

Key Clinical Message
We describe a rare case of homozygous inactivating calcium‐sensing receptor mutation detected during pregnancy and mimicking primary hyperparathyroidism. In pregnancy, the differential diagnosis of hypercalcaemia requires a cautious approach as physiological changes in calcium homeostasis may mask rare genetic conditions.
Keywords: Familial hypocalciuric hypercalcemia, homozygous mutation, hypercalcemia, pregnancy
Introduction
Hypercalcemia during pregnancy poses a high risk to both mother and baby, and the management can be challenging. Primary hyperparathyroidism (PHPT) is the most common cause of marked hypercalcemia in pregnancy 1 and may require surgery. Differential diagnosis of hypercalcemia includes familial hypocalciuric hypercalcemia (FHH), a lifelong benign condition, generally determined by heterozygous inactivating mutations of the calcium‐sensing receptor (CaSR) and biochemically characterized by mild hypercalcaemia, normal PTH, normal serum phosphorus, normal Vitamin D, low calcium urinary excretion. Homozygous mutations of the CaSR gene result in neonatal severe primary hyperparathyroidism (NSHPT), a life‐threatening condition presenting within 6 months of life with severe hypercalcemia (>3 mmol/L). Here, we present a case of novel homozygous mutation exceptionally detected in adulthood during pregnancy which mimicked PHPT.
Case History
In September 2012, a 28‐year‐old Tanzanian woman was found to have hypercalcemia in the 16th week of her second pregnancy and she was referred to our endocrine clinic for appropriate evaluation. She had previously had a healthy 3‐year‐old daughter born at term, following an uneventful first pregnancy with no identified hypercalcemia. She did not report any calcium‐related symptoms, and she denied history of kidney stones or fractures. She was not taking drugs that could influence her calcium status. There was no family history of calcium disturbances or endocrine disorders; her parents were from the same region in Tanzania, but they were not knowingly consanguineous. At the time of presentation, she was clinically well and her physical examination was unremarkable with no neck palpable masses or lymphadenopathy. An initial diagnostic workup demonstrated a corrected calcium of 3.01 mmol/L (normal 2.15–2.55), inappropriately “normal” PTH of 35 ng/L (10–65), vitamin D deficiency (<20 mmol/L), serum phosphorus of 0.7 mmol/L (0.9–1.4), and elevated urine calcium‐to‐creatinine ratio (UCCR) (0.019). Thyroid, cortisol, and renal function tests were unremarkable. Given her young age, fasting gut peptides and calcitonin were requested and found to be in the normal range. The differential diagnosis included PHPT and FHH with laboratory findings strongly suggestive of PHPT. She was initially managed with aggressive oral hydration and careful vitamin D supplementation, with no improvement in biochemical status. As sestamibi scan is contraindicated in pregnancy due to risk of irradiation, a neck ultrasound was performed but failed to localize a parathyroid adenoma. PHPT with possible parathyroid hyperplasia was considered the most likely diagnosis. She was assessed by an experienced endocrine surgeon and at week 24 of pregnancy, underwent full parathyroid exploration. Three glands were removed with a normal appearing gland left in situ. Histologically, each gland showed hyperplasia of the parathyroid tissue. The right and left upper parathyroid glands weighed 0.16 and 0.11 g and measured 12 × 7 × 3 and 10 × 4 × 3 mm, respectively. The right lower parathyroid gland weighed 0.07 g and measured 6 × 4 × 2 mm. To our surprise, postoperatively she remained hypercalcaemic with calcium levels fluctuating between 2.85 and 2.98 mmol/L. No further parathyroid intervention occurred during pregnancy. Following an uncomplicated delivery, her newborn was assessed for calcium disturbance and unexpectedly found hypercalcemic (3.07 mmol/L, normal range 1.9–2.6) with inappropriately normal PTH (46 ng/L) and normal UCCR. Persistent hypercalcemia, hyperplastic parathyroid glands, and the baby's calcium level indicated familial hyperparathyroidism. Sequence analysis of MEN1 and CDC73 genes did not detect a mutation. Analysis of the CaSR gene by the polymerase chain reaction revealed homozygosity for a transition cytosine>thymine at codon 1393 resulting in the missense mutation Arginine‐465‐Tryptophan in the extracellular domain of the gene encoding the CaSR. Genetic testing also revealed that the newborn and the first daughter were heterozygous for the same mutation (Fig. 1). Corrected calcium, PTH, serum P, 25‐hydroxy vitamin D levels of the patient and her two daughters are reported in the Table 1. The UCCR of the patient after pregnancy and urine from the baby and the previous daughter are not available.
Figure 1.
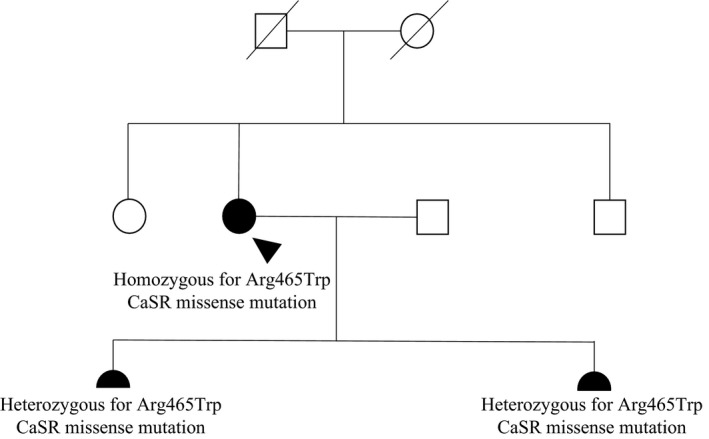
Family tree of the proband. Squares represent male family members and circles represent female family members. Clinical status is indicated by open symbol (unaffected) and solid symbol (affected). The proband is indicated by an arrowhead. The proband's daughters are indicated by filled semicircles.
Table 1.
Laboratory data of the patient and her two daughters
| Patient | 1st daughter | 2nd daughter | |
|---|---|---|---|
| Corrected calcium (mmol/L) | 3.01* | 2.63†† | 3.07# |
| PTH (ng/L) | 35** | 28** | 46** |
| Serum phosphorus (mmol/L) | 0.7*** | 1.4††† | 2.0## |
| 25‐hydroxy vitamin D (nmol/L) | <20† | <20† | <20† |
*Normal range 2.15–2.55 mmol/L; **normal range 10–65 ng/L; ***normal range 0.9–1.4 mmol/L; †normal range>50 nmol/L; ††normal range 2.19–2.69 mmol/L; †††normal range 0.8–1.0 mmol/L; #normal range 2.24–2.74 mmol/L; ##normal range 1.0–2.6 mmol/L.
Discussion
Our case confirms that differential diagnosis of hypercalcemia in pregnancy can be challenging. The patient displayed features more in keeping with PHPT rather than FHH, due to the homozygous state and probably because of the physiological changes in calcium homeostasis occurring in pregnancy (lower intact PTH level, increased glomerular filtration resulting in maternal hypercalciuria). Our patient had no symptoms, but this is not surprising. Patients with FHH or even PHPT, having moderate‐severe hypercalcaemia, may be asymptomatic 2, 3.
In hindsight, the therapeutic management of this case could have been more conservative, but at the time of presentation, surgery was considered as a sensible approach based on clinical judgement and current guidelines. Hypercalcemia (over 2.85 mmol/L) in pregnancy represents a condition potentially leading to maternal complications 4 and fetal loss and the second trimester is considered the safest time to surgically intervene. In our patient, hypercalcemia was not identified during her first pregnancy as calcium levels are not routinely measured during pregnancy or in women who wish to get pregnant. To our knowledge, this is one of very few cases of FHH in pregnancy described in the literature 5, 6 and the first case of homozygous CaSR mutation identified in a pregnant woman. The benign course and the first presentation in adult age of this mutation suggest that this woman's CaSR has residual activity and hypercalcaemia was not severe to cause life‐threatening consequences. Other cases of homozygous CaSR mutations have been diagnosed after infancy. Some authors have proposed a spectrum of phenotypes in FHH and NSHPT with the effects of the mutations dependent on their location 2. We have not carried out any in vitro (or in silico) functional analysis to establish the nature of the missense mutation; however, there is evidence that the position of the mutation can predict the functional behavior of the CaSR receptor. From an analysis of CaSR mutations with marked hypercalcaemia detected after infancy, it emerged that most of these mutations are located in the extracellular domain and are associated with a more severe hypercalcaemia than those located in the transmembrane and intracellular domains 2. Loss‐of‐function and gain‐of‐function caused by missense mutations have been described in all CaSR domains. We believe that this mutation is an inactivating mutation as the phenotype was associated with marked hypercalcaemia and has been previously identified as causing FHH 3. NSHPT has been diagnosed in patients having only one parent with FHH 7, 8 and in families with normocalcemic parents 9.
Beyond the rarity, our case highlights that distinguishing between FHH and PHPT during pregnancy can be challenging due to marked changes in calcium homeostasis and the restricted applicability of diagnostic tools. UCCR is the biochemical index of choice to differentiate between FHH (<0.01) and PHPT (>0.02) 10; however, during pregnancy, changes in maternal calcium handling and an increased calcium excretion make CCCR a less useful tool to distinguish between two conditions 11. Urinary calcium excretion has been reported to be 250%–300% higher during pregnancy 11.
We conclude that in pregnancy, diagnosis of hypercalcaemia requires a cautious approach and genetic tests should be considered when UCCR is between 0.01 and 0.02.
Authorship
PVC, GM, BMM, LI, KH, and JGH: patient management; PVC, GM, BMM, LI, and JGH: writing the report. Written consent to publish was obtained.
Conflict of Interests
The authors have no conflict of interests to disclose.
Acknowledgments
We thank Ms Eshika Haque for the genetic counseling provided.
Clinical Case Reports 2017; 5(10): 1587–1590
References
- 1. Kovacs, C. S. 2011. Calcium and bone metabolism disorders during pregnancy and lactation. Endocrinol. Metab. Clin. North Am. 40:795–826. [DOI] [PubMed] [Google Scholar]
- 2. Hannan, F. M. , Nesbit M. A., Christie P. T., Lissens W., Van der Schueren B., Bex M., et al. 2010. A homozygous inactivating calcium‐sensing receptor mutation, Pro339Thr, is associated with isolated primary hyperparathyroidism: correlation between location of mutations and severity of hypercalcaemia. Clin. Endocrinol. (Oxf) 73:715–722. [DOI] [PubMed] [Google Scholar]
- 3. Guarnieri, V. , Canaff L., Yun F. H., Scillitani A., Battista C., Muscarella L. A., et al. 2010. Calcium‐sensing receptor (CASR) mutations in hypercalcemic states: studies from a single endocrine clinic over three years. J. Clin. Endocrinol. Metab. 95:1819–1829. [DOI] [PubMed] [Google Scholar]
- 4. Norman, J. , Politz D., and Politz L.. 2009. Hyperparathyroidism during pregnancy and the effect of rising calcium on pregnancy loss: a call for earlier intervention. Clin. Endocrinol. (Oxf) 71:104–109. [DOI] [PubMed] [Google Scholar]
- 5. Ghaznavi, S. , Saad N., and Donovan L. E.. 2014. Familial hypocalciuric hypercalcemia and primary hyperparathyroidism: the diagnostic pitfalls encountered when determining the etiology of hypercalcemia during pregnancy. endocrine society's 96th Annual Meeting and Expo, June 21–24, 2014; June 22, 2014; Chicago.
- 6. Murthy, A. , Murthy N. P. N., Ashawesh K., Kulambil P. R. N., and Anwar A.. 2009. Familial hypocalciuric hypercalcaemia and pregnancy outcome. Society for Endocrinology BES 2009; Harrogate, UK.
- 7. Marx, S. J. , Attie M. F., Spiegel A. M., Levine M. A., Lasker R. D., and Fox M.. 1982. An association between neonatal severe primary hyperparathyroidism and familial hypocalciuric hypercalcemia in three kindreds. N. Engl. J. Med. 306:257–264. [DOI] [PubMed] [Google Scholar]
- 8. Eftekhari, F. , and Yousefzadeh D. K.. 1982. Primary infantile hyperparathyroidism: clinical, laboratory, and radiographic features in 21 cases. Skeletal Radiol. 8:201–208. [DOI] [PubMed] [Google Scholar]
- 9. Ujihara, M. , Sato K., Ohashi T., Tomori N., Kasono K., Tsushima T., et al. 1991. A case of hypocalciuric hypercalcemia without family history. Endocrinol. Jpn. 38:689–692. [DOI] [PubMed] [Google Scholar]
- 10. Eastell, R. , Brandi M. L., Costa A. G., D'Amour P., Shoback D. M., and Thakker R. V.. 2014. Diagnosis of asymptomatic primary hyperparathyroidism: proceedings of the Fourth International Workshop. J. Clin. Endocrinol. Metab. 99:3570–3579. [DOI] [PubMed] [Google Scholar]
- 11. Seely, E. W. , Brown E. M., DeMaggio D. M., Weldon D. K., and Graves S. W.. 1997. A prospective study of calciotropic hormones in pregnancy and post partum: reciprocal changes in serum intact parathyroid hormone and 1,25‐dihydroxyvitamin D. Am. J. Obstet. Gynecol. 176(1 Pt 1):214–217. [DOI] [PubMed] [Google Scholar]