic (Fig. 3B). Filgotinb and momelotinib potently suppressed JAK1 and STAT3 phosphorylation in a dose-dependent manner. Interestingly, these JAK/STAT3 inhibitors suppressed OSMR expression as well.
To determine whether co-targeting of a major oncogenic pathway and OSMRs/JAK1/STAT3 pathway molecules could efficiently inhibit drug resistance caused by rhOSM, we treated cells with agents targeting each pathway in combination with siRNA against OSMR/LIFR, JAK1, and STAT3, and selective JAK1 inhibitor filgotinib. The reason we chose to use filgotinib, which is in an ongoing clinical trial for the treatment of rheumatoid arthritis and Crohn disease (17, 34), over momelotinib, which is in an ongoing clinical trial in combination with trametinib (NCT02206763) or erlotinib (NCT02206763) for the treatment of NSCLC, is that momelotinib would have had off-target activities (35). In other words, by using filgotinib instead of momelotinib, we were able to ensure more complete inhibition of JAK1 activation at doses that did not exhibit off-target effects. As shown in Fig. 3C, all 6 cell lines treated with rhOSM showed resistance to targeted drugs, and siRNA-mediated downregulation of OSMR/LIFR, JAK1, and STAT3 prevented the targeted drug resistance caused by OSM treatment. In particular, siRNA-mediated JAK1 inhibition showed a potent antitumor effect in combination with targeted drugs. Filgotinib also inhibited the resistance in combination with each targeted drug. Western blotting showed that, after 48 hours of treatment, expression of apoptosis marker cleaved (c)-PARP was suppressed by rhOSM treatment, while co-targeting of the oncogenic pathway by a targeted drug and of JAK1 by filgotinib inhibited p-STAT3 and OSMR and induced strong expression of c-PARP (Fig. 3D).
Cross-talk between NSCLC cells and fibroblasts reduces sensitivity to molecularly targeted therapies, while JAK1 inhibition can prevent resistance to targeted drugs
Because CAFs reportedly influence tumor progression and are associated with resistance to molecularly targeted drugs via the secretion of extra-cellular matrix proteins, proteases, cytokines, and growth factors, we analyzed the effect of cross-talk of cancer cells and fibroblasts on resistance to targeted drugs (3, 36, 37). Both NLFs and CAFs showed high expression levels of IL6 and OSM mRNAs compared to normal bronchial epithelial HBEC3KT cells by qRT-PCR, and high concentration of the IL6 protein in cell culture media or OSM in whole-cell lysates by ELISA (Fig. 4A). Cancer cells co-cultured with fibroblasts in cell culture inserts showed upregulation of p-STAT3, OSMR, and LIFR, and downregulation of E-cadherin, suggesting that the OSMRs/STAT3 pathway was activated by the fibroblasts (Fig. 4B). In co-culture with CAFs, cancer cells were highly resistant to each targeted drug (Fig. 4C).
Figure 4. Cross-talk of oncogene-driven cancer cells and fibroblasts reduces sensitivity to targeted drugs, while inhibition of JAK1 can prevent resistance to targeted drugs.
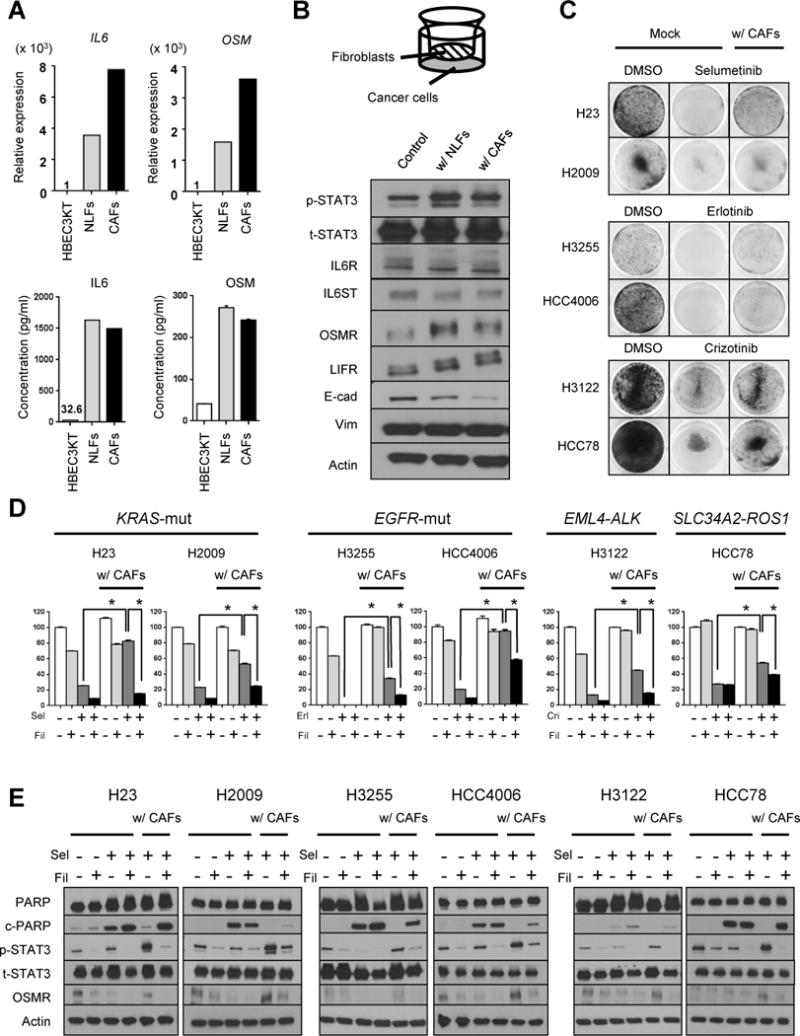
(A) (Upper) Relative expression levels of IL6 and OSM mRNA in normal lung fibroblasts (NLFs) and cancer-associated fibroblasts (CAFs) compared to HBEC3KT cells were analyzed by qRT-PCR. (Lower) Levels of IL6 protein in cell culture supernatants and OSM protein in whole-cell lysates from HBEC3KT, NLFs, and CAFs were analyzed by ELISA.
(B) H23 cells in the lower chamber were co-cultured with controls, NLFs, or CAFs in the upper chamber for 5 days in transwell polycarbonate membrane cell culture inserts as shown in the top-panel. Proteins harvested from the H23 cells were analyzed by Western blotting.
(C) Six NSCLC cell lines were co-cultured with CAFs or without (Mock) for 24 hours in transwell cell culture inserts. The cells were then treated with DMSO or a targeted drug: H23 and H2009, selumetinib (10 μM); H3255 and HCC4006, erlotinib (100 nM); H3122 and HCC78, crizotinib (500 nM), with or without CAFs. After 7 to 10 days of treatment, cancer cells were stained by crystal violet.
(D) NSCLC cells were co-cultured with or without CAFs for 24 hours in transwell cell culture inserts, and then treated with a targeted drug alones [same drug and dose as in panel (C)] or in combination with filgotinib (5 μM) in co-culture with or without CAFs for 7 to 10 days. The viabilities of the cancer cells compared with control cells (without co-culture and treatment) were analyzed by MTT assay. Sel, selumetinib; Fil, filgotinib; Erl, erlotinib; Cri, crizotinib. *P <0.05.
(E) NSCLC cells were treated with the indicated targeted drug and co-cultured with or without CAFs for 5 days. Lysates obtained from cancer cells were examined by Western blotting with the indicated antibodies.
In cancer cells treated with a targeted drug in combination with filgotinib (Fig. 4D), a single treatment with filgotinib had a weak to moderate antitumor effect, while co-targeting of each oncogenic pathway and JAK1 by targeted drugs and filgotinib prevented drug resistance caused by co-culturing with CAFs. Targeted drug monotherapy suppressed apoptosis marker c-PARP in cancer cells co-cultured with CAFs, while co-treatment with a targeted drug and filgotinib reversed this c-PARP suppression while including suppression of p-STAT3 and OSMR (Fig. 4E).
JAK1 inhibition improves acquired resistance caused by proinflammatory pathway activation
To explore whether co-targeting the oncogenic pathway and OSMRs/JAK1/STAT3 pathway molecules could be effective in ameliorating acquired resistance to targeted drugs, we treated cells with each molecularly targeted drug in combination with siRNA against OSMR/LIFR, JAK1, or STAT3, or in combination with filgotinib. As shown in Fig. 5A, siRNA-mediated downregulation of OSMR/LIFR, JAK1, or STAT3 in combination with a targeted drug was effective in both parental and acquired-resistance cell lines. Importantly, filgotinib also reduced resistance to targeted drugs in combination with a molecularly targeted drug. After 48 hours of treatment, expression of p-STAT3 and OSMR was suppressed in both parental and acquired-resistance cells that received co-targeted treatment, and expression of c-PARP was restored in acquired-resistance cells (Fig. 5B).
Figure 5. JAK1 inhibition reduces acquired resistance caused by proinflammatory pathway activation.
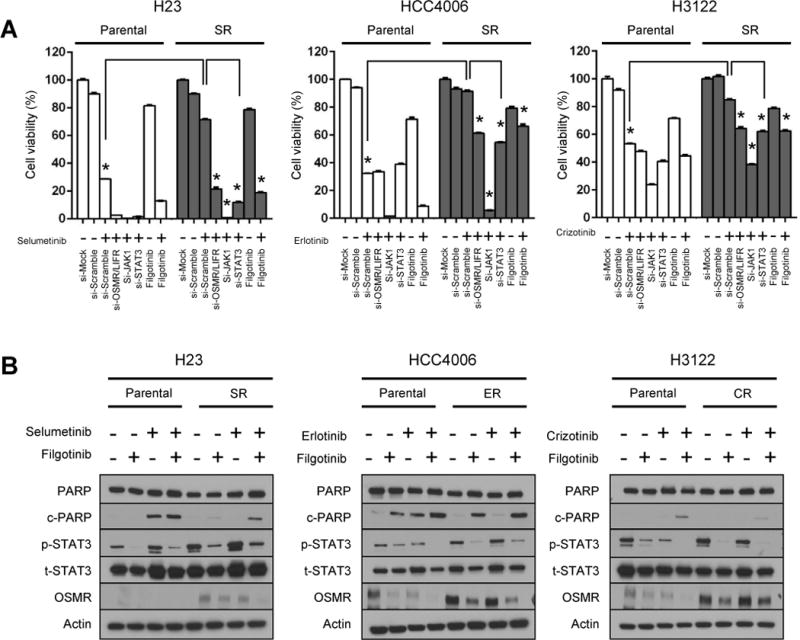
(A) Parental or acquired-resistance (SR, ER, CR) cells were seeded at 2×104 cells per well in 6-well plates and incubated for 24 hours. They were then transfected with siRNA specific for OSMR/LIFR, JAK1, STAT3, or scrambled negative control (Mock) for 48 hours, and treated with the respective targeted drug, with or without filgotinib, as follows: H23-parental and SR, selumetinib (10 μM); HCC4006-parental and ER, erlotinib (100 nM); H3122-parenat and CR, crizotinib (500 nM). After 7 to 10 days of treatment, cell viabilities compared to control cells (without siRNA and drugs) were measured by MTT assay. *P <0.05.
(B) Parental and acquired-resistance cells were treated with the indicated targeted drug, alone or in combination with filgotinib [same drug and dose as for panel (A)] for 72 hours, and lysates were examined by Western blotting with the indicated antibodies.
Correlation of each IL6 family cytokines and receptors and prognostic impact of OSMR in lung adenocarcinoma patients
To explore the correlation of each IL6 family cytokine and receptor in clinical lung adenocarcinoma samples, we analyzed gene expression levels of clinical samples in 515 TCGA lung adenocarcinoma samples. Fig. 6A shows the correlation of gene expression level between each IL6 family cytokine and receptor in TCGA samples. The gene expression levels of each IL6 family cytokines (IL6, OSM, and LIF) have positive correlation, in particular, between IL6 and OSM (Spearman ρ=0.56).
Figure 6. IL6 family cytokines and receptors have a role in treatment resistance in NSCLC.
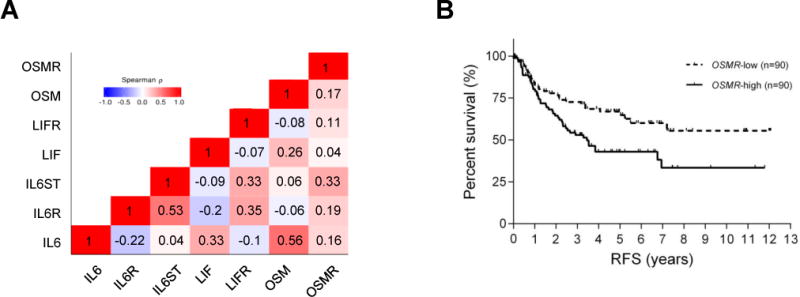
(A) The correlation between expression of each IL6 family cytokine and receptor in TCGA lung adenocarcinoma samples. Spearman ρ value is plotted in each box.
(B) Kaplan-Meier curves showing the recurrence-free survival (RFS) rates of the subset of PROSPECT patients with lung adenocarcinoma according to tumor expression of OSMR (upper median vs lower median).
Kaplan-Meier curves analysis of surgically resected 180 patients in PROSPECT with lung adenocarcinoma show that prognosis in the OSMR-high (upper median) patients was significantly poorer than that in the OSMR-low (lower median) patients (p=0.0096 for recurrent-free survival) (Fig. 6B). Univariate Cox proportional hazards regression analysis in surgically resected lung adenocarcinoma samples (TCGA and PROSPECT) and chemorefractory advanced lung adenocarcinoma samples (BATTLE-1 and BATTLE-2) was shown in Table S2. Interestingly, OSMR gene expression may be a prognostic factor in patients with surgically resected lung adenocarcinoma: TCGA (OS, hazard ratio [HR]: 1.18, 95% confidence interval [CI]: 1.00–1.40, p=0.050), and PROSPECT (RFS, HR: 1.25, 95% CI: 1.01–1.55, p=0.039), whereas it was not significant in patients with chemorefractory advanced lung adenocarcinoma. In multivariable Cox regression model of PROSPECT patients adjusting for age, tumor size, neoadjuvant treatment, pathologic-Stage, and taking into consideration of interaction between neoadjuvant treatment and pathologic-Stage, OSMR expression is an independent prognostic factor for RFS (HR: 1.25, 95% CI: 1.01–1.55, p=0.045) (Table S3).
Discussion
In this study, we found that activation of the IL6 family proinflammatory cytokine pathway in the cancer microenvironment can contribute to resistance to molecularly targeted drugs in oncogene-driven NSCLC cells. Our findings suggest that inhibition of MEK, functioning downstream of various receptor tyrosine kinases, triggers the feedback activation of STAT3 through not only IL6, as shown by Lee et al., but also OSM upregulation in cancer cells, significantly contributing to resistance to pathway-targeted therapies (9). STAT3 has been reported to be a major contributor to inflammation and also has been shown to promote the acquisition of treatment resistance (38). In most cells, STAT3 activation can suppress apoptosis through regulation of the expression of various cell survival gene products, including BCL-2, BCL-xl, MCL-1, survivin, and cIAP2 (38). However, effective targeted therapies generally induce apoptosis in oncogene-addicted cancers. This process involves the downstream BCL-2 family of proteins, which act as guardians of mitochondria-mediated apoptosis (39). Therefore, activation of STAT3 directly affects sensitivity to targeted drugs in oncogene-addicted cancers. These findings are compatible with our findings that activation of STAT3 by rhOSM treatment or co-culture with CAFs suppressed expression of apoptosis marker c-PARP induced by targeted drugs and that co-inhibition of major oncogenic pathways by targeted drugs and STAT3 by a JAK1 inhibitor restored c-PARP expression.
There seem to be several possibilities for how targeted drugs upregulate IL6 or OSM expression in cancer cells. Ishiguro et al. reported that IL6 promoter contains an activator protein-1 (AP-1) binding site and that EGFR-TKI treatment activated IL6 promoter transcription via regulation of AP-1 activation in cancer cells (40). Interestingly, OSM promoter also contains an AP-1 binding site (41). Thus, AP-1 activation by targeted drugs might be one of the mechanisms for IL6 or OSM upregulation. Indeed, the gene expression levels of IL6 and OSM has positive correlation in TCGA samples.
In addition to autocrine activation of STAT3 in NSCLC cells triggered by targeted drugs, tumor stromal CAFs secreted IL6 and OSM and also contributed to resistance to targeted drugs in cancer cells. Lapeire et al. reported that cancer-associated adipose tissue promotes breast cancer progression by paracrine OSM secretion and STAT3 activation (26). Caffarel et al. reported that the main source of OSM in cancers seems to be the tumor stroma, suggesting the existence of paracrine signaling between tumor stroma and cancer cells (42). At the same time, they mentioned that OSM production has been observed in several kinds of cancers, including breast and prostate cancers, indicating the potential for additional autocrine signaling (42). The stromal cells along with CAFs may give rise to local inflammation in the cancer microenvironment through IL6 and OSM secretion, thus contributing to resistance to targeted drugs. Therefore, reduction of inflammatory pathway activation in cancer cells by JAK1/STAT3 inhibition may improve the cells’ sensitivity to pathway-targeted drugs.
We found that JAK1 is a critical mediator of STAT3 activation by IL6 family cytokines in NSCLC cells. Many cytokines relying on JAK family members for intracellular signal transduction have key pathophysiological roles in autoimmune and inflammatory diseases (43). Our findings indicate that STAT3 activation by IL6 family cytokines could be effectively blocked by JAK1 inhibition in NSCLC cells. Therefore, we investigated the combination efficacy of combining pathway-targeted drugs and selective JAK1 inhibitor filgotinib in STAT3-activated cells resistant to targeted drugs. In clinical trial, the combination of JAK1/2 inhibitor ruxolitinib with erlotinib did not demonstrate dramatic effect in patients with acquired erlotinib resistance in recent study (44), while the sample size was small and the results were still controversial. On the other hand, filgotinib is an oral selective JAK1 inhibitor, and showed encouraging safety and efficacy in clinical studies in patients with rheumatoid arthritis (17, 34, 45). Selective inhibition of JAK1 also may offer favorable safety and clinical efficacy in cancer treatment. Interestingly, JAK1/STAT3 pathway inhibition by siRNA or filgotinib suppressed not only p-STAT3 but upstream OSMR protein expression. Although the mechanisms that cause this suppression are still unclear, inhibition of the JAK1/STAT3 pathway could block the feedback activation of OSMR.
We found that OSMR expression in surgically resected samples appeared to portend worse prognosis for patients with NSCLC. The OSM/OSMR pathway closely relates to inflammation, hematopoiesis, and development (46). Activation of this pathway is increasingly recognized as an important contributor to progression of many types of cancer, including lung cancer (27, 29, 32, 47, 48). In breast cancer, high OSMR expression correlated with shorter recurrence-free and overall survival-duration and with chemotherapy resistance in estrogen receptor-negative tumors (27). To our knowledge, the present study is the first report that OSMR could be a possible prognostic marker in patient with surgically resected NSCLC. In contrast, OSMR expression did not correlate with prognosis significantly in patients with chemorefractory advanced stage NSCLC. Although the detailed mechanism remains unclear, one possible explanation is that most of heavily treated chemorefractory NSCLC samples could harbor basal upregulation of inflammatory-related pathways, including OSM/OSMR pathway. The previous treatment caused inflammation in the cancer microenvironment and upregulation of OSMR in a subset of tumors, which could become resistant to targeted drugs.
Our study has some limitations. One is the inability to confirm the JAK1/STAT3 pathway activation associated with IL6 family cytokines upregulation in clinical samples that showed acquired resistance to targeted drugs. In order to confirm these pathway activations and to obtain a possible new biological profile that can be a treatment target, the comparative analysis of samples before and after acquired resistance to targeted therapies is required.
In summary, our findings indicate that activation of the OSMRs/JAK1/STAT3 pathway contributes to resistance to targeted drugs in oncogene-driven NSCLC cells. These findings suggest that co-treatment targeting the OSMRs/JAK1/STAT3 pathway could further improve the antitumor efficacy of oncogenic pathway inhibition by targeted therapies.
Supplementary Material
Acknowledgments
The authors would like to thank the Department of Scientific Publications, The University of Texas MD Anderson Cancer Center, for the English language review.
Financial supports: This work was supported by the NIH NCI grants R01 CA155196 (V. A. Papadimitrakopoulou, R. S. Herbst, and I. I. Wistuba) and 2P50CA070907-16A1 (J. D. Minna and I. I. Wistuba). In addition, this work was partially supported by the UT Lung Specialized Programs of Research Excellence grant P50CA70907, and MD Anderson’s Institutional Tissue Bank (ITB) Award Number 2P30CA016672 from the NIH NCI. K. Shien is supported by the Japan Society for the Promotion of Science (JSPS) Postdoctoral Fellowship for Research Abroad.
References
- 1.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–80. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 2.Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11:473–81. doi: 10.1038/nrclinonc.2014.104. [DOI] [PubMed] [Google Scholar]
- 3.Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9. doi: 10.1038/nature11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou BB, Peyton M, He B, Liu C, Girard L, Caudler E, et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell. 2006;10:39–50. doi: 10.1016/j.ccr.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yonesaka K, Zejnullahu K, Lindeman N, Homes AJ, Jackman DM, Zhao F, et al. Autocrine production of amphiregulin predicts sensitivity to both gefitinib and cetuximab in EGFR wild-type cancers. Clin Cancer Res. 2008;14:6963–73. doi: 10.1158/1078-0432.CCR-08-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res. 2008;68:9479–87. doi: 10.1158/0008-5472.CAN-08-1643. [DOI] [PubMed] [Google Scholar]
- 7.Kim SM, Kwon OJ, Hong YK, Kim JH, Solca F, Ha SJ, et al. Activation of IL-6R/JAK1/STAT3 signaling induces de novo resistance to irreversible EGFR inhibitors in non-small cell lung cancer with T790M resistance mutation. Mol Cancer Ther. 2012;11:2254–64. doi: 10.1158/1535-7163.MCT-12-0311. [DOI] [PubMed] [Google Scholar]
- 8.Vultur A, Villanueva J, Krepler C, Rajan G, Chen Q, Xiao M, et al. MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene. 2014;33:1850–61. doi: 10.1038/onc.2013.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ, Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014;26:207–21. doi: 10.1016/j.ccr.2014.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J. 2003;374:1–20. doi: 10.1042/BJ20030407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hibi M, Nakajima K, Hirano T. IL-6 cytokine family and signal transduction: a model of the cytokine system. J Mol Med (Berl) 1996;74:1–12. doi: 10.1007/BF00202068. [DOI] [PubMed] [Google Scholar]
- 12.Thoma B, Bird TA, Friend DJ, Gearing DP, Dower SK. Oncostatin M and leukemia inhibitory factor trigger overlapping and different signals through partially shared receptor complexes. J Biol Chem. 1994;269:6215–22. [PubMed] [Google Scholar]
- 13.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–54. [PubMed] [Google Scholar]
- 14.Wendt MK, Balanis N, Carlin CR, Schiemann WP. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAKSTAT. 2014;3:e28975. doi: 10.4161/jkst.28975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 16.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576–83. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 17.Menet CJ, Fletcher SR, Van Lommen G, Geney R, Blanc J, Smits K, et al. Triazolopyridines as selective JAK1 inhibitors: from hit identification to GLPG0634. J Med Chem. 2014;57:9323–42. doi: 10.1021/jm501262q. [DOI] [PubMed] [Google Scholar]
- 18.Burns CJ, Bourke DG, Andrau L, Bu X, Charman SA, Donohue AC, et al. Phenylaminopyrimidines as inhibitors of Janus kinases (JAKs) Bioorg Med Chem Lett. 2009;19:5887–92. doi: 10.1016/j.bmcl.2009.08.071. [DOI] [PubMed] [Google Scholar]
- 19.Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. 2006;13:1235–42. doi: 10.1016/j.chembiol.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 20.Shien K, Toyooka S, Yamamoto H, Soh J, Jida M, Thu KL, et al. Acquired Resistance to EGFR Inhibitors Is Associated with a Manifestation of Stem Cell-like Properties in Cancer Cells. Cancer Res. 2013;73:3051–61. doi: 10.1158/0008-5472.CAN-12-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Jr, Tsao A, et al. The BATTLE trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1:44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Papadimitrakopoulou V, Lee JJ, Wistuba II, Tsao AS, Fossella FV, Kalhor N, et al. The BATTLE-2 Study: A Biomarker-Integrated Targeted Therapy Study in Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016 doi: 10.1200/JCO.2015.66.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun Y, Nowak KA, Zaorsky NG, Winchester CL, Dalal K, Giacalone NJ, et al. ALK inhibitor PF02341066 (crizotinib) increases sensitivity to radiation in non-small cell lung cancer expressing EML4-ALK. Mol Cancer Ther. 2013;12:696–704. doi: 10.1158/1535-7163.MCT-12-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lovly CM, Heuckmann JM, de Stanchina E, Chen H, Thomas RK, Liang C, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71:4920–31. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergethon K, Shaw AT, Ou SH, Katayama R, Lovly CM, McDonald NT, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lapeire L, Hendrix A, Lambein K, Van Bockstal M, Braems G, Van Den Broecke R, et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res. 2014;74:6806–19. doi: 10.1158/0008-5472.CAN-14-0160. [DOI] [PubMed] [Google Scholar]
- 27.West NR, Murray JI, Watson PH. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene. 2014;33:1485–94. doi: 10.1038/onc.2013.105. [DOI] [PubMed] [Google Scholar]
- 28.Guo L, Chen C, Shi M, Wang F, Chen X, Diao D, et al. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013;32:5272–82. doi: 10.1038/onc.2012.573. [DOI] [PubMed] [Google Scholar]
- 29.Winder DM, Chattopadhyay A, Muralidhar B, Bauer J, English WR, Zhang X, et al. Overexpression of the oncostatin M receptor in cervical squamous cell carcinoma cells is associated with a pro-angiogenic phenotype and increased cell motility and invasiveness. J Pathol. 2011;225:448–62. doi: 10.1002/path.2968. [DOI] [PubMed] [Google Scholar]
- 30.Smith DA, Kiba A, Zong Y, Witte ON. Interleukin-6 and oncostatin-M synergize with the PI3K/AKT pathway to promote aggressive prostate malignancy in mouse and human tissues. Mol Cancer Res. 2013;11:1159–65. doi: 10.1158/1541-7786.MCR-13-0238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fossey SL, Bear MD, Kisseberth WC, Pennell M, London CA. Oncostatin M promotes STAT3 activation, VEGF production, and invasion in osteosarcoma cell lines. BMC Cancer. 2011;11:125. doi: 10.1186/1471-2407-11-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.David E, Tirode F, Baud’huin M, Guihard P, Laud K, Delattre O, et al. Oncostatin M is a growth factor for Ewing sarcoma. Am J Pathol. 2012;181:1782–95. doi: 10.1016/j.ajpath.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 33.Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal. 2015;8:ra82. doi: 10.1126/scisignal.aab1111. [DOI] [PubMed] [Google Scholar]
- 34.Namour F, Diderichsen PM, Cox E, Vayssiere B, Van der Aa A, Tasset C, et al. Pharmacokinetics and Pharmacokinetic/Pharmacodynamic Modeling of Filgotinib (GLPG0634), a Selective JAK1 Inhibitor, in Support of Phase IIB Dose Selection. Clin Pharmacokinet. 2015;54:859–74. doi: 10.1007/s40262-015-0240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4:452–65. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 37.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, et al. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Ann N Y Acad Sci. 2009;1171:59–76. doi: 10.1111/j.1749-6632.2009.04911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ebi H, Faber AC, Engelman JA, Yano S. Not just gRASping at flaws: finding vulnerabilities to develop novel therapies for treating KRAS mutant cancers. Cancer Sci. 2014;105:499–505. doi: 10.1111/cas.12383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ishiguro Y, Ishiguro H, Miyamoto H. Epidermal growth factor receptor tyrosine kinase inhibition up-regulates interleukin-6 in cancer cells and induces subsequent development of interstitial pneumonia. Oncotarget. 2013;4:550–9. doi: 10.18632/oncotarget.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma Y, Streiff RJ, Liu J, Spence MJ, Vestal RE. Cloning and characterization of human oncostatin M promoter. Nucleic Acids Res. 1999;27:4649–57. doi: 10.1093/nar/27.23.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caffarel MM, Coleman N. Oncostatin M receptor is a novel therapeutic target in cervical squamous cell carcinoma. J Pathol. 2014;232:386–90. doi: 10.1002/path.4305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–50. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu HA, Perez L, Chang Q, Gao SP, Kris MG, Riely GJ, et al. A Phase 1/2 Trial of Ruxolitinib and Erlotinib in Patients with EGFR-Mutant Lung Adenocarcinomas with Acquired Resistance to Erlotinib. J Thorac Oncol. 2017;12:102–9. doi: 10.1016/j.jtho.2016.08.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191:3568–77. doi: 10.4049/jimmunol.1201348. [DOI] [PubMed] [Google Scholar]
- 46.Tanaka M, Miyajima A. Oncostatin M, a multifunctional cytokine. Rev Physiol Biochem Pharmacol. 2003;149:39–52. doi: 10.1007/s10254-003-0013-1. [DOI] [PubMed] [Google Scholar]
- 47.Komyod W, Bohm M, Metze D, Heinrich PC, Behrmann I. Constitutive suppressor of cytokine signaling 3 expression confers a growth advantage to a human melanoma cell line. Mol Cancer Res. 2007;5:271–81. doi: 10.1158/1541-7786.MCR-06-0274. [DOI] [PubMed] [Google Scholar]
- 48.Lauber S, Wong S, Cutz JC, Tanaka M, Barra N, Lhotak S, et al. Novel function of Oncostatin M as a potent tumour-promoting agent in lung. Int J Cancer. 2015;136:831–43. doi: 10.1002/ijc.29055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.