

Abstract
Neurons in the adult mammalian CNS decrease in intrinsic axon growth capacity during development in concert with changes in Krüppel-like transcription factors (KLFs). KLFs regulate axon growth in CNS neurons including retinal ganglion cells (RGCs). Here, we found that knock-down of KLF9, an axon growth suppressor that is normally upregulated 250-fold in RGC development, promotes long-distance optic nerve regeneration in adult rats of both sexes. We identified a novel binding partner, MAPK10/JNK3 kinase, and found that JNK3 (c-Jun N-terminal kinase 3) is critical for KLF9's axon-growth-suppressive activity. Interfering with a JNK3-binding domain or mutating two newly discovered serine phosphorylation acceptor sites, Ser106 and Ser110, effectively abolished KLF9's neurite growth suppression in vitro and promoted axon regeneration in vivo. These findings demonstrate a novel, physiologic role for the interaction of KLF9 and JNK3 in regenerative failure in the optic nerve and suggest new therapeutic strategies to promote axon regeneration in the adult CNS.
SIGNIFICANCE STATEMENT Injured CNS nerves fail to regenerate spontaneously. Promoting intrinsic axon growth capacity has been a major challenge in the field. Here, we demonstrate that knocking down Krüppel-like transcription factor 9 (KLF9) via shRNA promotes long-distance axon regeneration after optic nerve injury and uncover a novel and important KLF9–JNK3 interaction that contributes to axon growth suppression in vitro and regenerative failure in vivo. These studies suggest potential therapeutic approaches to promote axon regeneration in injury and other degenerative diseases in the adult CNS.
Keywords: Jnk, KLFs, regeneration, survival
Introduction
Failure of adult CNS neurons to regenerate their axons after injury arises from both intrinsic and extrinsic regulation (Yiu and He 2006; Moore et al., 2009). For example, inhibitory environmental cues such as glial-derived factors (Yiu and He 2006) and cell-autonomous signaling pathways such as cAMP, Rho, Rho-associated kinase, and phosphatase and tensin homolog (PTEN) (Nicholls and Saunders, 1996; Blackmore and Letourneau, 2006; Park et al., 2008; Moore et al., 2009; Blackmore et al., 2012) all play a role in regenerative failure.
We found previously that a family of zinc-finger transcription factors, the Krüppel-like factor (KLF) family, regulates intrinsic axon growth in RGCs (Moore et al., 2009). The KLF family of transcription factors includes two neurite-growth-enhancing KLFs (KLF6 and KLF7) and nine neurite-growth-suppressive KLFs (KLF1, KLF2, KLF4, KLF5, KLF9, KLF13, KLF14, KLF15, and KLF16) (Moore et al., 2009, 2011; Blackmore et al., 2010). Expressing KLF7 or knocking out KLF4 in adult CNS neurons promotes axon regeneration after injury in vivo (Blackmore et al., 2012; Qin et al., 2013). KLF9, which was shown previously to suppress neurite growth in embryonic and postnatal RGCs and young postnatal cortical neurons in vitro, is another strong candidate for in vivo regulation of axon regeneration because its expression increases 250-fold in concert with the developmental loss of axon growth ability in RGCs (Moore et al., 2009). Functions ranging from improving survival to increasing neurite growth, branching, and elongation have been observed with KLF9 expression in some neuronal subtypes (Denver et al., 1999; Cayrou et al., 2002; Bonett et al., 2009; Lebrun et al., 2013).
Little is known about whether KLF9's ability to suppress axon growth is itself regulated by binding partners or posttranscriptional modification. Although KLFs are known to interact with binding partners such as the scaffold protein mSin3A to regulate their effects on transcription outside of the nervous system (Moore et al., 2011), regulatory binding partners have not yet been described in neurons. To address these issues, we designed shRNA knock-down constructs targeted against KLF9 and packaged them into adeno-associated viral serotype 2 (AAV2), described by us and others to transduce RGCs specifically when injected intravitreally (Shevtsova et al., 2005; Hellström et al., 2009). Here, we show that knocking down KLF9 promotes long-distance axon regeneration and now identify a novel binding partner, MAPK10/JNK3 kinase, and two novel phosphorylation sites, S106 and S110, both required for KLF9's ability to suppress neurite growth in vitro and regeneration in vivo. We propose a model in which JNK3 mediates the activation level of KLF9 through S106/S110 to suppress intrinsic regenerative capacity in CNS neurons.
Materials and Methods
Animals.
All use of animals conformed to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Research and all animal procedures were approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee of the University of Miami and the University of California–San Diego. Sprague Dawley rats and mice of varying ages and either sex were obtained from Harlan Laboratories.
Animal surgery.
All surgeries were performed under adequate anesthesia with an intraparietal injection of ketamine hydrochloride, 60 mg/kg, and xylazine hydrochloride, 8 mg/kg, according to body weight. After surgery, animals were allowed to recover on a heating pad and were given subcutaneous injections of buprenorphine hydrochloride, 0.1 mg/kg, twice a day for 3 consecutive days to minimize discomfort (Zhao et al., 2011).
Intravitreal injection of viruses and optic nerve crush.
To inhibit KLF9 mRNA expression, the left eye of Sprague Dawley rats of both sexes were injected intravitreally with a 4 μl volume of AAV2-anti KLF9 shRNA-EGFP or AAV2-anti-luciferase shRNA-EGFP at postnatal day 28 (titers ranged from 2.0 to 3.0 × 1013 genome copies/ml). Some postnatal day 28 (P28) Sprague Dawley rats were received 4 μl of AAV2-EGFP, AAV2-KLF9ΔJBD-EGFP, AAV2-KLF9S106/110A-EGFP, or AAV2-JBD-P-EGFP intravitreal injection (titers ranged from 5.0 to 8.9 × 1012 genome copies/ml). One week after virus injection, RGCs were labeled retrogradely with Fluorogold (FG; Fluorochrome) to investigate RGC survival, as described previously (Chiu et al., 2008). In brief, P35 Sprague Dawley rats were anesthetized and the skull was exposed by a midline incision through the skin and superficial fascia. Bilateral 2-mm-diameter craniotomies were placed 0.5 mm posterolateral to the sagittal and transverse sutures. A small piece of gelfoam (Gelfoam) soaked in 6% FG was then placed on the surface of the superior colliculus. For the optic nerve surgery, mice or rats of either sex were use. Two weeks after virus injection (rats) or at time 0 (mice), the left optic nerve was exposed from outer canthus and crushed for 5 s with a Dumont Fine Science Tools #5 forceps ∼1.5 mm behind the globe. Care was taken to avoid damaging the blood supply to the retina. All optic nerve crush procedures were performed by a surgeon blinded to the viral treatment. Intravitreal injection of 4 μl (rat) or 1 μl (mice) of cholera toxin subunit B-conjugated Alexa Fluor 594/555 (CTB-594/555, 10 μg/μl; Invitrogen) was performed 2 d before animals were killed as an anterograde tracer to visualize axons and nerve terminals originating from living RGCs. Animals with any significant postoperative complications (e.g., retinal ischemia, cataract) were excluded from further analysis. At different time points after the optic nerve injury, animals were deeply anesthetized and transcardially perfused with 4% PFA in PBS. Optic nerves and retinas were dissected and fixed in 4% PFA for 1 h and subsequently washed in PBS. Optic nerves were incubated in 20% sucrose at 4°C overnight before mounting in optimal cutting temperature mounting medium before sectioning. Longitudinal sections (10 μm) were made of the entire optic nerve and imaged with a 20× magnification objective as above. Photographs were taken, starting with the furthest regenerating axons and working backward toward the crush site. Lines were drawn perpendicular to the long axis of the optic nerve 1, 2, and 5 mm past the crush site. CTB-positive axons passing these lines were counted in every fourth section per optic nerve in a masked fashion. The width of the nerve at each line was measured and used to calculate the number of axons per millimeter of nerve width. The average number across all sections was combined as axons per millimeter width to control for volume. The total number of axons extending to distance (d) in a nerve with a radius of (r) was estimated by summing over all sections having a thickness t (10 μm)as follows: Σad = πr2 × (average axons/mm)/t, as described previously (Park et al., 2008).
In vivo KLF9 and luciferase shRNA delivery. To suppress KLF9 expression in vivo, an inducible RNA polymerase II promoter (Chung et al., 2006) was subcloned upstream of four target shRNAs against both mouse and rat KLF9 gene (Gene Bank accession numbers NM_010638 and NM_057211, respectively) using target sequences as follows: 5′-GGA GGC GCT GCC GGT TAC GTA-3′, 5′-TGG CTG CCC AGT GTC TGG TTT-3′, 5′-CGG GGG ACA CCT GGA AGG ATT-3′ and 5′-GCA AAT AAA TGC TTT TGG TAC-3′. These four shRNA sequences in a SIBR cassette (Chung et al., 2006) were concatenated into a single vector and inserted into pAAV2 vector backbone, which also expresses GFP as a transduction marker. Luciferase used the following SIBR cassette: TGGAGGCTTGCTGAAGGCTGTATGCTGTTTATGAGGATCTCTCTGATTTTTTTGGCCTCTGACTGAAAATCAAGAGTCCTCATAAACAGGACACAAGGCCTGTTACTAGCACTCACATGGAACAAATGGCCACCGTGGGAGGATGACAA. Both vectors were kind gifts from Vance P Lemmon, University of Miami. All constructs were verified by sequencing.
Immunohistochemistry.
For cultured neurons, cultures were fixed using prewarmed, 37°C 4% paraformaldehyde (PFA). For sections, frozen 15 μm sections were fixed with 4% PFA. For retinal flat mount, animals were killed by transcardial perfusion with 4% PFA and retinas were then postfixed with 4% PFA for 1 h at room temperature. After rinses in PBS, samples were blocked and permeabilized in 20% normal goat serum or 20% normal donkey serum with 0.02% Triton X-100 in antibody buffer containing 150 mm NaCl, 50 mm Tris base, 1% BSA, 100 mm l-lysine, and 0.04% Na azide, pH 7.4, for 1 h to reduce nonspecific binding. Samples were incubated overnight at 4°C in antibody buffer containing primary antibodies, washed with PBS, incubated in antibody buffer containing secondary antibodies and DAPI for 1 h at room temperature, and washed with PBS. Cultures were left in PBS for imaging and imaged for neurite growth.
Primary antibodies used for these experiments included anti-β III tubulin antibody from E7 hybridoma (1:500; Developmental Studies Hybridoma Bank), anti-FLAG (1:250; Sigma-Aldrich, F7425), anti-GFP (1:200; Aves Laboratories, GFP-1020), anti-Brn3a (1:100; Millipore, MAB1585) and anti-RBPMS (1:300; PhosphoSolutions, 1830-RBPMS). Secondary antibodies were Alexa Fluor 488-, 594-, or 647-conjugated, highly cross-adsorbed antibodies (1:500; Invitrogen).
RGC survival and viral efficiency analysis.
For RGC survival and viral efficacy analysis after optic nerve crush, the following techniques were used: briefly, the retinas were divided into 4 quadrants and each quadrant was subdivided into 3 areas (central, middle, and peripheral), which were 1, 2, and 3 mm from the optic nerve head, respectively. One digital micrograph was taken randomly from each of the 12 fields. Therefore, 12 images were quantified per retina. Three to four retinas were used for each condition. FG-positive, Brn3a-positive, RBPMS-positive, or GFP-positive cells were counted manually in a masked fashion and presented as cells per millimeter squared in each region of the retina. Whole retinal RGC density was estimated using the following formula: (DC + 3DM + 5DP)/9, where DC, DM, and DP represent densities of the central, middle, and peripheral regions, respectively (Wang et al., 2013).
Quantification of neurite length and neurite number.
For high-content screening of neuronal morphology, including average neurite length, maximum neurite length, neurite number, and neurite branching, automated microscopy (ArrayScan VTI or KSR; Cellomics) and image analysis software (Cellomics Neuronal Profiling BioApplication; Thermo Fisher Scientific) were used to image and trace neurons using a 5× or 10× objective after immunostaining. RGCs were traced using β III tubulin immunoreactivity to visualize neurites. Neurons with dim signal in neurites were excluded from analysis due to frequent tracing errors of faint processes; the threshold for exclusion was established using a population of negative control, no primary antibody-immunostained RGCs. We previously validated this approach's consistency and reliability relative to manually tracing (Moore et al., 2009; Blackmore et al., 2012). Images and tracing were spot checked to verify that the algorithms were correctly identifying neurites and quantifying growth. In Figure 4, bars represent average total neurite length of transfected/transduced neurons.
Figure 4.
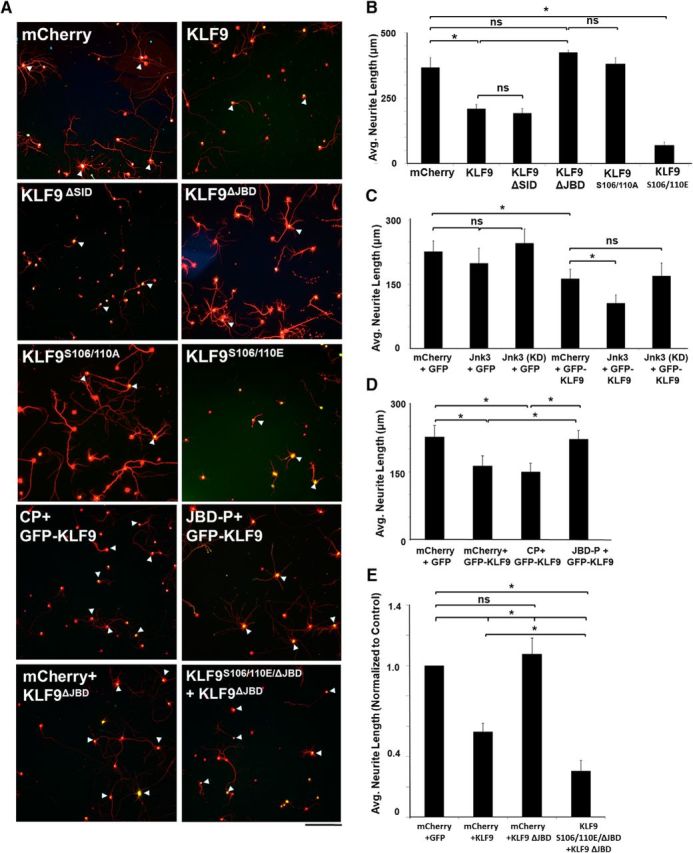
JNK3-binding domain and serines S106 and S110 are critical to KLF9's function in suppressing axon growth. A, Images of RGCs immunostained for GFP (transduced cells) and β-III tubulin (red). P0 RGCs were virally transduced with mCherry, KLF9 or mutants of KLF9 with GFP reporter. Scale bar, 50 μm. B–E, Quantification of neurite growth of RGCs in different conditions. The KLF9ΔJBD but not the KLF9ΔSID deletion abolished wild-type KLF9's growth-suppressive effect on P0 (B) and P8 (data not shown) RGCs. JNK3 potentiated KLF9's neurite growth-suppressive activity on P0 RGCs; however, JNK3 alone or a kinase-dead mutant (KD) showed no effect (C). JBD-P but not CP abolished KLF9's suppressive effect on P0 RGCs cotransduced with KLF9 (D). KLF9S106/110A substitutions abolished wild-type KLF9's growth-suppressive effect, whereas KLF9S106/110E enhanced KLF9's suppressive effect on P0 (A) and P8 (data not shown) RGCs. Phosphomimic S106/S110E substitutions rescued KLF9 suppression on P0 RGC neurite growth even in the absence of the R223-I233 JNK3-binding domain (E). *p < 0.05, one-way ANOVA with Bonferroni/Dunn correction; n = 3–5. Error bars indicate SD., NS, Not significant.
Cortical slice culture.
Cortical slice cultures were prepared as described previously (Blackmore et al., 2012). Briefly, P5 Sprague Dawley rats were anesthetized with ketamine on ice and killed in accordance with guidelines set by Institutional Animal Care and Use Committees. Brains were placed in dissection medium containing 37.7 ml of Hank's balanced salt solution (HBSS; Invitrogen), 0.8 ml of 1 m HEPES (Invitrogen), 1.1 ml of 1.2 m d-glucose (Sigma-Aldrich), and 0.4 ml of 1 m magnesium sulfate (Sigma-Aldrich). Meninges were removed and 350 μm coronal sections of cortex were prepared using a McIlwain tissue chopper. Sections were trimmed to prepare ∼2 × 2 mm sections of right and left cortex linked by an intact corpus callosum. Slices were cultured on 30 mm organotypic polytetrafluoroethylene 0.4 μm culture inserts (Millipore; PIC-MORG50) on 35 × 10 mm culture dishes (Falcon) with a culture medium consisting of 50% basal Eagle medium (Invitrogen), 18% HBSS (Invitrogen), 2% SM1 50× supplement (STEMCELL Technologies), 4 mm l-glutamine (Invitrogen), 6 mg/ml 1.2 m d-glucose (Sigma-Aldrich), and antibiotic/antimycotic (Invitrogen). Two hours later, a 0.5 μl drop of viral particles of each shRNA virus was applied to each hemisphere. After 2 h of treatment, sections were rinsed with culture medium to wash out virus. Sections were rinsed again the next day with culture medium. Forty-eight hours after transduction, RNA was collected and quantitative reverse transcriptase PCR (qRT-PCR) was performed using primers and methods described previously (Eaton et al., 2008; Jiang et al., 2012).
Purification, culture, transfection, and transduction of primary RGCs and hippocampal neurons.
RGCs were purified by immunopanning as described previously (Barres et al., 1988; Meyer-Franke et al., 1995; Goldberg, Espinosa et al., 2002; Hu et al., 2010; Trakhtenberg et al., 2014). RGCs were plated onto PDL- and laminin-coated 24-well or 6-well tissue culture plates in growth medium as described previously (Meyer-Franke et al., 1995) and including a homemade supplement similar to B27 (Chen et al., 2008), forskolin (5 mm), BDNF (50 ng/ml), and CNTF (10 ng/ml). RGCs were cultured in 10% CO2 for 48 h to analyze neurite growth.
For cotransfection electroporation experiments, after purification, 500,000 RGCs/condition were resuspended in electroporation solution containing 3.0 μg of total DNA in 1:1 ratio (1.5 μg gene-1 plus 1.5 μg of gene-2). Concentrations of DNA were adjusted to allow all 3 μg to occupy 2 μl of volume, which was then added to 27 μl of electroporation solution for resuspension of cells. Resuspended cells were placed in a small cell number cuvette (Sigma-Aldrich) and electroporated using Amaxa program SCN#1. Immediately after electroporation, growth medium was added and the whole solution was placed into a 1.5 ml Eppendorf tube. RGCs were centrifuged for 16 min at 1800 rpm (∼300 × g) in a standard 8.4 cm rotor Eppendorf tabletop centrifuge before media resuspension and plating. RGCs were cultured in 10% CO2 for 48 h to quantify cell neurite growth.
For viral transduction of RGCs, 10,000 P0 or P8 RGCs/well were plated on PDL/laminin-coated 24-well plates. For viral transduction of E18 hippocampal neurons for proteomic analysis [immunoprecipitation (IP)/Western blots/mass spectrometry (MS)/kinase assays), 1–2.5 million hippocampal neurons/well were plated on PDL/laminin-coated 6-well plates. In all cases, 16 h after plating, virus was diluted 1:250 for lentiviruses and 1:1000 for AAV2 viruses in medium and added to cultured cells. Where noted, multiple viruses were first mixed in a 1:1 or 1:1:1 ratio before dilution in medium. Full medium changes were performed 5.5 and 24 h after virus exposure. For neurite outgrowth assays, RGCs were fixed in paraformaldehyde 2 d after initial addition of lentiviruses and 5 d after application of AAV2 viruses. For protein analysis by IP and Western blots, cells were lysed 48 h after transduction. The titer of AAV2 viruses ranged from 2.0 to 3.0 × 1013 genome copies/ml. All KLF and JNK3 lentiviruses ranged in titer from 1.5 to 4 × 107 pg/ml by P24 titration ELISA (PerkinElmer). There are ∼2000 molecules of p24 per physical particle of HIV, so p24 values can be correlated to virus titer.
Cloning of KLF constructs for transfection and virus transduction.
EGFP-KLF9 was obtained in AAV expression vector and was subcloned into pLenti-MP2 by PCR using Xho1/Xba1 restriction sites 5-′ and 3-′ to EGFP-KLF9, respectively. PCR product was then gel purified and double digested with both enzymes to create the insert. pLenti-MP2 was double digested, treated with shrimp alkaline phosphatase, gel purified, and ligated with inserts with T4 DNA ligase. Once inserted into pLenti-MP2, EGFP-KLF9 was then mutated at S88/106/110/122 using the QuikChange II XL Site-Directed and QuikChange Lightning MultiSite-Directed Mutagenesis Kits (Agilent Technologies) for single and multiple mutations, respectively.
Primers used for mutations (italicized) include: KLF9S88A forward: 5′ CGACAGTCTGGAAGCTCCAGATGAGGATATGGG 3′; KLF9S88A reverse: 5′ CCCATATCCTCATCTGGAGCTTCCAGACTGTCG 3′; KLF9S88E forward: 5′GCGACAGTCTGGAAGAGCCAGATGAGGATATGGGATCCG3′; KLF9S88E reverse: 5′ CGGATCCCATATCCTCATCTGGCTCTTCCAGACTGTCGC 3′; KLF9S106A forward: 5′GAATCTGGGTCGGCCCCTTCCCACAGCCCGGAGGAGAG 3′; KLF9S106A reverse: 5′CTCTCCTCCGGGCTGTGGGAAGGGGCCGACCCAGATTC 3′; KLF9S106E forward: 5′GAATCTGGGTCGGAGCCTTCCCACAGCCCGGAGGAGAG 3′; KLF9S106E reverse: 5′CTCTCCTCCGGGCTGTGGGAAGGCTCCGACCCAGATTC 3′; KLF9S110A forward: 5′GAATCTGGGTCGAGTCCTTCCCACGCCCCGGAGGAGAG 3′; KLF9S110A reverse: 5′ CTCTCCTCCGGGGCGTGGGAAGGACTCGACCCAGATTC 3′; KLF9S110E forward: 5′GAATCTGGGTCGAGTCCTTCCCACGAGCCGGAGGAGAG 3′; KLF9S110E reverse: 5′CTCTCCTCCGGCTCGTGGGAAGGACTCGACCCAGATTC 3′; KLF9S122A forward: 5′GCAGCGCGCCCGCCCCGCTCTCCC 3′; KLF9S122A reverse: 5′ GGGAGAGCGGGGCGGGCGCGCTGC 3′; KLF9S122E forward: 5′ CTGGCAGCGCGCCCGAGCCGCTCTCCCTC 3′; KLF9S122E reverse: 5′GAGGGAGAGCGGCTCGGGCGCGCTGCCAG 3′; KLF9S106/110A forward: 5′GAATCTGGGTCGGCCCCTTCCCACGCCCCGGAGGAGAG 3′; KLF9S106/110A reverse: 5′CTCTCCTCCGGGGCGTGGGAAGGGGCCGACCCAGATTC 3′; KLF9S106/110E forward: 5′ACCGAATCTGGGTCGGAGCCTTCCCACGAGCCGGAGGAGAGACAG 3′; KLF9S106/110E reverse: 5′ CTGTCTCTCCTCCGGCTCGTGGGAAGGCTCCGACCCAGATTCGGT 3′; KLF9Δ223–233 Forward: 5′CCTCACAAAGCACGCCAAGCGATCGAAAAAGG 3′; KLF9Δ223–233 reverse: 5′CCTTTTTCGATCGCTTGGCGTGCTTTGTGAGG 3′; KLF9ΔSID(1–21)Forward:5′GTCGAAAGAATTCGGTACCAAGTCGAAAGAATTCGGTA 3′; and KLF9ΔSID(1–21) reverse: 5′TACCGAATTCTTTCGACTTGGTACCGAATTCTTTCGAC 3′.
Flag-tagged mCherry and flag-tagged JNK3α1 were kind gifts from V.P. Lemmon (University of Miami). JNK3α1 was subcloned into the SpeI/XbaI site in pLenti-MP2 expression vector with flag-mCherry upstream before packaging into lentivirus. Flag-mCherry and flag-mCherry-JNK3α1 lentiviruses were used between 1.5 and 4.0 × 107 pg/ml assayed as above.
Protein extraction, MS analysis, IP, Western blot, and in vitro kinase assays.
For MS analysis, we used purified P0 rat RGCs transduced with lentivirus to express flag-tagged KLF9. KLF9 is expressed at this age, although its expression continues to rise postnatally. Although it would be ideal to perform IP for endogenous KLF9 from older RGCs when its expression is high for MS, cell and protein yields from older animals are significantly reduced. For example, from 150 P0 animals, we could purify 10 million RGCs, which in turn yielded ∼200 μg of protein, which is still at the lowest recommended range for IP/MS experiments for most mass spectrometers. Therefore, we used purified P0 rat RGCs transduced with KLF9 for initial IP/MS, followed by validating hits with older RGCs. RGCs were purified by immunopanning and transduced with virus as above. Forty-eight hours after lentiviral exposure with Flag-KLF9, RGC proteins were extracted with a custom lysis buffer containing 125 mm Tris-HCl, 100 mm NaCl, 0.1% Genapol C-100 (EMD Biosciences), 0.1% trehalose, 0.1% n-octyl-b-d-glucopyranoside (EMD Biosciences), and protease and phosphatase inhibitor cocktails (Thermo Fisher Scientific). Flag-tagged protein was then immunoprecipitated using anti-Flag affinity M2 gel (Sigma-Aldrich, A2220), and resolved on a 4–10% gradient Phastgel (GE Healthcare). A strip of bands were excised and sent for MS analysis at the Scripps Research Institute. Sample was analyzed using an LC MS/MS LTQ Orbitrap mass spectrometer. MS data were searched with MASCOT and SEQUEST and peptide spectra were checked manually and blasted with SWISSPROT databases.
For hippocampal neuron IP and Western blots, hippocampal neurons were dissected and dissociated as described previously (Bradke and Dotti, 1997). Eight to 10 million cells were used per condition. Cells were virally transduced and cultured as described above. At 48 h, cells were lysed first with cytosolic extraction buffer (10 mm PIPES, 100 mm NaCl, 300 mm sucrose, 3 mm MgCl2, and protease/phosphatase inhibitor cocktails; Thermo Fisher Scientific). Supernatant was removed and nuclear material resuspended with 50–100 μl of custom lysis buffer (see above). Suspension was rotated for 5 min at 4°C. Nuclear suspension was then centrifuged at 600 RPM (∼34 × g) for 5 min at 4°C. Supernatant was collected and used for IP and Western blots.
For retina IP and Western blots, retinas were collected from 80–90 P10 rats and dissociated with papain as described previously (Barres et al., 1988; Meyer-Franke et al., 1995; Goldberg, Espinosa et al., 2002; Hu et al., 2010; Yu et al., 2014); cell suspensions were lysed immediately and nuclei fractionated directly in the same way as hippocampal neurons described above. Supernatant was collected and used for IP and Western blots.
IP was performed with protein G dynabeads (Invitrogen), BS3 cross0linking reagent (Thermo Fisher Scientific) and antibodies. Antibodies used included anti-FLAG (15 μg/condition for IP; 1:200 for Western blot; Sigma-Aldrich, F7425 or F3165), anti-GFP (15 μg/condition for IP; 1:200 for Western blot; Invitrogen, A11122). Cell extracts were separated by SDS-PAGE on 4–20% Tris-Glycine gels (Invitrogen) and electrotransferred to PVDF membranes (Millipore) at 25 V for 2 h using the XCell SureLock Mini-Cell (Invitrogen). Membranes were blocked with Blok-CH chemiluminescent blocker (Millipore) and the antibody reaction performed with the SNAP I.D. system (Millipore). Other primary antibodies used for these experiments included anti-KLF9 antibody (1:200; Santa Cruz Biotechnology, SC12994) and anti-JNK3 (1:200, Millipore, 05–893). Secondary antibodies were HRP-conjugated anti-mouse, anti-rabbit, and anti-goat IgGs (1:400; Santa Cruz Biotechnology Biotechnology or Rockland, TRUBLOT). Immunopositive bands were visualized by chemiluminescence with ECL (Thermo Fisher Scientific) and imaged on a LAS-3000 (Fujifilm). Densitometry was performed with Multi Gauge version 3.1 software (Fujifilm).
For in vitro kinase assays, E18 hippocampal neurons were transduced with flag-tagged wild-type KLF9, KLF9ΔJBD, or KLF9S85/88/95/106/110A mutants. One hundred million cells were used per condition. Cells were lysed and nuclei were fractionated and immunoprecipitated with magnetic Dynabeads (Life Technologies, 10007D) conjugated with anti-flag antibody. Eluents were combined with recombinant JNK3 (Millipore, 14–501) and radiolabeled [ϒ-32] ATP for a standard in vitro kinase assay. Briefly, 2.5 μl of 10× reaction buffer was added each tube. Next, 2.5 μl of ATF-2-positive control or 2.5 μl of 10× buffer for negative control was added to corresponding tubes. Then, 2.5 μl (13–69 ng) of active JNK3/SAPK1b was added to each sample tube and control, followed by 7.5 μl of dH2O and then 10 μl of diluted [γ-32P] ATP mixture to each tube. Tubes were then incubated for 10 min at 30°C and the reaction stopped by addition of 5 μl/tube of 3% phosphoric acid. Mixtures were then resolved on 4–20% Tris-glycine gradient SDS-PAGE gel (Invitrogen), vacuum dried, and the film developed for 3 d.
Experimental design and statistical analysis.
For all in vivo experiments, at least three animals (rats for shRNA KLF9 and mice for JNK3−/− experiments) of either sex were used per condition. Two-tailed Student's t test or one-way ANOVA was used for statistical analysis of distance traveled by regenerating axons and RGC survival. p < 0.05 was regarded as statistically significant using GraphPad Prism software. For in vitro neurite tracing, data analysis was performed using the JMP version 8 statistical package program (SAS Institute). Two-tailed Student's t test was used for statistical comparison of two samples. p < 0.05 was regarded as statistically significant. For KLF alignments, clustalW2 software was used on human KLF genes. Genes were grouped and colored by the presence of known motifs. In the figures, all data are displayed as means and SEM and significance is represented with asterisks.
Results
Anti-KLF9 shRNA promotes RGC survival and axon regeneration
We designed four concatenated SIBR shRNA constructs targeting both rat and mouse KLF9 mRNA transcripts. Knock-down efficiency was tested in cortical slice cultures and purified P10 RGCs in vitro by qRT-PCR and Western blot. More than 80% knock-down of mRNA and >50% knock-down of protein were observed with anti-KLF9 shRNA relative to an anti-luciferase control virus in both models (Fig. 1A,B). We then investigated whether KLF9 expression changes in vivo after optic nerve injury. In an adult rat optic nerve crush model, RGC death begins at 3–5 d and peaks at 1–2 weeks after injury (Berkelaar et al., 1994). Compared with uninjured controls, 3 d after optic nerve injury RGCs showed no significant change in KLF9 protein expression, quantified by both Western blot and immunofluorescence (Fig. 1C,D). Therefore, KLF9 expression is high in RGCs but does not change after optic nerve injury.
Figure 1.
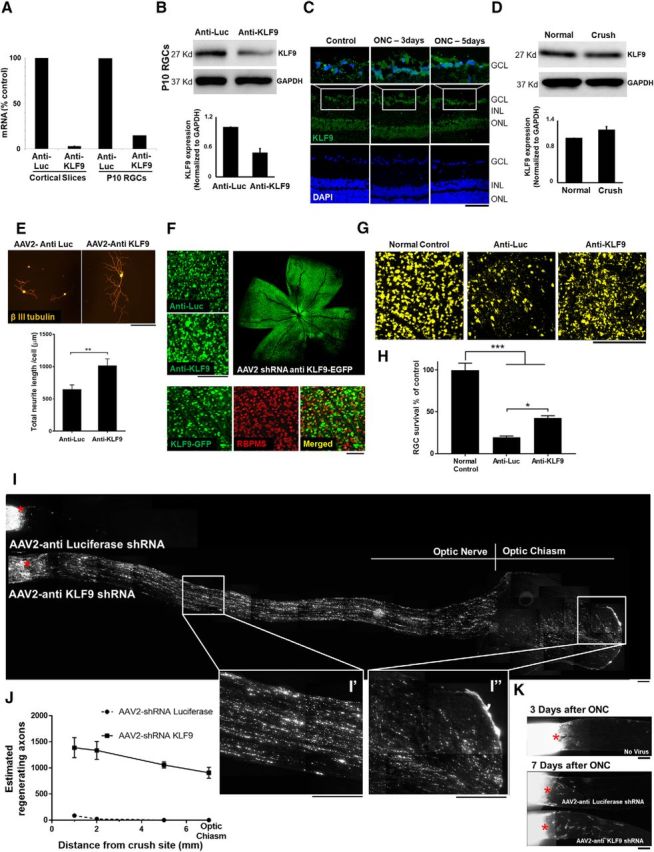
Anti-KLF9 shRNA promotes RGC survival and axon regeneration at 2 weeks after optic nerve injury. A, AAV2-shRNA anti-KLF9 decreased KLF9 expression levels in cortical slice culture and purified P10 RGCs. B, Western blot for KLF9 in RGCs purified after intravitreal injection of AAV2-KLF9 shRNA or AAV2-anti-luceferase control. C, Immunohistochemistry of KLF9 in retinal slides in control and at 3 d and 5 d after optic nerve injury. D, Western blot for KLF9 in RGCs purified from P18 rats 3 d after optic nerve injury and uninjured (normal) control. E, KLF9 knockdown increased neurite growth in purified P8 RGCs. RGCs were treated with AAV2-shRNA anti-KLF9-GFP or AAV2-shRNA anti-Luciferase-GFP and cultured in growth media for 5 d. Neurites were labeled by β-III-tubulin staining (red). F, Images of the retinal segments showing GFP-positive virus transduced cells were co-labelled with RGC-specific marker RBPMS. G, Images of the retinal segments showing FG labeled RGCs. H, RGC quantification demonstrated significantly higher RGC density in anti-KLF9 group than the injury control group. **p < 0.01, one-way analysis of variance; N = 3–4 per group. I, Images of the optic nerve sections showing CTB-labelled regenerating axons at 2 weeks after optic nerve injury. Asterisks, lesion sites. High-magnification images of the boxed area in (I′) showed fibers extending contralaterally through the optic chiasm of an anti-KLF9 virus-treated animal (I′′). J, Significantly more fibers regenerated after KLF9 knockdown compared to control group. **p < 0.01, Student t-test; N = 3–4 per group. I′,I′′, High-magnification images of the boxed area in I. K, Images of the optic nerve sections showing CTB-labelled regenerating axons at 3 d and 1 week after optic nerve injury for AAV2-shRNA anti-KLF9-GFP or AAV2-shRNA anti-Luciferase-GFP. Asterisks, lesion sites. Error bars, SEM. Scale bars, 200 μm.
Because KLF9 overexpression suppresses axon growth in vitro, we next examined the effects of KLF9 knock-down. Knocking down KLF9 by shRNA in purified P8 RGCs increased neurite outgrowth significantly (Fig. 1E), demonstrating that endogenous levels of KLF9 expression limit neurite growth actively in RGCs. To examine the effects of KLF9 knock-down in vivo, AAV2-shRNA-anti-KLF9 virus was injected intravitreally 2 weeks before and retrograde labeling of RGCs was performed 1 week before optic nerve injury. High RGC transduction specificity and efficiency as measured by colocalization with RGC-specific marker RBPMS (Kwong et al., 2010; Rodriguez et al., 2014) were observed in both control virus- and anti-KLF9 virus-injected eyes 2 weeks after intravitreal injection (Fig. 1F). Measurement of FG-positive RGCs 2 weeks after injury showed a significant increase in RGC survival after anti-KLF9 shRNA treatment relative to control (Fig. 1G,H). There was also a significant increase in the number of CTB-positive regenerating axons in the anti-KLF9 experimental group (Fig. 1I,J). Regenerating fibers passed 5 mm from the crush site, reaching the optic chiasm. Most of the fibers entering the optic chiasm were noted to cross to the contralateral side (Fig. 1I,I′′). Significantly fewer CTB-positive fibers were observed in the anti-KLF9 group at 1 week after injury (Fig. 1K) and many more GFP-positive degenerating fibers were observed than CTB-positive regenerating fibers, ruling out a sparing effect and confirming progressive regeneration after 2 weeks. Therefore, shRNA-mediated knock-down of KLF9 expression increased RGC survival and promoted axon regeneration after optic nerve injury in vivo.
KLF9 is bound and phosphorylated by JNK3
We next sought to identify KLF9-binding partners, hypothesizing that such proteins may contribute to KLF regulation of axon growth and regeneration. We first investigated whether mSin3A binds KLF9 in neurons in a manner similar to what was reported previously for non-neuronal cells (Moore et al., 2011), by immunoprecipitating KLF9 protein from whole retinal lysate and Western blotting for mSin3A. mSin3A was present in both input and unbound flow-through, but not in immunoprecipitated eluent (Fig. 2A), indicating that, although mSin3A is expressed in the retina, it does not bind KLF9. To probe for novel binding partners, we performed IP/MS on whole retinal lysates and purified P0 rat RGCs transduced with lentivirus to express flag-tagged KLF9. We did not identify mSin3a or any other cofactors that could regulate KLF9 function. However, MAPK10/JNK3 (Fig. 2B, box) was identified with six independent peptides detected from eight unique and 19 total spectra demonstrating 13% protein coverage. Multiple peptides unique to JNK3 protein suggested unequivocal identification (Fig. 2C). JNK3 expression increases during RGC development (Fig. 2D) and parallels the developmental increase in KLF9 expression (Moore et al., 2009), suggesting a possible connection between KLF9 and JNK3 and the RGCs' developmental decrease in intrinsic axon growth ability (Goldberg, Klassen et al., 2002).
Figure 2.

KLF9 is bound by JNK3 in neurons. A, mSin3a is present in the retina but does not bind KLF9. Retina lysate from P10 rats was immunoprecipitated for KLF9 and Western blotted for mSin3a or KLF9. B, Purified P0 rat RGCs virally transduced with flag-tagged KLF9 were lysed and immunoprecipitated for flag. The IP lane (box) was sent for MS analysis. C, JNK3 is identified from KLF9 IP/MS. MAPK10/JNK3 was detected with six independent peptides (identified from eight unique and 19 total spectra with >95% confidence, yellow) and with 13% coverage (oxidized residue in green). D, JNK3 mRNA expression (arbitrary units) in developing RGCs from a previously described microarray dataset (Wang et al., 2007) shows robust upregulation during development. E, Endogenous KLF9 co-IPs with JNK3. Retinas from P10 rats were immunoprecipitated for KLF9 and Western blotting for anti-KLF9, anti-JNK3, and anti-β-actin antibodies, as marked. F, Confocal nanoscopy imaging of GFP-KLF9- and Flag-JNK3-electroporated RGCs demonstrated nuclear colocalization. Asterisk indicates that not all cells were successfully cotransfected. Fluorescence intensity of KLF9, JNK3, and TOPRO-3 were calculated separately and compared along the line in the cell cotransfected with both proteins (F′, arrows). Intensities showed higher similarity between KLF9 and JNK3 thanTOPRO-3 (red lines). F′′, High-magnification images of the boxed area in F. Scale bar, 10 μm. I, input; E, eluent; F, flow-through.
The binding interaction between KLF9 and JNK3 was further verified by co-IP of endogenous KLF9 and Western blot for endogenous JNK3 in P10 rat RGCs (Fig. 2E). Without a commercially available JNK3 IP antibody, we were not able to perform a reverse IP of endogenous JNK3 to further confirm the binding interaction. To explore subcellular colocalization, we transfected tagged proteins in purified RGCs and immunostained for each. JNK3's nuclear localization has been well described (Song et al., 2007; Turjanski et al., 2007; Abdelli et al., 2009; Pan et al., 2009) and, consistent with previous research, JNK3 was primarily detected in the nucleus of RGCs (Fig. 2F). Coloc2 analysis showed colocalization (p = 1.00) and ImageJ plot analysis showed a higher degree of similarity between KLF9 and JNK3 than between KLF9 and a control nuclear protein, TOPRO-3 (Fig. 2F′,F′′), not only suggesting nuclear specificity, but also supporting colocalization at the limits of confocal light microscopy. Together, these data demonstrate that JNK3 is a novel binding partner of KLF9 in RGCs.
Where does JNK3 bind KLF9 and does this interaction lead to KLF9 phosphorylation? Because there is no complete x-ray crystal structure for KLF9, we performed a surface modeling analysis using Scansite (scansite.mit.edu). Analysis revealed a surface consensus JNK family docking domain sequence, arginine 223 to isoleucine 233 (R223-I233), near KLF9's C terminus (Fig. 3A), and four potential serine/threonine kinase phosphorylation sites on KLF9, S88, S95, S106, and S110 (Fig. 3B), all with predicted high surface accessibility (Fig. 3C). Although potential phospho-acceptor sites for MAPK family kinases appear throughout the KLF family, only KLF9, KLF13, KLF14, and KLF16 (BTEB subfamily of KLFs) and KLF10 and KLF11 have sequences closely conforming to the JNK consensus docking site (Fig. 3A). We assessed the biochemical relevance of these residues by transducing E18 hippocampal neurons (used for their similar neurite growth-inhibited response to KLF9 expression (Moore et al., 2009) and their greater cell and protein yields) with a wild-type KLF9, KLF9ΔSID deletion mutant lacking the mSin3a-binding site, a KLF9Δ223–233 deletion mutant lacking the JNK3 consensus binding site, or a KLF9S88/95/106/110A mutant substituting the putative phospho-acceptor sites with nonphosphorylatable alanines (Fig. 3D). When hippocampal neurons were transduced with lentiviral GFP, GFP-KLF9, or GFP-KLF9Δ223–233 along with flag-mCherry-JNK3 after nuclear fractionation and co-IP, only in the GFP-KLF9 elution did we detect flag-mCherry-JNK3 (Fig. 3E,F). Conversely, the putative JNK3-binding domain of KLF9 (R223-I233) peptide (JBD-P) fused with GFP and a nuclear localization sequence (NLS), but not the NLS control peptide construct (CP) (Fig. 3G), immunoprecipitated JNK3 from hippocampal nuclear extracts (Fig. 3H). Furthermore, JBD-P but not CP was able to decrease the interaction between KLF9 and JNK3 (Fig. 3H,I). Therefore, the R223–I233 domain of KLF9 is necessary and sufficient for JNK3 binding and acts as an inhibitor of the KLF9–JNK3 interaction. When recombinant JNK3 was added to immunoprecipitated KLF9 protein, only wild-type KLF9, but not either of the mutants, incorporated radiolabeled [ϒ-32] ATP (Fig. 3J). Therefore, recombinant JNK3 is capable of phosphorylating KLF9 protein and this phosphorylation depends on the JNK-binding domain (R223-I233) and the presence of one or more identified serine residues (S88/S95/S106/S110).
Figure 3.
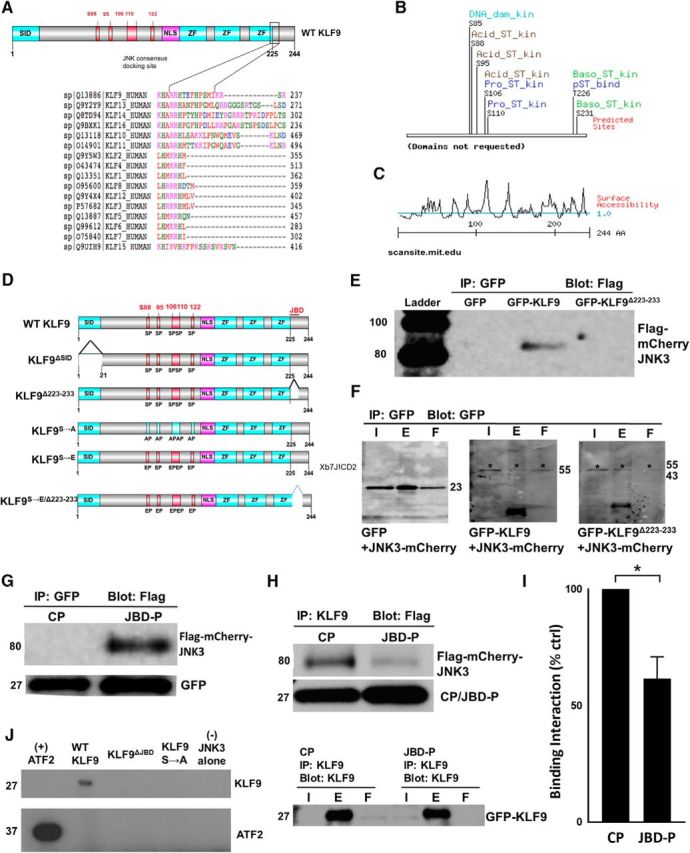
Identification of JNK3-binding domain and potential serine/threonine phosphorylation sites of KLF9. A, KLF9 structural analysis revealing potential JNK3-binding domains. ClustalW2 alignment shows region spanning R223 to I233 near C terminus of KLF9, as well as similar regions on subfamily relatives KLF13, KLF14, and KLF16, as well as KLF10 and KLF11, closely conforming to the known consensus JNK3 “DEJL” docking domain. B, MIT Scansite surface analysis of KLF9 full-length protein reveals potential phospho-acceptor sites at S88, S95, S106, and S110. C, MIT Scansite-predicted surface accessibility of KLF9 protein (>1 indicates exposed residues; <1 indicates buried or inaccessible residues). D, Schematic of a subset of mutant constructs for KLF9. ZF, Zinc finger domains. E, F, R223–I233 of KLF9 is necessary for KLF9–JNK3 interaction in neurons. E18 hippocampal neurons were cotransduced with Flag-mCherry-JNK3 and GFP, GFP-KLF9, or GFP-KLF9 R223-I233 deletion mutant (GFP-KLF9 Δ223–233) constructs using lentivirus. Cells were immunoprecipitated for GFP and Western blotted for flag (E) or GFP (F). G, H, JBD-P of KLF9 is sufficient for KLF9/JNK3 interaction in neurons. E18 hippocampal neurons were virally cotransduced with Flag-mCherry-JNK3 and either CP or JBD-P constructs (G), immunoprecipitated for GFP and Western blotted for flag. Flag-mCherry-JNK3 was only detected with the JBD-P construct (G). H, I, JBD-P reduces KLF9–JNK3 interaction. E18 hippocampal neurons were virally cotransduced with GFP-KLF9, Flag-mCherry-JNK3, and the CP or JBD-P, immunoprecipitated for KLF9, and Western blotted for flag or GFP (I). Densitometry quantification of blot bands showed an average of 38% decrease in the amount of flag-mCherry-JNK3 pulled down by the JBD-P coexpression group compared with the CP condition (I). J, KLF9 incorporation of radiolabeled [γ-32P] ATP depends on JNK-binding domain (JBD) and identified serine residues. E18 hippocampal neurons were transduced with flag-tagged wild-type KLF9, KLF9ΔJBD, or KLF9S85/88/95/106/110A mutants. Cells were immunoprecipitated for flag. Eluents were combined with recombinant JNK3 and radiolabeled [ϒ-32] ATP in a standard in vitro kinase assay. Incorporation of radioactivity was only observed in the eluents from wild-type KLF9 transduced neurons. *p < 0.05, 2-tailed Student's t test, n = 3. Error bars indicate SD. I, Input; E, eluent; F, flow-through.
JNK3-binding domain and S106 and S110 are critical to KLF9's function in suppressing axon growth
We next investigated whether the KLF9–JNK3 interaction regulates KLF9's ability to suppress neurite growth in RGCs. When KLF9–JNK3 binding was disrupted either by deleting the JNK3-binding domain (KLF9ΔJBD) or by delivering the JBD-P, KLF9's suppression of RGC neurite growth was abolished (Fig. 4A,B). Although the N-terminal mSin3A-interacting domain (SID) has been shown to be critical for KLF9's function in non-neuronal cells (Grzenda et al., 2009), its deletion did not affect KLF9's suppressive effect on neurite growth in RGCs (Fig. 4A,B). Furthermore, coexpressing JNK3 with KLF9 on P0 RGCs potentiated wild-type KLF9's neurite growth-suppressive activity, although JNK3 expression did not affect neurite growth on its own (Fig. 4C). The kinase activity of JNK3 is essential for this functional interaction because a kinase-dead JNK3 mutant (Ho et al., 2006) had no ability to potentiate KLF9's neurite growth suppression (Fig. 4C). Therefore, KLF9 and JNK3 interact functionally to suppress neurite growth in RGCs.
To address which of the four potential serine phosphorylation acceptor sites are crucial for KLF9's function in neurons, different serine-to-alanine nonphosphorylatable and serine-to-glutamate phosphomimic mutants of KLF9 were transduced into RGCs by lentivirus at P0 and P8, examining both ages to assess function in the context of lower (P0) or higher (P8) levels of endogenous KLF9 (Moore et al., 2009). In all cases, the KLF9 constructs were found to localize to RGC nuclei after transduction (Fig. 4A), suggesting that none of the mutations affected nuclear localization. Of all of the phosphomutants tested, only S106/S110 proved functionally relevant in neurite growth assays. When both residues were mutated to phospho-null alanines (S106/110A), KLF9's ability to suppress neurite growth was abolished in both P0 and P8 RGCs (P0, Fig. 4A,B; P8, data not shown). Conversely, when both were mutated to phosphomimic glutamates (S106/110E), KLF9's suppression of neurite growth was potentiated at both ages (P0, Fig. 4A,B; P8, data not shown). Furthermore, phosphomimic substitutions were able to rescue the JNK3-binding domain deletion: RGCs transduced with KLF9ΔJBD/S106E/S110E demonstrated a suppressed neurite growth capacity similar to the full-length S106/110E mutant (Fig. 4A,E), demonstrating that phosphorylation of these two serines can rescue KLF9's neurite growth suppression in the absence of JNK3 binding. Therefore, the phosphorylation state of S106/S110 modulates KLF9's suppressive effect on neurite growth, suggesting a novel mode of KLF9 regulation, consistent with the JNK3 binding and phosphorylation shown above.
Disrupting KLF9–JNK3 interaction or expressing KLF9 S106A/S110A mutants enhance optic nerve axon regeneration
We first examined JNK phosphorylation after optic nerve injury, which was described previously to increase phosphorylated JNK (Fernandes et al., 2012). In the absence of pJNK3-specific antibodies, we used JNK knock-out mice to determine isoform contribution to expression after injury. In wild-type mice, immunofluorescence against pJNK showed a low baseline level that increased after optic nerve injury (Fig. 5A,B). A similar increase after optic nerve injury was detected in JNK2-null mice, but not in JNK2/3 double-null mice (Fig. 5A,B), indicating that the pJNK increase seen after optic nerve injury reflects JNK3's contribution to phosphorylation.
Figure 5.

JNK3 knock-out promoted RGC survival but not axon regeneration. A, Immunofluorescence for retinal ganglion cells using β-III tubulin (green) and phosphorylated JNK (red) in wild-type control retinal sections and after optic nerve injury in wild-type mice, Jnk2−/−, and Jnk2 and 3−/−. B, pJNK quantifications for control and optic nerve injury. C, Images of the retina showing RGC-specific markers RBPMS (green) and Brn3a (yellow) in JNK3 knock-out (KO) and age-matched wild-type (WT) mice before and 2 weeks after optic nerve crush. D, RGC quantification demonstrated similar RGC density in JNK3 KO and WT control mice before the optic nerve injury. Two weeks after optic nerve injury, however, RGC density in JNK3 KO mice was significantly higher than in WT animals (*p < 0.05, **p < 0.001, one-way ANOVA; n = 5 per group; NS, Not significant). E, Images of the optic nerve sections showing CTB-labeled axons 2 weeks after optic nerve injury. Asterisks indicate lesion sites. F, A similar number of regenerating fibers was observed in JNK3 KO and WT control animals. Error bars indicate SE. Scale bar, 200 μm.
We next studied whether knocking out JNK3, which likely has many substrate targets, contributes to RGC survival or axon regeneration. We found that JNK3 KO promoted RGC survival (Fig. 5C,D), but not axon regeneration, significantly compared with wild-type animals (Fig. 5E,F), indicating that JNK3 regulates RGC survival and axon regeneration through different mechanisms and likely has many targets other than KLF9. This is consistent with recent data on a kinase upstream of JNKs, dual leucine zipper kinase (DLK) because deletion of DLK promotes RGC survival but reduces the number of regenerating axons induced by PTEN deletion after optic nerve injury (Watkins et al., 2013; Welsbie et al., 2013; Welsbie et al., 2017).
We narrowed in on the more specific question of whether disrupting the KLF9–JNK3 interaction or altering S106/110 phosphorylation enhances RGC survival or axon regeneration after optic nerve injury in vivo. AAV2 packaged with GFP, JBD-P, KLF9S106/110A, or KLF9ΔJBD were injected intravitreally 2 weeks before optic nerve injury to allow adequate expression in RGCs; axon regeneration was assessed 2 weeks after optic nerve injury. The transduction efficiencies of all the constructs were similar and >80% (data not shown, similar to the AAV2 shRNA anti KLF9; Fig. 1E). None of the constructs affected RGC survival after injury (Fig. 6A,B). An increased number of regenerating axons were observed up to 1000 μm from the site of injury in animals treated with JBD-P, KLF9S106/110A, or KLF9ΔJBD AAV2 (Fig. 6C,D). Therefore, disrupting the KLF9–JNK3 interaction by introducing JBD-P or KLF9ΔJBD or expressing KLF9 with nonphosphorylatable alanine substitutions at S106 and S110 increases the regenerative potential of adult RGCs in vivo.
Figure 6.

Disrupting the KLF9–JNK3 interaction or expressing KLF9 S106/110A mutants enhance optic nerve axon regeneration 2 weeks after optic nerve injury. A, Images of Brn3a-positive cells in retinal segments from different groups. Scale bar, 100 μm. B, Quantification of RGC survival showed no significant differences among all groups. C, Images of merged optic nerve sections showing CTB-labeled regenerating axons. Scale bar, 200 μm. Asterisks indicate lesion sites. D, Significantly more regenerating fibers were observed in JBD-P, KLF9S106/110A, and KLF9ΔJBD treated animals than in control animals. *p < 0.05, **p < 0.01, one-way ANOVA; n = 4. Error bars indicate SEM.
Discussion
Together, these data demonstrate three important, related findings. First, KLF9 expression contributes to the cell-intrinsic suppression of axon regeneration in vivo and knock-down by RNAi promotes long-distance axon regeneration. Axon regeneration promoted by KLF9 genomic excision using a floxed KLF9 allele or CRISPR/Cas9 technology could further validate these results; however, in contrast to gene deletion, inhibition of KLF9 via virally delivered shRNA could be a more useful tool for potential future translational application. The mechanisms for KLF actions in different neurons and on different cellular phenotypes have not been well studied, but KLFs may have opposing effects in different neurons, implying differing mechanisms. For example, inconsistent with its suppressive effect on RGC survival and axon growth, KLF9 improved Purkinje cell survival in organotypic cultures (Lebrun et al., 2013) and increased neurite growth, branching, and elongation in Neuro-2a and XTC-2 cell lines and dissociated cerebral hemisphere cultures (Denver et al., 1999; Cayrou et al., 2002; Ronald Bonett et al., 2009). We hypothesize that such differences may be due to differences in primary neurons versus cell lines and in KLF cofactor expression and note the in vivo validation of our findings. Here, we find a robust effect on both RGC survival and axon regeneration of KLF9 knock-down in the adult rat delivered just before optic nerve injury. These data suggest that although many KLFs are developmentally regulated at the time RGC axon growth declines early postnatally, decreasing KLF activity acutely in adult rat and mice is sufficient to promote regeneration and this KLF-mediated decline in axon growth capacity is reversible.
Interestingly, we showed previously that KLF4 knock-down during early development promoted axon regeneration but not survival after optic nerve injury in the adult mice (Moore et al., 2009), suggesting that KLF4 and KLF9 may regulate neuron survival and perhaps axon regeneration by different mechanisms. It is likely that these differences are mediated by the structural and binding partner differences between the two proteins. For example, KLF4 was not able to pull down JNK3 from whole retinal lysates (data not shown). Further studies on the differences in KLF biology across cells and tissues throughout the CNS will lead to better understanding of their regulation of survival and axon regeneration.
Second, these data reveal an important interaction between KLF9 and a novel KLF9-binding partner, JNK3, which phosphorylates KLF9 in neurons. Activated MAPKs in injured neurons of both the CNS and PNS mediate changes in the expression and/or activation levels of transcription factors such as c-Jun, STAT-3, and ATF2 (Herdegen and Leah, 1998; Tsujino et al., 2000; Schweizer et al., 2002; Powell et al., 2014)—and now KLFs. JNK3's activation after axon injury and role in promoting RGC death has been well studied (Fernandes et al., 2012), although some prior work is not consistent with our observation of increased RGC survival in JNK3 knock-out animals after optic nerve injury (Quigley et al., 2011). Conditionally knocking out c-jun, one of JNK3's substrates, was found to improve survival significantly (Fernandes et al., 2012). Paradoxically, c-jun expression has also been shown to be an important regulator of successful regeneration after injury (Raivich and Makwana, 2007; Ruff et al., 2012). This dual functionality has also been observed at the kinase level with DLK, a kinase upstream of JNKs, which was shown to have both proregenerative and proapoptotic effects on RGCs after optic nerve injury (Watkins et al., 2013; Welsbie et al., 2013). JNK3's direct role in axon regeneration, however, has not been well characterized previously. Some previous research on cultured neurons showed that lack of JNK3 delayed neurogenesis (Barnat et al., 2010) and impaired regenerative neurite outgrowth (Tonges et al., 2011). In the current study, we showed that JNK3 played a role in regulating intrinsic regenerative capacity in neurons. It interacts with KLF9 to suppress neurite growth in RGCs and the disruption of their interaction promotes axon regeneration after optic nerve injury in vivo. Interestingly, globally knocking out JNK3 throughout development does not promote axon regeneration after optic nerve injury. JNK2, which is highly structurally and functionally related to JNK3 (Fernandes et al., 2012), could compensate for JNK3's loss during development. Acute knock-down of JNK3 in adult animals would be useful to confirm the role of JNK3 on axon regeneration and overexpression of exogenous KLF9 should suppress any such regenerative response. Another likely hypothesis is that global knock-out of JNK3 influences regeneration negatively through other KLF9-independent downstream signaling events. Supporting this, previous studies have shown that JNK phosphorylates JUN and DLK after optic nerve crush (Huntwork-Rodriguez et al., 2013), both of which are known to exert a proregenerative role (Ruff et al., 2012; Tedeschi and Bradke, 2013; Watkins et al., 2013). Therefore, disrupting the KLF9–JNK3 interaction lessens axon growth suppression and promotes axon regeneration, whereas knocking out JNK3 may lessen both growth suppression and growth promotion pathways and result in a net nonregeneration phenotype, as seen in our data. Additional exploration of JNK3 and KLF9 protein and gene targets will be an interesting direction for future work.
Together, our data may suggest that injured RGCs initiate stimulus-dependent pathways, triggering upstream kinases such as DLK, which then respond by activating JNK family kinases. These kinases not only mediate an orderly cell death program, but also play a role in regulating regenerative neurite growth capacity (both pro-growth and anti-growth) depending on the particular balance of JNK isoforms involved, their localization, and the particular cellular context. This would explain why KLF9 is a more potent neurite growth suppressor after it has been phosphorylated by JNK3, which itself is activated by upstream kinases as well as cellular stress. Indeed, KLF9 expression itself has also been shown to be induced directly by stressors through a glucocorticoid-dependent mechanism in Xenopus brain (Bonett et al., 2009). Whether JNK3 regulates KLFs in their diverse roles outside of the CNS will be an important area for future study.
Finally, in characterizing the importance of KLF9's JNK3-binding domain and of two novel phosphorylation sites, S106 and S110, in KLF9's suppressive activity on axon growth, these data suggest that it is possible to interfere with the axon-suppressive effects of KLF9 through dominant-negative constructs. The effects of KLF9 knock-down are stronger than JNK3 interference, as expected, and may be explained by a larger pool of endogenous KLF9 protein than can be readily inhibited by blocking the KLF9–JNK3 interaction. It may also be due to other reasons, such as degradation of the short inhibiting peptide, compensation from JNK2, or other modes of regulation of KLF9 activity, all of which could be quite interesting to address in future study. Furthermore, all four BTEB subfamily KLFs (KLF9, KLF13, KLF14, and KLF16) suppress axon growth in CNS neurons (Moore et al., 2009) and contain a similar JNK3-binding domain, indicating a structure–function relationship for these KLFs in neurons. Although, in our preliminary experiments, KLF13 was not able to pull down JNK3 from whole retinal lysates (data not shown), it remains possible that the relatively low expression of KLF13 compared with KLF9 made it difficult to detect an interaction. Future experiments could explore this hypothesis more thoroughly by creating epitope-tagged versions of multiple KLF family members, overexpressing them with epitope-tagged JNK3, and conducting IP experiments from both sides using respective antibodies in purified RGCs. Manipulations such as these involving the expression of interfering peptides that disrupt multiple KLF interactions with regulatory partners could prove to be a useful strategy to promote axon regeneration and contribute to therapeutic approaches for addressing CNS injury and degenerative disease.
Footnotes
Author contributions: A.A., J.M.G., Y.W., D.L.M., R.T.L., and J.L.G. designed research; A.A., J.M.G., Y.W., M.G.B., A.T., K.I., D.P.B., K.A.F., A.H., T.N., M.A., X.Z., P.X.S., N.J.K., and D.L.M. performed research; J.L.G. contributed unpublished reagents/analytic tools; A.A., J.M.G., Y.W., M.G.B., K.I., D.P.B., M.A., N.J.K., and D.L.M. analyzed data; A.A., J.M.G., Y.W., and J.L.G. wrote the paper.
This work was funded by the National Eye Institute–National Institutes of Health (NIH Grant EY022129 to J.L.G., Grant EY018606 to R.T.L., and Grants P30-EY022589 and P30-EY014801), the National Institute of Neurological Disorders and Stroke–NIH (Grant NS074490), the American Heart Association, the James and Esther King Foundation, the Department of Defense (Grant W81XWH-12-1-0254), and Research to Prevent Blindness (unrestricted grant). We thank V.P. Lemmon for access to a cDNA library and for anti-Luciferase shRNA pAAV2 plasmid, D. Turner (University of Michigan) for the shRNA backbone, and Yan Shi, Pingping Jia, Eleut Hernandez, and Gabriel Gaidosh for technical assistance.
The authors declare no competing financial interests.
References
- Abdelli S, Puyal J, Bielmann C, Buchillier V, Abderrahmani A, Clarke PG, Beckmann JS, Bonny C (2009) JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia 52:1871–1880. 10.1007/s00125-009-1431-7 [DOI] [PubMed] [Google Scholar]
- Barnat M, Enslen H, Propst F, Davis RJ, Soares S, Nothias F (2010) Distinct roles of c-Jun N-terminal kinase isoforms in neurite initiation and elongation during axonal regeneration. J Neurosci 30:7804–7816. 10.1523/JNEUROSCI.0372-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Silverstein BE, Corey DP, Chun LL (1988) Electrophysiological variation among retinal ganglion cells purified by panning. Neuron 1:791–803. 10.1016/0896-6273(88)90127-4 [DOI] [PubMed] [Google Scholar]
- Berkelaar M, Clarke DB, Wang YC, Bray GM, Aguayo AJ (1994) Axotomy ganglion results in delayed death and apoptosis of retinal cells in adult rats. J Neurosci 14:4368–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, Lee JK, Goldberg JL, Lemmon VP, Bixby JL (2012) Krüppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A 109:7517–7522. 10.1073/pnas.1120684109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore MG, Moore DL, Smith RP, Goldberg JL, Bixby JL, Lemmon VP (2010) High content screening of cortical neurons identifies novel regulators of axon growth. Mol Cell Neurosci 44:43–54. 10.1016/j.mcn.2010.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackmore M, Letourneau PC (2006) Changes within maturing neurons limit axonal regeneration in the developing spinal cord. J Neurobiol 66:348–360. [DOI] [PubMed] [Google Scholar]
- Bonett RM, Hu F, Bagamasbad P, Denver RJ (2009) Stressor and glucocorticoid-dependent induction of the immediate early gene kruppel-like factor 9: implications for neural development and plasticity. Endocrinology 150:1757–1765. 10.1210/en.2008-1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradke F, Dotti CG (1997) Neuronal polarity: vectorial cytoplasmic flow precedes axon formation. Neuron 19:1175–1186. 10.1016/S0896-6273(00)80410-9 [DOI] [PubMed] [Google Scholar]
- Cayrou C, Denver RJ, Puymirat J (2002) Suppression of the basic transcription element-binding protein in brain neuronal cultures inhibits thyroid hormone-induced neurite branching. Endocrinology 143:2242–2249. 10.1210/endo.143.6.8856 [DOI] [PubMed] [Google Scholar]
- Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW (2008) NS21: re-defined and modified supplement B27 for neuronal cultures. J Neurosci Methods 171:239–247. 10.1016/j.jneumeth.2008.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu K, Lau WM, Yeung SC, Chang RC, So KF (2008) Retrograde labeling of retinal ganglion cells by application of fluoro-gold on the surface of superior colliculus. J Vis Exp 16: pii: 819. 10.3791/819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KH, Hart CC, Al-Bassam S, Avery A, Taylor J, Patel PD, Vojtek AB, Turner DL (2006) Polycistronic RNA polymerase II expression vectors for RNA interference based on BIC / miR-155. Nucleic Acids Res 34:e53. 10.1093/nar/gkl143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver RJ, Ouellet L, Furling D, Kobayashi A, Fujii-Kuriyama Y, Puymirat J (1999) Basic transcription element-binding protein (BTEB) is a thyroid hormone-regulated gene in the developing central nervous system: Evidence for a role in neurite outgrowth. J Biol Chem 274:23128–23134. 10.1074/jbc.274.33.23128 [DOI] [PubMed] [Google Scholar]
- Eaton SA, Funnell AP, Sue N, Nicholas H, Pearson RC, Crossley M (2008) A network of Krüppel-like Factors (Klfs). Klf8 is repressed by Klf3 and activated by Klf1 in vivo. J Biol Chem 283:26937–26947. 10.1074/jbc.M804831200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang IH, John SW, Libby RT (2012) JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis 46:393–401. 10.1016/j.nbd.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA (2002) Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron 33:689–702. 10.1016/S0896-6273(02)00602-5 [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA (2002) Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science 296:1860–1864. 10.1126/science.1068428 [DOI] [PubMed] [Google Scholar]
- Grzenda A, Lomberk G, Zhang JS, Urrutia R (2009) Sin3: master scaffold and transcriptional corepressor. Biochim Biophys Acta 1789:443–450. 10.1016/j.bbagrm.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström M, Ruitenberg MJ, Pollett MA, Ehlert EM, Twisk J, Verhaagen J, Harvey AR (2009) Cellular tropism and transduction properties of seven adeno-associated viral vector serotypes in adult retina after intravitreal injection. Gene Therapy 16:521–532. 10.1038/gt.2008.178 [DOI] [PubMed] [Google Scholar]
- Herdegen T, Leah JD (1998) Inducible and constitutive transcription factors in the mammalian nervous system: Control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Rev 28:370–490. 10.1016/S0165-0173(98)00018-6 [DOI] [PubMed] [Google Scholar]
- Ho DT, Bardwell AJ, Grewal S, Iverson C, Bardwell L (2006) Interacting JNK-docking sites in MKK7 promote binding and activation of JNK mitogen-activated protein kinases. J Biol Chem 281:13169–13179. 10.1074/jbc.M601010200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Cho S, Goldberg JL (2010) Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Invest Ophthalmol Vis Sci 51:1747–1754. 10.1167/iovs.09-4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork-Rodriguez S, Wang B, Watkins T, Ghosh AS, Pozniak CD, Bustos D, Newton K, Kirkpatrick DS, Lewcock JW (2013) JNK-mediated phosphorylation of DLK suppresses its ubiquitination to promote neuronal apoptosis. J Cell Biol 202:747–763. 10.1083/jcb.201303066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Chang Q, Zhao Z, Yan S, Wang L, Cai J, Xu G (2012) Melatonin-mediated cytoprotection against hyperglycemic injury in Müller cells. PLoS One 7:e50661. 10.1371/journal.pone.0050661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JM, Caprioli J, Piri N (2010) RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci 51:1052–1058. 10.1167/iovs.09-4098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrun C, Avci HX, Wehrlé R, Doulazmi M, Jaudon F, Morel MP, Rivals I, Ema M, Schmidt S, Sotelo C, Vodjdani G, Dusart I (2013) Klf9 is necessary and sufficient for Purkinje cell survival in organotypic culture. Mol Cell Neurosci 54:9–21. 10.1016/j.mcn.2012.11.010 [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA (1995) Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron 15:805–819. 10.1016/0896-6273(95)90172-8 [DOI] [PubMed] [Google Scholar]
- Moore DL, Apara A, Goldberg JL (2011) Kruppel-like transcription factors in the nervous system: novel players in neurite outgrowth and axon regeneration. Mol Cell Neurosci 47:233–243. 10.1016/j.mcn.2011.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL (2009) KLF family members regulate intrinsic axon regeneration ability. Science (New York), 326, 298–301. Science 326:298–301. 10.1126/science.1175737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls J, Saunders N (1996) Regeneration of immature mammalian spinal cord after injury. Trends Neurosci 19:229–234. 10.1016/0166-2236(96)10021-7 [DOI] [PubMed] [Google Scholar]
- Pan J, Xiao Q, Sheng CY, Hong Z, Yang HQ, Wang G, Ding JQ, Chen SD (2009) Blockade of the translocation and activation of c-Jun N-terminal kinase 3 (JNK3) attenuates dopaminergic neuronal damage in mouse model of Parkinson's disease. Neurochem Int 54:418–425. 10.1016/j.neuint.2009.01.013 [DOI] [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult cns by modulation of the PTEN/mTOR pathway. Science 322:963–966. 10.1126/science.1161566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell AE, Vlacich G, Zhao ZY, McKinley ET, Washington MK, Manning HC, Coffey RJ (2014) Inducible loss of one Apc allele in Lrig1-expressing progenitor cells results in multiple distal colonic tumors with features of familial adenomatous polyposis. Am J Physiol Gastrointest Liver Physiol 307:G16–G23. 10.1152/ajpgi.00358.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Zou Y, Zhang CL (2013) Cross-talk between KLF4 and STAT3 regulates axon regeneration. Nat Commun 4:2633. 10.1038/ncomms3633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Cone FE, Gelman SE, Yang Z, Son JL, Oglesby EN, Pease ME, Zack DJ (2011) Lack of neuroprotection against experimental glaucoma in c-Jun N-terminal kinase 3 knockout mice. Exp Eye Res 92:299–305. 10.1016/j.exer.2011.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivich G, Makwana M (2007) The making of successful axonal regeneration: genes, molecules and signal transduction pathways. Brain Res Rev 53:287–311. 10.1016/j.brainresrev.2006.09.005 [DOI] [PubMed] [Google Scholar]
- Rodriguez AR, de Sevilla Müller LP, Brecha NC (2014) The RNA binding protein RBPMS is a selective marker of ganglion cells in the mammalian retina. J Comp Neurol 522:1411–1443. 10.1002/cne.23521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruff CA, Staak N, Patodia S, Kaswich M, Rocha-Ferreira E, Da Costa C, Brecht S, Makwana M, Fontana X, Hristova M, Rumajogee P, Galiano M, Bohatschek M, Herdegen T, Behrens A, Raivich G (2012) Neuronal c-Jun is required for successful axonal regeneration, but the effects of phosphorylation of its N-terminus are moderate. J Neurochem 121:607–618. 10.1111/j.1471-4159.2012.07706.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer U, Gunnersen J, Karch C, Wiese S, Holtmann B, Takeda K, Akira S, Sendtner M (2002) Conditional gene ablation of Stat3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J Cell Biol 156:287–297. 10.1083/jcb.200107009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevtsova Z, Malik JM, Michel U, Bähr M, Kügler S (2005) Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol 90:53–59. 10.1113/expphysiol.2004.028159 [DOI] [PubMed] [Google Scholar]
- Song X, Gurevich EV, Gurevich VV (2007) Cone arrestin binding to JNK3 and Mdm2: conformational preference and localization of interaction sites. J Neurochem 103:1053–1062. 10.1111/j.1471-4159.2007.04842.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Bradke F (2013) The DLK signalling pathway–a double-edged sword in neural development and regeneration. EMBO Rep 14:605–614. 10.1038/embor.2013.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönges L, Planchamp V, Koch JC, Herdegen T, Bähr M, Lingor P (2011) JNK isoforms differentially regulate neurite growth and regeneration in dopaminergic neurons in vitro. J Mol Neurosci 45:284–293. 10.1007/s12031-011-9519-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trakhtenberg EF, Wang Y, Morkin MI, Fernandez SG, Mlacker GM, Shechter JM, Liu X, Patel KH, Lapins A, Yang S, Dombrowski SM, Goldberg JL (2014) Regulating set-β's subcellular localization toggles its function between inhibiting and promoting axon growth and regeneration. J Neurosci 34:7361–7374. 10.1523/JNEUROSCI.3658-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino H, Kondo E, Fukuoka T, Dai Y, Tokunaga A, Miki K, Yonenobu K, Ochi T, Noguchi K (2000) Activating transcription factor 3 (ATF3) induction by axotomy in sensory and motoneurons: a novel neuronal marker of nerve injury. Mol Cell Neurosci 15:170–182. 10.1006/mcne.1999.0814 [DOI] [PubMed] [Google Scholar]
- Turjanski AG, Vaqué JP, Gutkind JS (2007) MAP kinases and the control of nuclear events. Oncogene 26:3240–3253. 10.1038/sj.onc.1210415 [DOI] [PubMed] [Google Scholar]
- Wang JT, Kunzevitzky NJ, Dugas JC, Cameron M, Barres BA, Goldberg JL (2007) Disease gene candidates reveal by expression profiling of retinal ganglion cell development. J Neurosci 32:8593–8603. 10.1523/jneurosci.4488-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Brown DP Jr, Duan Y, Kong W, Watson BD, Goldberg JL (2013) A novel rodent model of posterior ischemic optic neuropathy. JAMA Ophthalmol 131:194–204. 10.1001/2013.jamaophthalmol.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins TA, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, Modrusan Z, Kaminker JS, Tessier-Lavigne M, Lewcock JW (2013) DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci U S A 110:4039–4044. 10.1073/pnas.1211074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, et al. (2013) Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci U S A 110:4045–4050. 10.1073/pnas.1211284110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsbie DS, et al. (2017) Enhanced functional genomic screening identifies novel mediators of dual leucine zipper kinase-dependent injury signaling in neurons. Neuron 94:1142–1154. 10.1016/j.neuron.2017.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z (2006) Glial inhibition of CNS axon regeneration. Nat Rev Neurosci 7:617–627. 10.1038/nrn1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Xu P, Zhao Z, Cai J, Sternberg P, Chen Y (2014) Subcellular distribution and activity of mechanistic target of rapamycin in aged retinal pigment epithelium. Invest Ophthalmol Vis Sci 55:8638–8650. 10.1167/iovs.14-14758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Chen Y, Wang J, Sternberg P, Freeman ML, Grossniklaus HE, Cai J (2011) Age-related retinopathy in NRF2-deficient mice. PLoS One 6:e19456. 10.1371/journal.pone.0019456 [DOI] [PMC free article] [PubMed] [Google Scholar]