

Signal transducer and activator of transcription 3 (STAT3) signaling is often activated during cancer development and has been linked to different hallmarks of cancer.1 A study conducted by Caetano et al. has showed the therapeutic potential of inhibiting the STAT3 pathway by targeting interleukin-6 (IL-6), a potent activator of STAT3, using anti-IL-6 antibodies in the CCSPCre; LSL-KrasG12D mouse model of lung adenocarcinoma.2 After 8 weeks of treatment there was a significant reduction in the number of adenomas and carcinomas. Anti-IL-6 treated tumors exhibited a reduction in cell proliferation, angiogenesis and altered immune infiltration including a reduction in the overall number of macrophages and regulatory T cells with a concomitant increase in cytotoxic CD8+ T cells and T helper (Th1) cells. Interestingly, treatment of lung cancer cells in vitro with anti-IL-6 antibodies did not result in decreased proliferation or increased cell death, suggesting that the therapeutic effect of IL-6 blockade is not cell autonomous but through reprogramming of the tumor microenvironment. Increased IL-6 production in the tumor microenvironment can result in increased serum levels of IL-6, thus activating STAT3 in distant organs. Indeed, Masri et al. have shown that in the LSL-KrasG12D; p53Flox/Flox (KP) conditional mouse model of lung adenocarcinoma, the serum levels of IL-6 were elevated resulting in altered hepatic circadian homeostasis.3 Livers of tumor bearing mice showed altered metabolic parameters, without disturbing the core circadian clock. Metabolic analyses of the livers revealed reduced levels of oscillating lipids, NAD+, ATP and acetyl-coA. The sterol regulatory element binding protein (SREBP) pathway is known to regulate lipid metabolism in the liver in a circadian manner. This pathway is inhibited by the energy sensor AMPK. Livers from tumor-bearing mice had elevated AMPK levels, reduced nuclear SREBP1c protein, and an increase in the levels of Il6ra, Il1r1, Tnfrsf1b, Il17ra, Stat3 and phospho-Stat3. Socs3, a prototypic Stat3 target, has been shown to regulate insulin signaling in liver and adipose tissues. These livers showed reduced insulin-dependent phosphorylation of AKT and decreased IRS-1 levels. Analysis of the liver metabolome after treating KP mice with anti-IL-6 antibodies could provide further insight into the effects of systemic IL-6.
Recent studies aimed to understand mechanisms involved in cancer dedifferentiation and metastasis identified multiple non-canonical activators of STAT3 (Fig. 1). In an attempt to uncover novel regulators of metastasis, Chuang et al. infected KP mice with barcoded lentiviral-Cre vectors to match primary tumors with their metastases.4 When comparing the transcriptional profile of non-metastatic primary tumors and metastases, the authors identified a glycosylphosphatidylinositol-linked cell surface protein, Cd109, as a potential mediator of metastasis. Silencing Cd109 reduced the phosphorylation of Stat3 and hence impaired the metastatic potential of the lung cancer cells in vivo. Silencing Cd109 or Stat3 did not decrease the ability of these cells to grow in vitro or to form subcutaneous tumors, highlighting the Cd109-Jak-Stat3 pathway as specific driver of the metastatic process. Loss of epithelial differentiation of lung adenocarcinoma is an indicator of highly aggressive disease and is associated with metastases. Our study showed that receptor-interacting serine/threonine protein kinase (Rip4) altered tumor differentiation in KP mice.5 Rip4 knockdown in vivo resulted in the development of tumors that progressed toward poorly differentiated adenocarcinoma. In contrast, overexpression of RIP4 in lung cancer cell lines reduced their invasiveness in vitro and the number of lung colonies after tail vein injection. Analysis of the transcriptional profile of tumors expressing shRNA against Rip4 revealed an activation of genes related to extracellular matrix remodeling and Stat3 signaling. Our mechanistic analyses highlighted a bimodal inhibitory effect of Rip4 on Stat3: first, RIP4 knockout cells showed an accelerated STAT3 nuclear import upon stimulation with IL-6. Second, overexpression of RIP4 promoted the nuclear export of STAT3. This suggests that RIP4 acts primarily by altering the nuclear localization of STAT3. Overexpression of RIP4 also reduced the levels of lysyl oxidase (LOX), a STAT3 target that is involved in collagen cross-linking, whereas tumors with sustained Rip4 knockdown showed elevated levels of Lox and increased collagen deposition. Comparatively, another study aiming to identify factors responsible for multi-organ metastasis of breast cancer cells demonstrated that collagen-mediated activation of STAT3 is crucial for metastasis.6 In that study, Gao et al. showed that tetraspanin TM4SF1 promotes the formation of clusters of discoidin domain receptor (DDR)-1, a collagen receptor tyrosine kinase, upon binding to collagen I6. This further activated JAK2 in a PKCα-dependent manner resulting in STAT3 activation. Genetic or pharmacologic interference with DDR1, TM4SF1, JAK2, PKCα or STAT3 significantly reduced the metastatic potential of these cells. Taken together, these and other studies reveal that STAT3 activation is a central feature in the development of highly aggressive cancers, where different signaling axes can converge on STAT3 stimulation. Hence, targeting the IL-6 receptor or ligand might not be sufficient for efficient STAT3 blockade. Future studies aimed to discover new compounds that prevent the phosphorylation, dimerization or nuclear localization of STAT3 could prove to be therapeutically useful against multiple malignancies.
Figure 1.
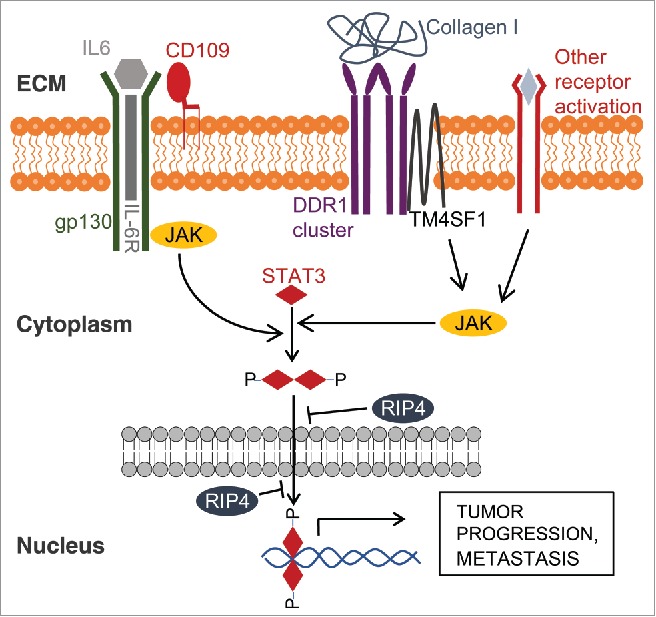
Regulation of STAT3 activation in tumor cells: On the extracellular matrix (ECM) side, multiple ligands bind and engage different membrane receptors. This results in activation of JAK family members, which phosphorylate STAT3 resulting in its dimerization, nuclear translocation and regulation of target gene transcription. RIP4 reduces nuclear STAT3 levels and hence inhibits STAT3-mediated responses, counteracting tumor progression.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Wake MS, Watson CJ. STAT3 the oncogene - still eluding therapy? FEBS J. 2015;282:2600-11. doi: 10.1111/febs.13285. PMID:25825152 [DOI] [PubMed] [Google Scholar]
- [2].Caetano MS, Zhang H, Cumpian AM, Gong L, Unver N, Ostrin EJ, Daliri S, Chang SH, Ochoa CE, Hanash S, Behrens C, Wistuba II, Sternberg C, Kadara H, Ferreira CG, Watowich SS, Moghaddam SJ.. IL6 blockade reprograms the lung tumor microenvironment to limit the development and progression of K-ras-mutant lung cancer. Cancer Res. 2016;76:3189-99. doi: 10.1158/0008-5472.CAN-15-2840. PMID:27197187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Masri S, Papagiannakopoulos T, Kinouchi K, Liu Y, Cervantes M, Baldi P, Jacks T, Sassone-Corsi P. Lung adenocarcinoma distally rewires hepatic circadian homeostasis. Cell. 2016;165:896-909. doi: 10.1016/j.cell.2016.04.039. PMID:27153497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chuang CH, Greenside PG, Rogers ZN, Brady JJ, Yang D, Ma RK, Caswell DR, Chiou SH, Winters AF, Grüner BM, Ramaswami G, Spencley AL, Kopecky KE, Sayles LC, Sweet-Cordero EA, Li JB, Kundaje A, Winslow MM.. Molecular definition of a metastatic lung cancer state reveals a targetable CD109-Janus kinase-Stat axis. Nat Med. 2017;23:291-300. doi: 10.1038/nm.4285. PMID:28191885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kopparam J, Chiffelle J, Angelino P, Piersigilli A, Zangger N, Delorenzi M, Meylan E. RIP4 inhibits STAT3 signaling to sustain lung adenocarcinoma differentiation. Cell Death Differ. 2017; 24(10):1761-1771. doi: 10.1038/cdd.2017.81. PMID:28574510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gao H, Chakraborty G, Zhang Z, Akalay I, Gadiya M, Gao Y, Sinha S, Hu J, Jiang C, Akram M, Brogi E, Leitinger B, Giancotti FG.. Multi-organ site metastatic reactivation mediated by non-canonical discoidin domain receptor 1 signaling. Cell. 2016;166:47-62. doi: 10.1016/j.cell.2016.06.009. PMID:27368100 [DOI] [PMC free article] [PubMed] [Google Scholar]