

Abstract
How can you use a ruthenium isomerization catalyst twice? A ruthenium-catalyzed sequence for the formal two-carbon scission of allyl groups to carboxylic acids has been developed. The reaction includes an initial isomerization step using commercially available ruthenium catalysts followed by in situ transformation of the complex to a metal-oxo species, which is capable of catalyzing subsequent oxidation reactions. The method enables enantioselective syntheses of challenging α-tri- and tetrasubstituted α-amino acids including an expedient total synthesis of the antiepileptic drug levetiracetam.
SYNOPSIS TOC
If you are submitting your paper to a journal that requires a synAuthors are required to submit a graphic entry for the Table of Contents (TOC) that, in conjunction with the manuscript title, should give the reader a representative idea of one of the following: A key structure, reaction, equation, concept, or theorem, etc., that is discussed in the manuscript. Consult the journal’s Instructions for Authors for TOC graphic specifications.

INTRODUCTION
The use of a single metal catalyst for several chemical transformations in a one-pot and/or a sequential manner1 is highly desirable due to high material cost and limited resources of transition metals.2 Moreover, new tools for modifying prevalent functional groups, which are easily installed in a chemo- and stereoselective fashion, are needed to expand the application of novel methodology in both academic and industrial settings.3 In this context, allyl groups are ubiquitous and their introduction is very well established including asymmetric additions.4 In general, three modes for their installation can be distinguished (Figure 1): As a nucleophile5,6 (e.g. Grignard addition7), as an electrophile8 (e.g. Tsuji-Trost allylation9) or as a formally neutral species10 (e.g. Keck radical allylation11).
Figure 1.

General modes for the introduction of allyl groups.
Most of these methods not only find widespread use in organic synthesis, but also allow for the installation of allyl groups in a highly stereoselective fashion, even to build challenging chiral quaternary stereocenters.12 However, tools for the formal scission of two carbons of an allyl group leading to a truncated carboxylic acid using a single transition metal catalyst have, to our knowledge, not been described (Scheme 1A, M1=M2). The use of only one catalyst is in strong contrast to the field of one-pot catalysis, where for instance two transition metal catalysts are used for isomerization/metathesis reactions. 13
Scheme 1.

Sequences and methods for the introduction of chiral carboxylic acid groups.
Although a sequence involving vinylation followed by oxidative cleavage of the double bond would in theory lead to the same carboxylic acid product, asymmetric vinylation reactions are far less established than allylations (Scheme 1B).4 In particular, catalytic enantioselective vinylation of carbonyl enolates is still highly challenging and limited in substrate scope and, moreover, methods for the construction of quaternary stereocenters are very rare.12,14 Even more scarce and far less established are catalytic enantioselective carboxylations (Scheme 1C).15
As a result, we focused our attention on the catalytic oxidative scission of two carbons from the tail of an allyl unit in order to render such groups as carboxylic acid synthesis equivalents (Scheme 2). Herein, we present the results of that study and provide illustrative examples of this new method in the context of high value and unusual amino acid synthesis.
Scheme 2.

Ruthenium-catalyzed two-carbon truncation of allyl groups to carboxylic acids.
RESULTS AND DISCUSSION
After exploring a variety of transition metal catalysts known for alkene isomerization (iridium, rhodium, palladium, ruthenium; for details see the Supporting Information), we found that ruthenium catalysts performed best and were amenable to further oxidative catalysis (Scheme 2).
In particular, the commercially available Grubbs second generation catalyst 1a (Figure 2) under Nishida’s conditions16 was highly chemoselective for alkene isomerization and displayed good conversion (>90%) without noticeable side reactions. Moreover, we discovered that the in situ formed Ru–H complex17 1b, after solvent exchange, can be oxidized with NaIO4 to an oxidized ruthenium species,18 which is capable of catalyzing subsequent oxidation reactions (e.g. oxidative cleavage of alkenes, Scheme 2).19 It has to be reinforced at this point that we do not add a new external ruthenium source for the second step. Instead, the crude transition metal catalyst from the previous isomerization step is modified in situ and reused for the catalytic oxidation reactions. Moreover, the only operation between the two steps is evaporation of the solvent from the isomerization reaction (toluene for 1a, acetone for 2) followed by dissolution of the crude reaction mixture in the solvent mixture used for oxidation (CCl4, MeCN, H2O).
Figure 2.

Identified ruthenium catalysts for olefin isomerization/ oxidation sequences – An in situ formed ruthenium-hydride complex 1b derived from Grubbs second generation catalyst (1a) and Grotjahn’s catalyst (2).
Even though NaIO4 mediated oxidations of ruthenium alkylidene complexes and their use in sequential and tandem catalysis19b,20 have been described previously (e.g. RCM/dihydroxylation),21 the modification and multiple use13 of a ruthenium-hydride complex17 1b (derived from precatalyst 1a) or a ruthenium phosphine-imidazole complex22 2 in an isomerization/oxidation sequence was not known, until now (Figure 2).
Hence, chiral homoallylic amine 3, which was readily synthesized via diastereoselective allylation of Ellman’s sulfinimine, 5e was subjected to the optimized conditions using Grubbs catalyst 1a giving the isomerized alkene in 98% conversion (Scheme 3A). After oxidation of the crude catalyst, an alkene cleavage/sulfinamide oxidation duet23 took place furnishing Bus-protected24 D-tert-leucine (4) in 72% yield over 2 steps, notably without racemization (99% ee). The Bus protected amino acid 4 can be employed directly for a peptide coupling as reported by Hanessian,24 or the Bus group is readily cleaved under acidic conditions to afford D-tert-leucine (5) in 96% yield (98% ee) after ion exchange chromatography.
Scheme 3.

a) 1. Grubbs 1a (5 mol%), VTMS, toluene, reflux, 21 h, 98% conv., E/Z 4.4:1; 2. NaIO4, CCl4, MeCN, H2O, 23°C, 2.5 d, 72% yield over 2 steps, 99% ee; b) 1. TfOH, CH2Cl2, 0°C, 2.5 h; 2. IEC, 96% yield, 98% ee; c) RuCl3•H2O (5 mol%), NaIO4, CCl4, MeCN, H2O, 23°C, 16 h, 93% yield; d) 1. TfOH, CH2Cl2, 0°C→23°C, 2 d; 2) IEC, 91% yield, 98% ee; e) 1. Grubbs 1a (5 mol%), VTMS, toluene, 128°C, 16 h, 97% conv., E/Z 4:1; 2. NaIO4, CCl4, MeCN, H2O, 23°C, 22 h, 88% yield over 2 steps; f) 1. 4 M aq. HCl, MeOH, reflux, 24 h; 2. IEC, 57% yield; g) 1. Grubbs 1a (5 mol%), VTMS, toluene, 128°C, 18 h, 92% conv., E/Z 4.5:1; 2. NaIO4, CCl4, MeCN, H2O, 23°C, 24 h, 62% yield over 2 steps; h) ClCO2Et, NEt3, THF, then NH4OH, 0°C→23°C, 19 h, 67% yield, >98% ee. IEC=ion exchange chromatography, VTMS=vinyloxy trimethylsilane.
Alternatively, by leaving out the olefin isomerization, sulfinamide 3 was subjected to a similar alkene cleavage/ sulfinamide oxidation sequence with a catalytic amount of RuCl3 hydrate giving Bus-protected β-amino acid 6 in 93% yield (Scheme 3B). After deprotection and ion exchange chromatography, (S)-β-neopentylglycine (7) was obtained in 91% yield and 98% ee. Benzoyl protected amine 8, which is accessible in enantiopure form using Schaus’ allylboration,25 performed equally well in our sequential ruthenium catalysis, affording carboxylic acid 9 in 88% yield over 2 steps (Scheme 3C). Benzoyl deprotection with aqueous HCl in methanol provided racemic tert-leucine (5) in 57% yield.
The sequential ruthenium catalysis was then used for an enantioselective total synthesis of levetiracetam (12), the active pharmaceutical ingredient of the antiepileptic medicine Keppra ® (Scheme 3D).26 The synthesis commenced from homoallylic amide 10, which was prepared in four steps from commercially available propionaldehyde and Ellman’s auxiliary5e (see the Supporting Information for details). Sequential ruthenium catalysis of alkene 10 with Grubbs catalyst 1a provided access to carboxylic acid 11 in 62% yield over two steps. Amidation of 11 was achieved via the corresponding mixed anhydride and reaction with ammonium hydroxide as described previously by Sánchez.27 Levetiracetam (12) was isolated in 67% yield (>98% ee) and, after one recrystallization, was enriched to excellent enantiopurity (>99.9% ee).
We next applied our sequential ruthenium catalysis conditions to α-quaternary lactams (Scheme 4), which were readily available in enantiopure form using palladium-catalyzed decarboxylative allylic alkylation.8a,28 The unprotected methyl- and ethyl-substituted lactams 13 and 14 were treated with a catalytic amount of Grubbs catalyst 1a and the internal alkene was subsequently cleaved to give the crude carboxylic acids. A Curtius rearrangement29 then furnished isocyanates 15 and 16 in 27% and 47% yield over 3 steps, respectively.30 Likewise, we found that 2 mol% of Grotjahn’s catalyst22 (2, Figure 2) worked equally well31 for the ruthenium-catalyzed isomerization/oxidation sequence of lactam 14 to give, after Curtius rearrangement, isocyanate 16 in a comparable 52% yield over 3 steps.32 Hydrolysis of the isocyanate and the amide bond of 15 and 16 were achieved under acidic conditions to give access to enantiopure (R)-α-methylornithine33 (17) and (R)-α-ethylornithine34 (18) in 61% and 95% yield, respectively. The latter has been synthesized for the first time as a single enantiomer. 34 Biologically, both α-alkyl ornithine analogs are known to be ornithine decarboxylase inhibitors.33a,34
Scheme 4.

a) 15: 1. Grubbs 1a (5 mol%), VTMS, toluene, 130°C, 18 h, 88% conv.; 2. NaIO4, CCl4, MeCN, H2O, 23°C; 3. DPPA, NEt3, MeCN, 0°C→65°C, 3 h, 27% yield over 3 steps; 16: 1. Grubbs 1a (5 mol%), VTMS, toluene, 125°C, 16 h, 96% conv. or Grotjahn 2 (2 mol%), acetone-d6, 70°C, 63 h, 95% conv.; 2. NaIO4, CCl4, MeCN, H2O, 23°C, 3. DPPA, NEt3, MeCN, 0°C→65°C, 47% yield over 3 steps (with 1a) and 52% yield over 3 steps (with 2); b) 17: 1. 4 M aq. HCl, 1,4-dioxane, 120°C, 12 h; 2. IEC, 61% yield; 18: 1. 2 M aq. HCl, THF, reflux, 24 h; 2. IEC, 95% yield; c) 1. Grubbs 1a (5 mol%), VTMS, toluene, 129°C, 21 h, 96% conv., 2. NaIO4, CCl4, MeCN, H2O, 23°C, 24 h; 3. DPPA, NEt3, MeCN, 0°C→65°C, 3 h, 65% yield over 3 steps; d) 1. 4 M aq. HCl, dioxane, reflux, 18 h; 2. IEC, 92% yield; e) 1. Grubbs 1a (5 mol%), VTMS, toluene, 129°C, 21 h, 93% conv.; 2. NaIO4, CCl4, MeCN, H2O, 23°C, 25 h; 3. DPPA, NEt3, MeCN, 0°C→65°C, 3 h, 34% yield over 3 steps; f) 1. 4 M aq. HCl, dioxane, reflux, 18 h; 2. IEC, 99% yield. DPPA=diphenyl phosphoryl azide, IEC=ion exchange chromatography, VTMS=vinyloxy trimethylsilane.
Treatment of caprolactam 19 and glutarimide 22 with catalyst 1a under our optimized conditions gave, after Curtius rearrangement, hydantoin 20 and isocyanate 23 in 65% and 34% yield over 3 steps, respectively. Anti-Bredt bicycle35–37 20 results from an intramolecular cyclization of the lactam NH to the isocyanate. Its structure was unambiguously confirmed by X-ray crystallography (see the Supporting Information). Subsequent hydrolysis of 20 and 23 with aqueous HCl furnished (R)-α-methyllysine33d (21) and (R)-α-methylglutamic acid33c,38 (24), respectively, in excellent yields (92% and 99%). Given the recent availability of α-quaternary lactams and imides,28 it is possible to envision the synthesis of a wide range of α-substituted amino acid derivatives for a multitude of applications.
The enantioenriched carboxylic acid 25, obtained by our Ru-catalyzed isomerization oxidation method on lactam 14 (Scheme 4), is not only an intermediate in Padwa’s racemic synthesis of desacetoxy-4-oxo-6,7-dihydrovindoro-sine39 (29), but can also be used for the synthesis of Cbz-protected α-amino lactam 26 (98% ee) by trapping the intermediate isocyanate 16 with benzyl alcohol following the Curtius rearrangement (Scheme 5). Hydrogenolysis of the Cbz group gave amino lactam 27 in quantitative yield, which renders Knabe’s racemic synthesis of 3-ethyl thalidomide40 (30) enantioselective. Reduction of lactam 27 with LiAlH4, followed by precipitation as the bishydrochloride salt furnished chiral diamine 28 in 89% yield. The racemic, mono-Boc protected version of 28 was used recently by Nishio et al.41 for the synthesis of dipeptidyl peptidase IV (DPP-4) inhibitors.
Scheme 5.

a) DPPA, NEt3, DCE, 23°C→reflux, 5 h, then BnOH, reflux, 38 h, 29% yield, 98% ee; b) H2, Pd/C, MeOH, 23°C, 4 h, >99% yield; c) LiAlH4, THF, 0°C→reflux, 24 h, then HCl/dioxane, 89% yield. DCE=1,2-dichloroethane, DPPA= diphenyl phosphoryl azide.
CONCLUSIONS
In summary, we have developed a highly efficient ruthenium catalyzed isomerization/oxidation sequence, which enabled the syntheses of challenging unnatural amino acids such as D-tert-leucine (5), (S)-β-neopentylglycine (7), (R)-α-methyl- and ethylornithine (17 and 18), (R)-α-methyllysine (21) and (R)-α-methylglutamic acid (24). The reaction sequence performs well with not only ruthenium-hydride complex 1b (derived from Grubbs catalyst 1a), but also with the less common Grotjahn catalyst 2. We found that both isomerization catalysts 1b and 2 can be used to perform one or several subsequent oxidation steps after NaIO4 treatment of the crude reaction mixtures. To demonstrate the utility of our method, we have completed an enantioselective total synthesis of the antiepileptic drug levetiracetam (12) and enantioselective formal syntheses of a vindorosine derivative 29 and of (R)-3-ethyl thalidomide (30). Given the importance and ubiquity of the amino acid subunit in organic chemistry, we believe that our method will inspire many creative permutations and applications. Further usages of the method are currently under investigation.
Supplementary Material
Acknowledgments
Dedicated to Professor Albert Padwa on the occasion of his 80th birthday. The authors wish to thank NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation and Caltech for financial support. M.L. thanks the Swiss National Science Foundation (SNSF) for a postdoctoral fellowship (P2EZP2_148751). Dr. Scott Virgil and the Caltech Center for Catalysis and Chemical Synthesis are acknowledged for the generous donation of catalyst 2 and Materia Inc. for the donation of catalyst 1a. The authors thank Dr. Michael Takase and Larry Henling for X-ray structural determinations. The Hsieh-Wilson group is acknowledged for nanopure water and the Dougherty group for using their freeze-drying equipment. Lukas Hilpert, Kyle Virgil and Katerina Korch are thanked for experimental assistance. Beau P. Pritchett is gratefully acknowledged for recording analytical data and for proofreading this manuscript.
Footnotes
ASSOCIATED CONTENT
Experimental procedures, characterization data, and crystallographic information files. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.For the definition of sequential transformations see: Tietze LF, Beifuss U. Angew Chem Int Ed Engl. 1993;32:131.
- 2.(a) North M. In: Sustainable Catalysis: Without Metals or Other Endangered Elements, Part 1. North M, editor. The Royal Society of Chemistry; Cambridge: 2016. pp. 1–2. [Google Scholar]; (b) Holzwarth MS, Plietker B. ChemCatChem. 2013;5:1650. [Google Scholar]; (c) Roeder MH, Eigenbrodt BC, Paul JJ. In: Catalysis for Sustainability: Goals, Challenges, and Impacts. Umile TP, editor. CRC Press; Boca Raton: 2016. pp. 149–176. [Google Scholar]
- 3.(a) Carreira EM, Yamamoto H, editors. Comprehensive Chirality. Vol. 9. Elsevier; Amsterdam: 2012. Industrial Applications of Asymmetric Synthesis; pp. 1–506. [Google Scholar]; (b) Brown DG, Boström J. J Med Chem. 2016;59:4443. doi: 10.1021/acs.jmedchem.5b01409. [DOI] [PubMed] [Google Scholar]
- 4.(a) Carreira EM, Kvaerno; L. Classics in Stereoselective Synthesis. Chapter 5. Wiley-VCH; Weinheim: 2008. pp. 153–186. [Google Scholar]; (b) Carreira EM, Kvaerno; L. Classics in Stereoselective Synthesis. Chapter 14. Wiley-VCH; Weinheim: 2008. pp. 457–482. [Google Scholar]
- 5.For selected seminal publications of nucleophilic allylation see: Hosomi A, Sakurai H. Tetrahedron Lett. 1976;17:1295.Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092.Roush WR, Walts AE, Hoong LK. J Am Chem Soc. 1985;107:8186.Keck GE, Tarbet KH, Geraci LS. J Am Chem Soc. 1993;115:8467.Liu G, Cogan DA, Ellman JA. J Am Chem Soc. 1997;119:9913.Cogan DA, Liu G, Ellman J. Tetrahedron. 1999;55:8883.Kinnaird JWA, Ng PY, Kubota K, Wang X, Leighton JL. J Am Chem Soc. 2002;124:7920. doi: 10.1021/ja0264908.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:6340. doi: 10.1021/ja802001b.Kim IS, Ngai MY, Krische MJ. J Am Chem Soc. 2008;130:14891. doi: 10.1021/ja805722e.Beeson TD, Mastracchio A, Hong JB, Ashton K, MacMillan DWC. Science. 2007;316:582.Vieira EM, Snapper ML, Hoveyda AH. J Am Chem Soc. 2011;133:3332. doi: 10.1021/ja200311n.Silverio DL, Torker S, Pilyugina T, Vieira EM, Snapper ML, Haeffner F, Hoveyda AH. Nature. 2013;494:216. doi: 10.1038/nature11844.
- 6.For recent reviews see: Denmark SE, Fu J. Chem Rev. 2003;103:2763. doi: 10.1021/cr020050h.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed. 2009;48:34. doi: 10.1002/anie.200802938.Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774. doi: 10.1021/cr1004474.
- 7.(a) Grignard V. C R Hebd Acad Sci. 1900;130:1322. [Google Scholar]; (b) Hutchison DA, Beck KR, Benkeser RA, Grutzner JB. J Am Chem Soc. 1973;95:7075. [Google Scholar]; (c) Benkeser RA. Synthesis. 1971:347. [Google Scholar]
- 8.For selected references of electrophilic allylation see: Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x.Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065. doi: 10.1126/science.1237068.Liu WB, Reeves CM, Virgil SC, Stoltz BM. J Am Chem Soc. 2013;135:10626. doi: 10.1021/ja4052075.Pupo G, Properzi R, List B. Angew Chem Int Ed. 2016;55:6099. doi: 10.1002/anie.201601545.For selected reviews see: Trost BM, Van Vranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.Trost BM, Crawley ML. Chem Rev. 2003;103:2921. doi: 10.1021/cr020027w.Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461. doi: 10.1021/ar100047x.Hong AY, Stoltz BM. Eur J Org Chem. 2013:2745. doi: 10.1002/ejoc.201201761.Zhuo CX, Zheng C, You SL. Acc Chem Res. 2014;47:2558. doi: 10.1021/ar500167f.
- 9.(a) Tsuji J, Takahashi H, Morikawa M. Tetrahedron Lett. 1965;6:4387. [Google Scholar]; (b) Trost BM, Fullerton TJ. J Am Chem Soc. 1973;95:292. [Google Scholar]
- 10.For examples of tin-free radical allylations see: Quiclet-Sire B, Zard SZ. J Am Chem Soc. 1996;118:1209.Schaffner AP, Renaud P. Angew Chem Int Ed. 2003;42:2658. doi: 10.1002/anie.200351171.Debien L, Quiclet-Sire B, Zard SZ. Acc Chem Res. 2015;48:1237. doi: 10.1021/acs.accounts.5b00019.
- 11.Keck GE, Yates JB. J Am Chem Soc. 1982;104:5829. [Google Scholar]
- 12.Quasdorf KW, Overman LE. Nature. 2014;516:181. doi: 10.1038/nature14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For selected examples of one-pot catalysis with 1a and 2 using a second catalyst, see: Baader S, Podsiadly PE, Cole-Hamilton DJ, Goossen LJ. Green Chem. 2014;16:4885.Dobereiner GE, Erdogan G, Larsen CR, Grotjahn DB, Schrock RR. ACS Catal. 2014;4:3069.Higman CS, De Araujo MP, Fogg DE. Catal Sci Technol. 2016;6:2077.
- 14.(a) Chieffi A, Kamikawa K, Åhman J, Fox JM, Buchwald SL. Org Lett. 2001;3:1897. doi: 10.1021/ol0159470. [DOI] [PubMed] [Google Scholar]; (b) Taylor AM, Altman RA, Buchwald SL. J Am Chem Soc. 2009;131:9900. doi: 10.1021/ja903880q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y, Virgil SC, Grubbs RH, Stoltz BM. Angew Chem Int Ed. 2015;54:11800. doi: 10.1002/anie.201505161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.The only example we could find for a homogeneous carboxylation using CO2 is a nickel-catalyzed asymmetric carboxylative cyclization of bis-1,3-dienes: Takimoto M, Nakamura Y, Kimura K, Mori M. J Am Chem Soc. 2004;126:5956. doi: 10.1021/ja049506y.
- 16.(a) Arisawa M, Terada Y, Nakagawa M, Nishida A. Angew Chem Int Ed. 2002;41:4732. doi: 10.1002/anie.200290031. [DOI] [PubMed] [Google Scholar]; (b) Arisawa M, Terada Y, Takahashi K, Nakagawa M, Nishida A. J Org Chem. 2006;71:4255. doi: 10.1021/jo060308u. [DOI] [PubMed] [Google Scholar]
- 17.(a) Trnka TM, Morgan JP, Sanford MS, Wilhelm TE, Scholl M, Choi TL, Ding S, Day MW, Grubbs RH. J Am Chem Soc. 2003;125:2546. doi: 10.1021/ja021146w. [DOI] [PubMed] [Google Scholar]; (b) Dinger MB, Mol JC. Eur J Inorg Chem. 2003:2827. [Google Scholar]; (c) Hong SH, Day MW, Grubbs RH. J Am Chem Soc. 2004;126:7414. doi: 10.1021/ja0488380. [DOI] [PubMed] [Google Scholar]
- 18.This oxidized ruthenium species might be similar to RuO4. However, the exact nature has not been unambiguously established.
- 19.For reviews about RuO4 mediated oxidations see: Gore ES. Platinum Metals Rev. 1983;27:111.Piccialli V. Molecules. 2014;19:6534. doi: 10.3390/molecules19056534.
- 20.For reviews about sequential and tandem Ru catalysis see: Alcaide B, Almendros P. Chem Eur J. 2003;9:1258. doi: 10.1002/chem.200390142.Alcaide B, Almendros P, Luna A. Chem Rev. 2009;109:3817. doi: 10.1021/cr9001512.
- 21.For the use of Ru complexes in sequential and tandem catalysis coupled to an oxidation step see: Louie J, Bielawski CW, Grubbs RH. J Am Chem Soc. 2001;123:11312. doi: 10.1021/ja016431e.Beligny S, Eibauer S, Maechling S, Blechert S. Angew Chem Int Ed. 2006;45:1900. doi: 10.1002/anie.200503552.Scholte AA, An MH, Snapper ML. Org Lett. 2006;8:4759. doi: 10.1021/ol061837n.Neisius NM, Plietker B. J Org Chem. 2008;73:3218. doi: 10.1021/jo800145x.Schmidt B, Krehl S. Chem Commun. 2011;47:5879. doi: 10.1039/c1cc11347j.Kato H, Ishigame T, Oshima N, Hoshiya N, Shimawaki K, Arisawa M, Shuto S. Adv Synth Catal. 2011;353:2676.Schmidt B, Krehl S, Hauke S. J Org Chem. 2013;78:5427. doi: 10.1021/jo4005684.Dornan PK, Wickens ZK, Grubbs RH. Angew Chem Int Ed. 2015;54:7134. doi: 10.1002/anie.201501505.Malik M, Ceborska M, Witkowski G, Jarosz S. Tetrahedron: Asymmetry. 2015;26:29.Dornan PK, Lee D, Grubbs RH. J Am Chem Soc. 2016;138:6372. doi: 10.1021/jacs.6b02653.
- 22.(a) Grotjahn DB, Larsen CR, Gustafson JL, Nair R, Sharma A. J Am Chem Soc. 2007;129:9592. doi: 10.1021/ja073457i. [DOI] [PubMed] [Google Scholar]; (b) Erdogan G, Grotjahn DB. J Am Chem Soc. 2009;131:10354. doi: 10.1021/ja903519a. [DOI] [PubMed] [Google Scholar]; (c) Larsen CR, Grotjahn DB. J Am Chem Soc. 2012;134:10357. doi: 10.1021/ja3036477. [DOI] [PubMed] [Google Scholar]; (d) Erdogan G, Grotjahn DB. Org Lett. 2014;16:2818. doi: 10.1021/ol500327k. [DOI] [PubMed] [Google Scholar]
- 23.For this classification of tandem reactions see: Jones AC, May JA, Sarpong R, Stoltz BM. Angew Chem Int Ed. 2014;53:2556. doi: 10.1002/anie.201302572.
- 24.Hanessian S, Wang X. Synlett. 2009:2803. [Google Scholar]
- 25.Lou S, Moquist PN, Schaus SE. J Am Chem Soc. 2007;129:15398. doi: 10.1021/ja075204v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyseng-Williamson KA. Drugs. 2011;71:489. doi: 10.2165/11204490-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 27.Boschi F, Camps P, Comes-Franchini M, Muñoz-Torrero D, Ricci A, Sánchez L. Tetrahedron: Asymmetry. 2005;16:3739. [Google Scholar]
- 28.(a) Behenna DC, Mohr JT, Sherden NH, Marinescu SC, Harned AM, Tani K, Seto M, Ma S, Novák Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem Eur J. 2011;17:14199. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Behenna DC, Liu Y, Yurino T, Kim J, White DE, Virgil SC, Stoltz BM. Nat Chem. 2012;4:130. doi: 10.1038/nchem.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Similar stereoselective Curtius rearrangements have been used previously to prepare α-tertiary amines, isocyanates and α-quaternary amino acids: Leuser H, Perrone S, Liron F, Kneisel FF, Knochel P. Angew Chem Int Ed. 2005;44:4627. doi: 10.1002/anie.200500672.Iosub V, Haberl AR, Leung J, Tang M, Vembaiyan K, Parvez M, Back TG. J Org Chem. 2010;75:1612. doi: 10.1021/jo902584r.Banerjee S, Wiggins WJ, Geoghegan JL, Anthony CT, Woltering EA, Masterson DS. Org Biomol Chem. 2013;11:6307. doi: 10.1039/c3ob41282b.
- 30.It should be noted that the isocyanates were stable to purification by column chromatography and could be stored as solids under high vacuum. However, upon exposure to air moisture, they decomposed and polymerized quickly (within hours).
-
31.The major difference between the reactivity of catalyst 1b and 2 is that ruthenium hydride complex 1b is highly selective for the isomerization of just one position to give the 2-ene product. In contrast, catalyst 2 is more reactive and less selective, which may lead to over isomerization for substrates containing a proton in position 4 of the allyl group such as for 3, 8 and 10. This difference in reactivity is in agreement with a lower catalyst loading used for 2 (2 mol%) compared to 1b (5 mol%). Since we had large quantities of 1a available in our lab and catalyst 2 was rather expensive at the time (commercially available from Strem Chemicals and abcr GmbH, CAS 930601-66-4), we preferred to use 1a for Scheme 4. However, similar yields are expected for both catalysts 1a and 2 as we have shown for substrate 14.
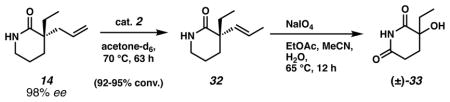
- 32.When we used forcing conditions for the second step with catalyst 2 (5 and 10 mol%), we observed a “lactam oxidation/alkene cleavage/decarboxylation/hydroxylation” reaction quartet for 32. The tertiary alcohol 33 was isolated in 16% and 32% yield over 2 steps, respectively. Unfortunately, the reaction led to a racemic product.
- 33.(a) Bey P, Danzin C, Van Dorsselaer V, Mamont P, Jung M, Tardif C. J Med Chem. 1978;21:50. doi: 10.1021/jm00199a009. [DOI] [PubMed] [Google Scholar]; (b) Kirihata M, Mihara S, Ichimoto I, Ueda H. Agric Biol Chem. 1978;42:185. [PubMed] [Google Scholar]; (c) Yamamoto Y, Kirihata M, Ichimoto I, Ueda H. Agric Biol Chem. 1985;49:1761. [PubMed] [Google Scholar]; (d) Gander-Coquoz M, Seebach D. Helv Chim Acta. 1988;71:224. [Google Scholar]
- 34.For the only racemic synthesis see: Abdel-Monem MM, Newton NE, Ho BC, Weeks CE. J Med Chem. 1975;18:600. doi: 10.1021/jm00240a015.
- 35.For a recent review on the synthesis and reactivity of bridged lactams, see: Szostak M, Aubé J. Chem Rev. 2013;113:5701. doi: 10.1021/cr4000144.
- 36.For previous studies from our group in the topic of twisted lactams, see: Tani K, Stoltz BM. Nature. 2006;441:731. doi: 10.1038/nature04842.Ly T, Krout M, Pham DK, Tani K, Stoltz BM, Julian RR. J Am Chem Soc. 2007;129:1864. doi: 10.1021/ja067703m.Liniger M, VanderVelde DG, Takase MK, Shahgholi M, Stoltz BM. J Am Chem Soc. 2016;138:969. doi: 10.1021/jacs.5b11750.
- 37.A few 1,7-diazabicyclo[4.2.1]nonane-8,9-dione derivatives are known: Akhtar MS, Brouillette WJ, Waterhous DV. J Org Chem. 1990;55:5222.Brouillette WJ, Jestkov VP, Brown ML, Akhtar MS, DeLorey TM, Brown GB. J Med Chem. 1994;37:3289. doi: 10.1021/jm00046a013.Knaup G. Ger Offen. 2001:DE10020818.
- 38.(a) Aebi JD, Seebach D. Helv Chim Acta. 1985;68:1507. [Google Scholar]; (b) Acher F, Azerad R. Tetrahedron: Asymmetry. 1994;5:731. [Google Scholar]; (c) Obrecht D, Bohdal U, Daly J, Lehmann J, Schönholzer P, Müller K. Tetrahedron. 1995;51:10883. [Google Scholar]; (d) Sawamura M, Nakayama Y, Tang WM, Ito Y. J Org Chem. 1996;61:9090. [Google Scholar]; (e) Chinchilla R, Galindo N, Nájera C. Synthesis. 1999:704. [Google Scholar]; (f) Abellán T, Chinchilla R, Galindo N, Nájera C, Sansano JM. J Heterocyclic Chem. 2000;37:467. [Google Scholar]
- 39.Padwa A, Price AT. J Org Chem. 1998;63:556. doi: 10.1021/jo971424n. [DOI] [PubMed] [Google Scholar]
- 40.Knabe J, Omlor G. Arch Pharm. 1989;322:499. doi: 10.1002/ardp.19893220809. [DOI] [PubMed] [Google Scholar]
- 41.Nishio Y, Kimura H, Sawada N, Sugaru E, Horiguchi M, Ono M, Furuta Y, Sakai M, Masui Y, Otani M, Hashizuka T, Honda Y, Deguchi J, Nakagawa T, Nakahira H. Bioorg Med Chem. 2011;19:5490. doi: 10.1016/j.bmc.2011.07.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.