

Abstract
Familial melanoma-astrocytoma syndrome is a tumor predisposition syndrome caused by inactivating germline alteration of the CDKN2A tumor suppressor gene on chromosome 9p21. While some families with germline CDKN2A mutations are prone to development of just melanomas, other families develop both melanomas, astrocytomas, and occasionally other nervous-system neoplasms including peripheral nerve sheath tumors and meningiomas. The histologic spectrum of the astrocytomas that arise as part of this syndrome is not well described, nor are the additional genetic alterations that drive these astrocytomas apart from the germline CDKN2A inactivation. Herein, we report the case of a young man with synchronous development of a pleomorphic xanthoastrocytoma, diffuse astrocytoma, and paraspinal mass radiographically consistent with a peripheral nerve sheath tumor. His paternal family history is significant for melanoma, glioblastoma, and oral squamous cell carcinoma. Genomic profiling revealed that he harbors a heterozygous deletion in the germline of chromosome 9p21.3 encompassing the CDKN2A and CDKN2B tumor suppressor genes. Both the pleomorphic xanthoastrocytoma and diffuse astrocytoma were found to have homozygous deletion of CDKN2A/B due to somatic loss of the other copy of chromosome 9p containing the remaining intact alleles. Additional somatic alterations included BRAF p.V600E mutation in the pleomorphic xanthoastrocytoma and PTPN11, ATRX, and NF1 mutations in the diffuse astrocytoma. The presence of germline CDKN2A/B inactivation together with the presence of multiple anatomically, histologically, and genetically distinct astrocytic neoplasms, both with accompanying somatic loss of heterozygosity for the CDKN2A/B deletion, led to a diagnosis of familial melanoma-astrocytoma syndrome. This remarkable case illustrates the histologic and genetic diversity that astrocytomas arising as part of this rare glioma predisposition syndrome can demonstrate.
Keywords: glioma predisposition syndrome, CDKN2A, p16INK4a, diffuse astrocytoma, pleomorphic xanthoastrocytoma, nerve sheath tumor, melanocytic nevi
Introduction
Family history of brain tumors is an important risk factor in a small fraction of patients with primary glial neoplasms of the central nervous system [1, 2]. Among these patients, a subset harbor pathogenic germline alterations as part of well-characterized tumor predisposition syndromes [3]. These include germline mutations or deletions affecting the NF1 tumor suppressor gene as part of neurofibromatosis type 1 syndrome, TP53 tumor suppressor gene as part of Li-Fraumeni syndrome, and mismatch repair genes (e.g. MLH1, MSH2, MSH6, PMS2) as part of Lynch/Turcot syndrome. Multiple additional glioma susceptibility genes have been more recently identified. These include POT1, which encodes a protein involved in telomere maintenance, in which inactivating germline mutations have been identified in families with multiple members affected by oligodendroglioma [4]. Consortiums, such as GLIOGENE, are currently performing genome-wide association studies on large numbers of patients with primary glial neoplasms in order to determine the complete spectrum of genetic variants that increase risk of glioma development [5].
In addition to the aforementioned genetic syndromes, familial melanoma-astrocytoma syndrome has emerged as a rare cause of inherited glioma predisposition. Kaufman et al. [6] first described in 1993 a family in which cutaneous malignant melanoma or cerebral astrocytoma were seen in eight members over three generations and therefore suggested the presence of a possible new genetic syndrome. A study in 1995 examined the incidence of tumors of the nervous system as second cancers or in family members of 904 patients with cutaneous melanoma, which identified 15 families with cutaneous melanoma in which one or more family members had nervous system tumors including astrocytoma, glioblastoma, meningioma, and “acoustic neurilemmoma” (now referred to as vestibular schwannoma) [7]. Then a third study in 1997 described a family with a cancer syndrome including cutaneous melanoma, dysplastic nevi, astrocytoma, neurofibroma, schwannoma, and meningioma [8]. Together, these epidemiologic studies identified the presence of a distinct tumor predisposition syndrome that includes increased risk for both melanoma and nervous system tumors, predominantly astrocytomas.
The genetic basis of this tumor predisposition syndrome was elucidated in 1998 by Bahuau et al. [9] who reported identification of deletions of the INK4 locus at chromosome 9p21.3 in both of the families described in the original 1993 and 1997 reports. The INK4 locus contains, in close proximity, both the CDKN2A and CDKN2B tumor suppressor genes. CDKN2A encodes the p16INK4a cyclin-dependent kinase inhibitor and in an alternative reading frame also encodes p14ARF, an inhibitor of p53 signaling, while CDKN2B encodes p15INK4b, a cyclin-dependent kinase inhibitor with a high degree of homology to p16INK4a. CDKN2A had already been recognized as one of the major susceptibility genes for familial cutaneous melanoma, most commonly due to inactivating point mutations but also occasionally gene deletions [10, 11]. Familial melanoma-astrocytoma syndrome is now appreciated to represent an autosomal-dominant variant of the familial melanoma syndrome caused by heterozygous germline CDKN2A inactivation that also includes development of astrocytomas and occasionally other neural tumors including peripheral nerve sheath tumors and meningioma (Online Mendelian Inheritance in Man, entry # 155755).
Since the initial reports, a few additional families with genetically-confirmed familial melanoma-astrocytoma syndrome have been described [12, 13, 14, 15, 16, 17]. However, the histologic features of the astrocytomas that arise as part of this syndrome are not well described, nor are the somatic genetic alterations that drive these astrocytomas in addition to the germline CDKN2A inactivation. Herein, we report the case of a young man with a family history of melanoma, glioblastoma, and oral squamous cell carcinoma who was found to have synchronous development of a pleomorphic xanthoastrocytoma, diffuse astrocytoma, and a paraspinal mass radiographically consistent with a peripheral nerve sheath tumor. Pathologic and genomic assessment demonstrated the presence of germline CDKN2A/B deletion diagnostic of familial melanoma-astrocytoma syndrome, and also revealed the diversity of histologic features and genetic alterations that can be seen in astrocytomas arising as part of this rare glioma predisposition syndrome.
Case report
A 23-year-old Caucasian man initially presented with new-onset generalized tonic-clonic seizures. He was found to have a non-enhancing lesion in the left frontal lobe and underwent resection which demonstrated a low-grade astrocytoma. Evaluation of IDH1/2, ATRX, TP53, and BRAF status was not performed, and this specimen is not currently available for our pathologic review and genetic analysis. Dermatologic evaluation did not demonstrate any café-au-lait macules or axillary or inguinal freckling but did reveal scattered melanocytic nevi. Additionally, no Lisch nodules or cutaneous neurofibromas were present. His mother is alive without a personal or significant family history of neoplasia. His father has a history of multifocal high-grade epithelial dysplasia of the oropharynx as well as resection of squamous cell carcinoma from the oral cavity. His sister died of glioblastoma at age 14, and his paternal grandfather had a history of cutaneous melanoma and died of brain cancer. Two paternal uncles are currently alive, one with a history of cutaneous melanoma and the other with a history of oral cancer. A pedigree of the paternal lineage is shown in Figure 1.
Figure 1. Pedigree of the patient’s family and paternal lineage. The proband is marked with an arrow. SCC = squamous cell carcinoma of the oropharynx; LGG = low-grade glioma (diffuse astrocytoma); PXA = pleomorphic xanthoastrocytoma.
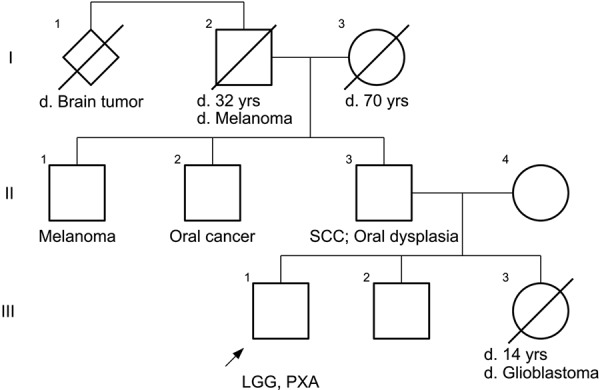
After resection, no additional adjuvant therapy was administered, and he was monitored by regular MR imaging of the brain over the next several years. Aside from suffering from occasional seizures associated with poor compliance to anticonvulsant medications, the patient remained otherwise neurologically intact. A surveillance scan 8 years after his initial surgery, now at 31 years of age, revealed a new area of nodular enhancement with associated hemorrhage adjacent to the prior resection cavity in the left frontal lobe (Figure 2A, B, C). Additionally, a new T2- and fluid-attenuated inversion recovery (FLAIR) hyperintense mass lesion in the right cerebellar hemisphere was seen that did not enhance after contrast administration (Figure 3A, B). The patient underwent a gross total resection of the enhancing nodule in the left frontal lobe. Pathology demonstrated a solid, non-infiltrative astrocytic neoplasm with marked nuclear pleomorphism, occasional xanthomatous tumor cells with foamy cytoplasm, and numerous eosinophilic granular bodies (Figure 2D, E). The mitotic index was low, and high-grade histologic features including necrosis and microvascular proliferation were not identified. An immunostain for type IV collagen revealed abundant intercellular collagen deposition (Figure 2F). Additional immunostains revealed that the tumor cells had intact/retained expression of ATRX protein and were negative for IDH1-R132H mutant protein. Ki67 labeling was seen in ~ 2% of tumor cells. A diagnosis of pleomorphic xanthoastrocytoma (PXA), WHO grade II, was rendered. Given this diagnosis and the potential for PXA to disseminate in the cerebrospinal fluid throughout the neuraxis, the right cerebellar lesion was considered worrisome for disseminated disease versus possibly representing a second primary tumor. To further evaluate the patient, MR imaging of the spinal cord was also performed, which demonstrated an expansile, contrast-enhancing mass within the right C6-7 neural foramen tracking along the C7 nerve root, consistent with a peripheral nerve sheath tumor (Figure 4). A resection of the right cerebellar lesion was performed 1 month following the left frontal craniotomy. Pathology demonstrated a diffuse astrocytoma composed of neoplastic fibrillary astrocytes with elongate and irregular, hyperchromatic nuclei infiltrating through the subcortical white matter and internal granular layer of the cerebellum (Figure 3C). The tumor cells demonstrated absence of ATRX immunostaining with intact staining in entrapped non-neoplastic neurons and endothelial cells, consistent with ATRX loss (Figure 3D). The tumor cells were negative for IDH1-R132H and histone H3-K27M mutant proteins by immunohistochemistry. Ki67 labeling was present in ~ 5% of tumor cells. A diagnosis of diffuse astrocytoma, WHO grade II, was rendered. Given the significant family history and the presence of multiple histologically-distinct brain tumors and a peripheral nerve sheath tumor, genomic testing was recommended.
Figure 2. Imaging and histologic features of the pleomorphic xanthoastrocytoma resected from the left frontal lobe. A, B, C: Surveillance MR imaging 8 years after initial resection of a low-grade astrocytoma from the left frontal lobe demonstrated interval development of a new 7-mm focus of nodular enhancement adjacent to the prior resection cavity within the left medial orbital gyrus. Coronal T2/FLAIR-weighted image (A), coronal T1-weighted post-gadolinium image (B), and axial T1-weighted post-gadolinium image (C). D, E: Hematoxylin and eosin stained sections showing an astrocytic neoplasm with marked nuclear pleomorphism, numerous eosinophilic granular bodies (arrowheads), and occasional xanthomatous tumor cells with foamy cytoplasm. F: Immunostain for type IV collagen demonstrating abundant intercellular collagen deposition amongst the neoplastic astrocytes. Scale bar, 40 μm.
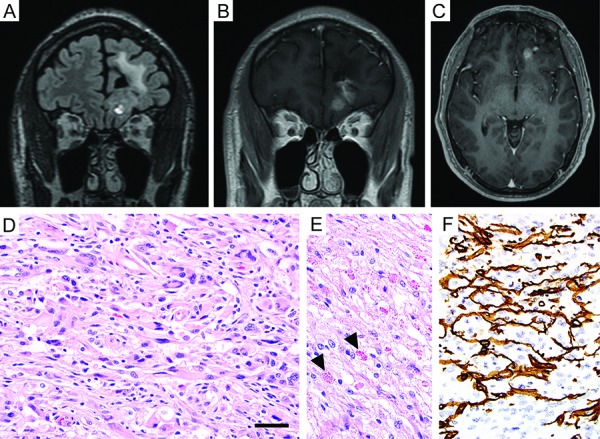
Figure 3. Imaging and histologic features of the diffuse astrocytoma resected from the right cerebellum. A, B: T2/FLAIR-weighted MR images demonstrating a hyperintense mass lesion in the right cerebellar hemisphere (coronal, A; sagittal, B). C: Hematoxylin and eosin stained section showing a diffuse glial neoplasm composed of neoplastic fibrillary astrocytes with elongate and irregular, hyperchromatic nuclei infiltrating through the cerebellar subcortical white matter. D: Immunohistochemistry for ATRX protein demonstrating absence of staining in the tumor cells with intact staining in entrapped non-neoplastic neurons and endothelial cells, consistent with somatic ATRX loss. Scale bar, 40 µm.
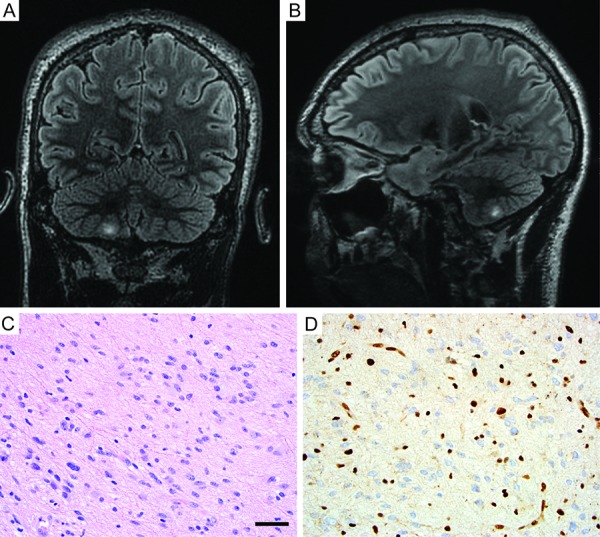
Figure 4. MR imaging of the cervical spine demonstrating an intradural, extramedullary mass centered in the right C6-C7 neural foramen and extending along the C7 nerve root (coronal T2-weighted, left; axial T1-weighted post-gadolinium, right).
After informed consent was obtained, targeted next-generation sequencing was performed on genomic DNA isolated from a peripheral blood sample and tumor tissue from the left frontal PXA and right cerebellar diffuse astrocytoma. This sequencing was performed on the UCSF500 Cancer Gene Panel as previously described, which utilizes capture-based next-generation sequencing targeting the coding regions of 479 cancer-associated genes along with select introns from 47 of these genes as well as the promoter region of the TERT gene [18].
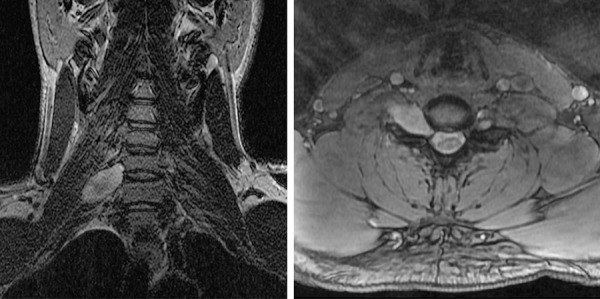
This analysis identified a focal heterozygous deletion on chromosome 9p21.3 spanning approximate coordinates chr9: g.21,700,000 – 22,800,000 (human genome assembly GRCh38) within the peripheral blood sample (Figure 5). This deletion contains both the CDKN2A and CDKN2B tumor suppressor genes. The left frontal PXA demonstrated homozygous deletion of CDKN2A/B due to loss of the other copy of chromosome 9 containing the remaining intact CDKN2A/B alleles. This was accompanied by the p.V600E somatic hotspot mutation in BRAF (Table 1). Alterations involving IDH1, IDH2, TP53, and ATRX were not identified. The right cerebellar diffuse astrocytoma demonstrated homozygous deletion of CDKN2A/B due to copy-neutral loss of heterozygosity of chromosome 9p. This was accompanied by a hotspot activating missense mutation in PTPN11, two inactivating frameshift mutations in the NF1 tumor suppressor gene, and an inactivating frameshift mutation in the ATRX tumor suppressor gene. No alterations involving IDH1, IDH2, or TP53 were identified, nor was the BRAF p.V600E mutation found in the PXA. Apart from the copy number changes involving chromosome 9, no other chromosomal copy number aberrations were seen in either tumor.
Figure 5. Genomic copy number analysis derived from targeted-capture next-generation sequencing data demonstrating heterozygous deletion on chromosome 9p21.3 containing the CDKN2A and CDKN2B tumor suppressor genes in the patient’s germline with somatic loss of chromosome 9p containing the remaining CDKN2A/B alleles in both astrocytic neoplasms. A: Genome-wide copy number plots of DNA extracted from peripheral blood (top), the left frontal pleomorphic xanthoastrocytoma (middle), and right cerebellar diffuse astrocytoma (bottom) demonstrating the focal deletion on chromosome 9p21, loss of chromosome 9 in the pleomorphic xanthoastrocytoma, and copy-neutral loss of heterozygosity of chromosome 9p in the diffuse astrocytoma, but no additional copy number alterations. B: Copy number plots for chromosome 9p highlighting identical boundaries of the focal 9p21.3 deletion within the peripheral blood and both tumors. The deletion is heterozygous within the peripheral blood and homozygous in both tumors due to somatic loss of the other copy of chromosome 9p.
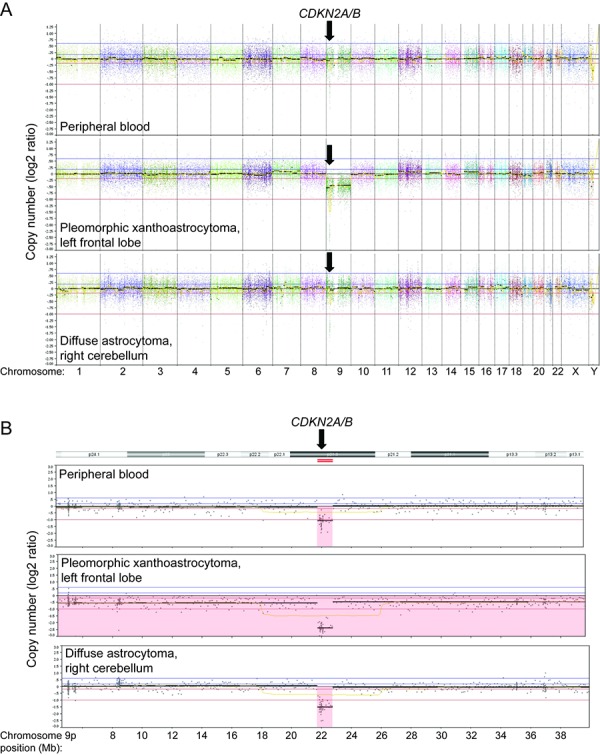
Table 1. Somatic alterations identified in the left frontal pleomorphic xanthoastrocytoma and right cerebellar diffuse astrocytoma.
| Pleomorphic xanthoastrocytoma, left frontal lobe | ||
| Variant | Reference transcript | Classification |
| CDKN2A/B homozygous deletion | N/A | Pathogenic |
| BRAF p.V600E | NM_004333 | Pathogenic |
| CDH1 p.V55G | NM_004360 | VUS |
| SPTA1 p.T1953I | NM_003126 | VUS |
| Diffuse astrocytoma, right cerebellum | ||
| Variant | Reference transcript | Classification |
| CDKN2A/B homozygous deletion | N/A | Pathogenic |
| ATRX p.G1152fs | NM_000489 | Pathogenic |
| NF1 p.L1246fs | NM_001042492 | Pathogenic |
| NF1 p.W1685fs | NM_001042492 | Pathogenic |
| PTPN11 p.T73I | NM_002834 | Pathogenic |
fs = frameshift; N/A = not applicable; VUS = variant of unknown significance.
The presence of germline CDKN2A/B inactivation together with the presence of multiple anatomically, histologically, and genetically distinct astrocytic neoplasms, both with accompanying somatic loss of the remaining CDKN2A/B alleles, is diagnostic of familial melanoma-astrocytoma syndrome. This genetic diagnosis is further supported by the significant family history of both glioblastoma and melanoma in the paternal lineage, suggesting that this is likely an inherited rather than de novo germline alteration in this patient. The patient and his family have been referred to a medical geneticist with expertise in cancer risk evaluation; however, no genetic evaluation of any family members has yet been performed. Given his newly-identified tumor predisposition syndrome, the patient has initiated the recommended cancer screening, including annual dermatologic, ophthalmologic, and dental examinations to monitor for the development of cutaneous, ocular, and mucosal melanoma. He will continue to undergo regular surveillance imaging of the brain. Additionally, imaging of the chest, abdomen, and pelvis was performed to rule out the presence of visceral malignancy, which was unrevealing except for the presence of the paraspinal peripheral nerve sheath tumor that remains asymptomatic and is being radiographically monitored at present.
Discussion
This is the first report, to our knowledge, to demonstrate multiple distinct histologic subtypes of astrocytoma in a patient with familial melanoma-astrocytoma syndrome. The histologic features of the astrocytomas that arise as part of this syndrome are not well described, and pleomorphic xanthoastrocytoma has not been previously described in association with familial melanoma-astrocytoma syndrome. We document the presence of two anatomically-distinct neoplasms, one with histologic features diagnostic of pleomorphic xanthoastrocytoma and one with histologic features diagnostic of diffuse astrocytoma. The pathologic findings in this patient thus expand the histologic diversity of astrocytic neoplasms that can be seen as part of this syndrome, which can range from pleomorphic xanthoastrocytoma to diffuse or anaplastic astrocytoma to glioblastoma. We are not aware of any reports describing tumors histologically resembling other glial neoplasms, including pilocytic astrocytoma, subependymal giant cell astrocytoma, ganglioglioma, or oligodendroglioma in patients with this syndrome.
Additionally, we describe for the first time the cooperating somatic alterations that drive gliomagenesis in those astrocytomas arising as part of familial melanoma-astrocytoma syndrome in addition to germline inactivation of CDKN2A. In the pleomorphic xanthoastrocytoma, we show that the germline CDKN2A/B deletion was accompanied by somatic loss of the remaining CDKN2A/B alleles as well as the activating p.V600E hotspot mutation in BRAF. Together, CDKN2A deletion and BRAF p.V600E are the two characteristic somatic alterations seen in the majority of sporadic pleomorphic xanthoastrocytomas [19]. In the diffuse astrocytoma, we show that the germline CDKN2A/B deletion was accompanied by somatic loss of the remaining CDKN2A/B alleles as well as an activating hotspot mutation in PTPN11 and inactivating frameshift mutations in the ATRX and NF1 tumor suppressor genes. ATRX mutations are present in greater than 90% of diffuse lower-grade astrocytomas and are frequently accompanied by TP53 and IDH1 or IDH2 mutations [20]. The diffuse astrocytoma in this patient lacked alterations in TP53, IDH1, and IDH2. Instead, the additional cooperating mutations involved PTPN11 and NF1. Amongst sporadic gliomas, PTPN11 mutations are rare and most commonly found in pilocytic astrocytomas, whereas NF1 mutations are very common in primary glioblastomas but not typically seen in diffuse lower-grade astrocytomas [19, 20, 21]. Somatic CDKN2A deletions are common in both diffuse lower-grade astrocytomas and primary glioblastomas [20, 21]. Thus, while the genetic alterations seen in this patient’s pleomorphic xanthoastrocytoma are typical for this tumor type, the genetic alterations seen in the diffuse astrocytoma are somewhat unusual and make prognostic classification based on the molecular profile challenging [22].
Regarding management, optimal treatment strategies for patients with familial melanoma-astrocytoma syndrome are unknown. While specific recommendations regarding disease surveillance have not been established given the rarity of this syndrome, these patients require regular brain imaging to monitor for astrocytoma development; dermatologic and ophthalmologic evaluation to monitor for cutaneous and ocular melanoma development; dental exam to monitor for oropharyngeal dysplasia and mucosal melanoma; and body imaging to monitor for development of visceral malignancies, in particular pancreatic carcinoma. Additionally, genetic counseling for the patient and their families is essential.
In summary, we present a rare case of a patient with familial melanoma-astrocytoma syndrome who was found to have synchronous development of two anatomically, histologically, and genetically distinct astrocytomas as well as a presumed peripheral nerve sheath tumor. Our report highlights the histologic spectrum of astrocytic neoplasms that can be seen as part of this syndrome, as well as defining the cooperating genetic alterations that drive these astrocytomas.
Acknowledgment
D.A.S. is supported by the NIH Director’s Early Independence Award (DP5 OD021403).
Conflict of interest
The authors have no conflicts of interest related to this case report to disclose.
References
- 1. Wrensch M Lee M Miike R Newman B Barger G Davis R Wiencke J Neuhaus J Familial and personal medical history of cancer and nervous system conditions among adults with glioma and controls. Am J Epidemiol. 1997; 145: 581–593. [DOI] [PubMed] [Google Scholar]
- 2. Malmer B Grönberg H Bergenheim AT Lenner P Henriksson R Familial aggregation of astrocytoma in northern Sweden: an epidemiological cohort study. Int J Cancer. 1999; 81: 366–370. [DOI] [PubMed] [Google Scholar]
- 3. Kyritsis AP Bondy ML Rao JS Sioka C Inherited predisposition to glioma. Neuro-oncol. 2010; 12: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bainbridge MN Armstrong GN Gramatges MM Bertuch AA Jhangiani SN Doddapaneni H Lewis L Tombrello J Tsavachidis S Liu Y Jalali A Plon SE Lau CC Parsons DW Claus EB Barnholtz-Sloan J Il’yasova D Schildkraut J Ali-Osman F Sadetzki S Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2014; 107: 384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Malmer B Adatto P Armstrong G Barnholtz-Sloan J Bernstein JL Claus E Davis F Houlston R Il’yasova D Jenkins R Johansen C Lai R Lau C McCarthy B Nielsen H Olson SH Sadetzki S Shete S Wiklund F Wrensch M GLIOGENE – an international consortium to understand familial glioma. Cancer Epidemiol Biomarkers Prev. 2007; 16: 1730–1734. [DOI] [PubMed] [Google Scholar]
- 6. Kaufman DK Kimmel DW Parisi JE Michels VV A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology. 1993; 43: 1728–1731. [DOI] [PubMed] [Google Scholar]
- 7. Azizi E Friedman J Pavlotsky F Iscovich J Bornstein A Shafir R Trau H Brenner H Nass D Familial cutaneous malignant melanoma and tumors of the nervous system. A hereditary cancer syndrome. Cancer. 1995; 76: 1571–1578. [DOI] [PubMed] [Google Scholar]
- 8. Bahuau M Vidaud D Kujas M Palangié A Assouline B Chaignaud-Lebreton M Prieur M Vidaud M Harpey JP Lafourcade J Caille B Familial aggregation of malignant melanoma/dysplastic naevi and tumours of the nervous system: an original syndrome of tumour proneness. Ann Genet. 1997; 40: 78–91. [PubMed] [Google Scholar]
- 9. Bahuau M Vidaud D Jenkins RB Bièche I Kimmel DW Assouline B Smith JS Alderete B Cayuela JM Harpey JP Caille B Vidaud M Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998; 58: 2298–2303. [PubMed] [Google Scholar]
- 10. Kamb A Shattuck-Eidens D Eeles R Liu Q Gruis NA Ding W Hussey C Tran T Miki Y Weaver-Feldhaus J McClure M Aitken JF Anderson DE Bergman W Frants R Goldgar DE Green A MacLennan R Martin NG Meyer LJ Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat Genet. 1994; 8: 23–26. [DOI] [PubMed] [Google Scholar]
- 11. Hayward NK Genetics of melanoma predisposition. Oncogene. 2003; 22: 3053–3062. [DOI] [PubMed] [Google Scholar]
- 12. Tachibana I Smith JS Sato K Hosek SM Kimmel DW Jenkins RB Investigation of germline PTEN, p53, p16(INK4A)/p14(ARF), and CDK4 alterations in familial glioma. Am J Med Genet. 2000; 92: 136–141. [DOI] [PubMed] [Google Scholar]
- 13. Petronzelli F Sollima D Coppola G Martini-Neri ME Neri G Genuardi M CDKN2A germline splicing mutation affecting both p16(ink4) and p14(arf) RNA processing in a melanoma/neurofibroma kindred. Genes Chromosomes Cancer. 2001; 31: 398–401. [DOI] [PubMed] [Google Scholar]
- 14. Goldstein AM Chan M Harland M Gillanders EM Hayward NK Avril MF Azizi E Bianchi-Scarra G Bishop DT Bressac-de Paillerets B Bruno W Calista D Cannon Albright LA Demenais F Elder DE Ghiorzo P Gruis NA Hansson J Hogg D Holland EA High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006; 66: 9818–9828. [DOI] [PubMed] [Google Scholar]
- 15. Pasmant E Laurendeau I Héron D Vidaud M Vidaud D Bièche I Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007; 67: 3963–3969. [DOI] [PubMed] [Google Scholar]
- 16. Frigerio S Disciglio V Manoukian S Peissel B Della Torre G Maurichi A Collini P Pasini B Gotti G Ferrari A Rivoltini L Massimino M Rodolfo M A large de novo 9p21.3 deletion in a girl affected by astrocytoma and multiple melanoma. BMC Med Genet. 2014; 15: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baker MJ Goldstein AM Gordon PL Harbaugh KS Mackley HB Glantz MJ Drabick JJ An interstitial deletion within 9p21.3 and extending beyond CDKN2A predisposes to melanoma, neural system tumours and possible haematological malignancies. J Med Genet. 2016; 53: 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kline CN Joseph NM Grenert JP van Ziffle J Talevich E Onodera C Aboian M Cha S Raleigh DR Braunstein S Torkildson J Samuel D Bloomer M Campomanes AGA Banerjee A Butowski N Raffel C Tihan T Bollen AW Phillips JJ Targeted next-generation sequencing of pediatric neuro-oncology patients improves diagnosis, identifies pathogenic germline mutations, and directs targeted therapy. Neuro-oncol. 2017; 19: 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang J Wu G Miller CP Tatevossian RG Dalton JD Tang B Orisme W Punchihewa C Parker M Qaddoumi I Boop FA Lu C Kandoth C Ding L Lee R Huether R Chen X Hedlund E Nagahawatte P Rusch M Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet. 2013; 45: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brat DJ Verhaak RG Aldape KD Yung WK Salama SR Cooper LA Rheinbay E Miller CR Vitucci M Morozova O Robertson AG Noushmehr H Laird PW Cherniack AD Akbani R Huse JT Ciriello G Poisson LM Barnholtz-Sloan JS Berger MS Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015; 372: 2481–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brennan CW Verhaak RG McKenna A Campos B Noushmehr H Salama SR Zheng S Chakravarty D Sanborn JZ Berman SH Beroukhim R Bernard B Wu CJ Genovese G Shmulevich I Barnholtz-Sloan J Zou L Vegesna R Shukla SA Ciriello G The somatic genomic landscape of glioblastoma. Cell. 2013; 155: 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eckel-Passow JE Lachance DH Molinaro AM Walsh KM Decker PA Sicotte H Pekmezci M Rice T Kosel ML Smirnov IV Sarkar G Caron AA Kollmeyer TM Praska CE Chada AR Halder C Hansen HM McCoy LS Bracci PM Marshall R Glioma groups based on 1p/19q, IDH, and TERT promoter mutations in tumors. N Engl J Med. 2015; 372: 2499–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]