

ABSTRACT
Anti-cancer small molecule ONC201 upregulates the integrated stress response (ISR) and acts as a dual inactivator of Akt/ERK, leading to TRAIL gene activation. ONC201 is under investigation in multiple clinical trials to treat patients with cancer. Given the unique imipridone core chemical structure of ONC201, we synthesized a series of analogs to identify additional compounds with distinct therapeutic properties. Several imipridones with a broad range of in vitro potencies were identified in an exploration of chemical derivatives. Based on in vitro potency in human cancer cell lines and lack of toxicity to normal human fibroblasts, imipridones ONC206 and ONC212 were prioritized for further study. Both analogs inhibited colony formation, and induced apoptosis and downstream signaling that involves the integrated stress response and Akt/ERK, similar to ONC201. Compared to ONC201, ONC206 demonstrated improved inhibition of cell migration while ONC212 exhibited rapid kinetics of activity. ONC212 was further tested in >1000 human cancer cell lines in vitro and evaluated for safety and anti-tumor efficacy in vivo. ONC212 exhibited broad-spectrum efficacy at nanomolar concentrations across solid tumors and hematological malignancies. Skin cancer emerged as a tumor type with improved efficacy relative to ONC201. Orally administered ONC212 displayed potent anti-tumor effects in vivo, a broad therapeutic window and a favorable PK profile. ONC212 was efficacious in vivo in BRAF V600E melanoma models that are less sensitive to ONC201. Based on these findings, ONC212 warrants further development as a drug candidate. It is clear that therapeutic utility extends beyond ONC201 to include additional imipridones.
KEYWORDS: ATF4, cancer therapy, CHOP, DR5, imipridone, ONC201, ONC206, ONC212, TRAIL
Introduction
ONC201, an orally active first-in-class imipridone small molecule compound that upregulates the endogenous tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and the cell surface receptor death receptor (DR5) genes is in clinical trials for various malignancies.1-3 When activated by the ligand TRAIL, DR5 triggers the extrinsic cell death pathway that selectively induces apoptosis in a variety of tumor and transformed cells, including cancer stem cells, without affecting normal cells.4,5
The unique ability of ONC201 to induced TRAIL-based signaling to induce apoptosis in cancer cells and not normal cells leads to a wide therapeutic index and favorable characteristics as an anti-cancer therapeutic.1-3,6,7 The TRAIL pathway is a powerful effector cytokine in the innate host immune response that suppresses tumor development, progression and metastasis. Early results from a Phase I clinical trial in advanced solid tumors demonstrated that ONC201 has a therapeutic PK profile, exceptional safety, induction of pharmacodynamics (PD) markers, and preliminary evidence of efficacy in various types of cancers.8,9
Given the unique imipridone core,10 we sought to explore chemical derivates of ONC201 to identify compounds with distinct therapeutic properties. We synthesized ONC201 analogs to identify compounds with distinct therapeutic properties that target ONC201-resistant tumor types or possess distinct signaling properties. We present here an exploration of ONC201 chemical derivatives, including screening in colorectal cancer and normal cell lines, selection and comparisons of lead candidates to ONC201, and in vivo evaluations of lead candidates.
Results
Generation of ONC201 chemical derivatives yielded several imipridones with potent anti-cancer activity
Based on the importance of the imidazopyridopyrimidone core structure of ONC201, now referred to as the imipridone core, we focused our exploration of chemical derivatives on manipulating substituents on the peripheral benzyl moieties while preserving the imipridone structure (Fig. 1a-b). Each analog was evaluated in a primary cell viability screen in CRC cell line HCT116-p53−/− and normal cell line MRC5 (Fig. 1c). Chemical changes to the R1 group produced compounds with a wide range of potency, many of which were much more potent than ONC201. Halogens substituted within the benzyl R1 group replacing the 2-methylbenzyl group, such as with ONC212 (4-CF3-benzyl group at R1), were more potent than ONC201, resulting in some cases in up to a 1000-fold reduction in GI50 on tumor cells without increased cytotoxicity toward normal cells. Other analogs with halide substituents in the R1 group such as ONC206 (2,4-diF-benzyl) and ONC219 (2,4-diCl−benzyl) were also potent in the colorectal cancer (CRC) cell line screen, and had a large in vitro therapeutic window. This suggests that replacing the R1 group with halide-benzyl groups increases potency. The widest separation between toxicity toward the tumor cells versus the normal cells was observed for compounds in which a group at the 2-position of the benzyl substituent was absent, such as ONC212, ONC213 (3,4-diF-benzyl) and ONC211 (3,4-diCl−benzyl). Variants at R2 were also prepared, with ONC207 (-H) having no anti-cancer activity while potency was restored with manipulation of the R1 group in ONC221 (R2-H; R1-4- CF3-benzyl group).
Figure 1.
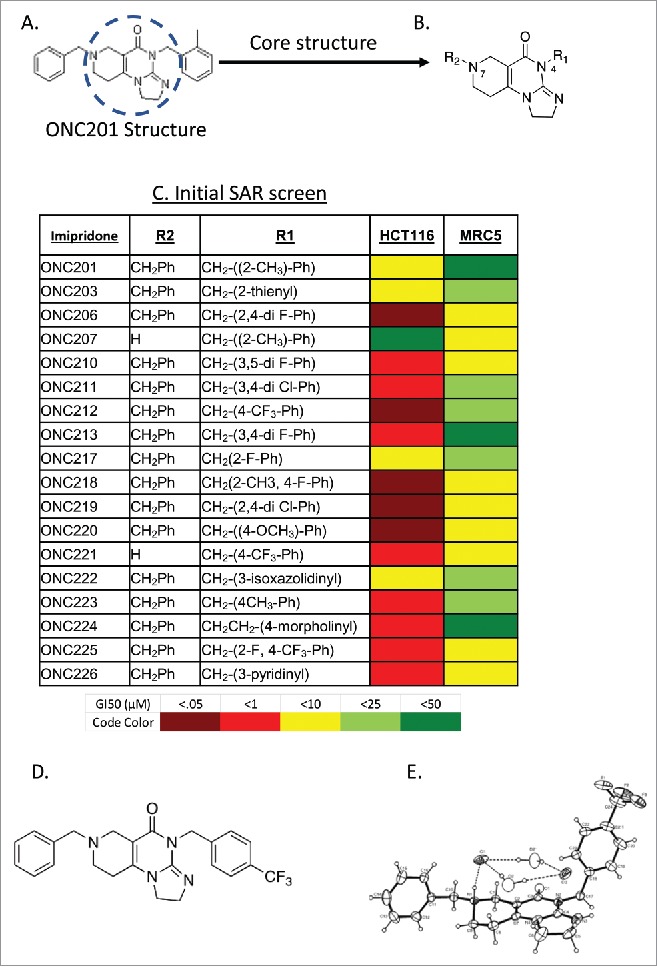
Exploration of chemical derivatives of ONC201. (A) Structure of ONC201. (B) Core pharmacophore of ONC201 used for exploration of chemical derivatives. (C) ONC201 analogs including ONC identifier, R1 and R2 groups, and GI50 in HCT116 cells. N = 3; GI50 collected after 72 hours and calculated by PRISM. (D) Chemical structure and (E) ORTEP drawing of ONC212 crystals from X-ray data. X-ray crystallography was determined as described in methods.
ONC212, a trifluoromethylbenzyl imipridone (Fig. 1d-e), and ONC206, a difluorobenzyl imipridone, were initially selected based on their GI50 in HCT116 cells compared to their effects on normal cells (an approximation of in vitro therapeutic window). The potencies of both analogs were evaluated in cancer cell lines across 10 tissue types and in an additional four normal cell lines (Fig. 1c). These experiments demonstrated that ONC212 and ONC206 have nanomolar activity that is consistently more potent than ONC201 across several tumor types (Fig. 2a-b).
Figure 2.
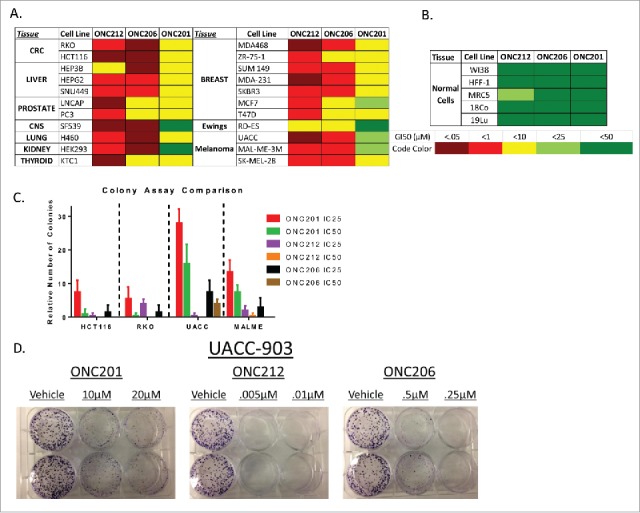
In vitro analysis of select analogs among several cell lines. A) Screen of GI50 values performed in lab of ONC212 and ONC206 compared to ONC201. B) GI50 in normal cell lines to determine toxicity in vitro. C) Colony assay of ONC201, ONC212, and ONC206 in select tissue types D) Representative colony assay in UACC-903. N = 3 for all experiments. ONC201: IC50 10 μM, IC25 2.5 μM; ONC212: IC50 0.01 μM, IC25 0.005 μM; ONC206: IC50 0.05 μM; IC25 0.01 μM) IC50 collected after 72 hours and calculated by PRISM. Colony assays where monitored until colonies formed.
Both compounds also demonstrated the ability to inhibit colony growth of both colorectal and melanoma cancer cell lines. The results prompted further investigation of these analogs (Fig. 2c-d).
Lead ONC201 analogs ONC212 and ONC206 engage the ISR and TRAIL pathway leading to tumor growth arrest and cell death
ONC212 and ONC206 activated similar signaling pathways as ONC201; namely, activation of the integrated stress response pathway leading to death receptor 5 (DR5) upregulation, TRAIL upregulation (Fig. 3a-c). Further, ONC212 displayed similar suppression of p-ERK and p-Akt as ONC201 (Fig. 3d). However, ONC212 demonstrated distinct kinetics – with upregulation of CHOP and DR5 mRNA occurring at 12 hours and an increase in surface TRAIL expression occurring at 24 hours. This contrasts with ONC201 and ONC206-mediated induction of CHOP and DR5 mRNA at 24 hours, and TRAIL expression after 72 hours of treatment (Fig. 3a-c, Fig. S1a-b). Western blot analyses also showed an earlier inhibition of ERK and Akt phosphorylation with ONC212 treatment as compared to ONC201 in HCT116 cells (Fig. 3d).
Figure 3.

ONC212 and ONC206 downstream mechanism of action is similar to ONC201. A) CHOP and B) DR5 Gene expression data from ONC201, ONC206, and ONC212 treated in HCT116 cells over time relative to GAPDH and Vehicle. C) ONC201, ONC212, and ONC206 TRAIL surface expression analysis at indicated time points in HCT116 and TRAIL-resistant HCT116-Bax−/−. D) Western blot analysis results of HCT116 cells treated with ONC201 and ONC212 treated overtime. E) Quantitation of western blots (ONC201: 10 μM; ONC212: 0.01 μM; ONC206: 0.05 μM). N = 3; Western blot representative of 3 biological replicates.
We previously described ONC201's ability to induce cell cycle arrest.1,2 Similarly, ONC212 and ONC206 significantly induced Sub-G1 apoptotic cells and/or cell cycle arrest. Interestingly, ONC212 and ONC206 did not induce cell cycle arrest in a colorectal cell line with acquired ONC201-resistance (RKO-ONC201 resistant2), suggesting cross-resistance between the compounds. (compare Fig. 4a-c, Fig. S1c-d).
Figure 4.
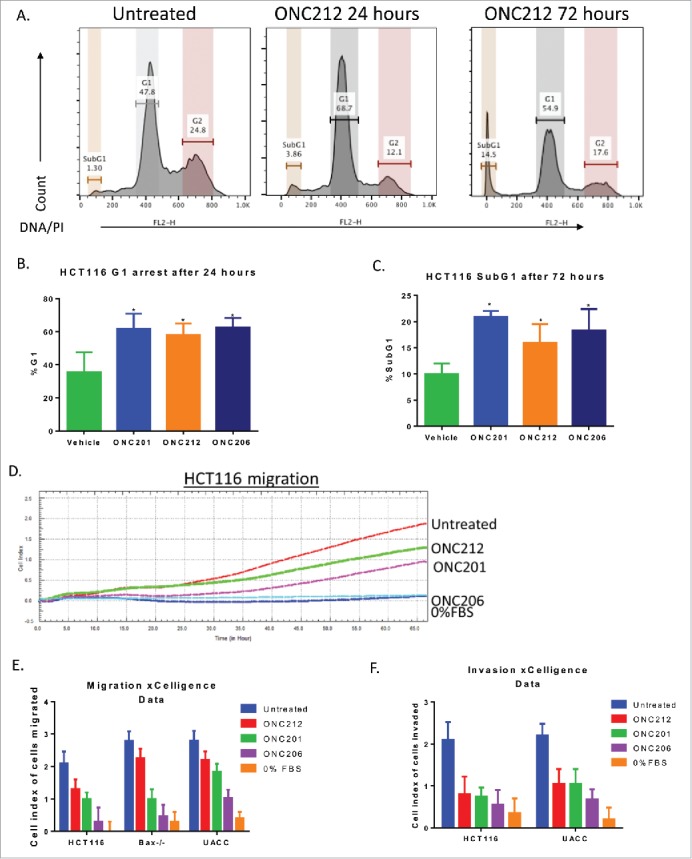
ONC212 and ONC06 induce cytotoxicity and inhibit migration and invasion similarly to ONC201. A) Representative cell cycle images in HCT116 cells. B) G1 and C) subG1 arrest in HCT116 cells at selected time points of ONC201 (10 μM), ONC212 (0.01 μM), and ONC206 (0.05 μM). D) Representative xCelligence migration assay. E) Cumulative Migration and F) Invasion using xCelligence software in select cell lines at GI50 values over 48 hours. ONC201: 10 μM; ONC212: 0.01 μM; ONC206: 0.05 μM. N = 3. Migration and cell cycle representative image of 3 biological replicates.
Imipridone ONC201 and analogs inhibit tumor cell migration and invasion
Prior studies have shown that TRAIL can inhibit metastasis.11,12 We therefore examined the ability of ONC201 and selected analogs to inhibit cell migration and invasion. Boyden-chamber xCelligence migration assays showed that ONC201 and ONC206 significantly inhibited tumor cell migration and invasion in vitro. In contrast, ONC212 inhibited only invasion. Furthermore, ONC201 and ONC206 inhibited migration of ONC201- and TRAIL-resistant HCT116 Bax−/− cells without inducing cell death or inhibiting cell proliferation (Fig. 4d-f).
Imipridone ONC212 is orally active and possesses favorable safety and PK profiles in mice
In vivo toxicity assessment experiments showed that ONC212 was well tolerated up to 250 mg/kg when administered by intra-peritoneal injection (I.P.) or orally administered through a gavage (P.O.). Oral administration yielded similar results, with no gross observations of toxicity noted until the maximum tolerated dose (MTD) was reached at 250 mg/kg through both I.P. and P.O. administration. Adverse events at the maximum tolerated dose of 250 mg/kg were alleviated within 24 hours included noticeable gate issues and perfuse sweating. These toxicities increased in severity when ONC212 was administered up to a lethal dose of 300 mg/kg (I.P. and P.O). 300 mg/kg of ONC212 by both methods of administration caused splenic damage and elevated liver enzymes (Fig. S2a-b). Other organs remained unaffected by a 300 mg/kg dose (Fig. S2c-d). The MTD was identified by taking into account the ability of mice to recover quickly from these adverse events and no significant concern arose from the toxicology studies.
A pharmacokinetic (PK) analysis using mass spectrometry was performed at a single oral dose of 125 mg/kg (P.O.) in C57/BL6 mice. ONC212 had a slightly shorter half-life than ONC201, with a clearance from the blood at 12 hours (Fig. 5a), T1/2 of 4.3 hours, and Cmax of 1.4 µg/mL (Fig. 5a, Fig. S3a).3 Despite the rapid PK characteristics of ONC212, its high MTD and relatively rapid and potent in vitro anti-tumor activity prompted in vivo efficacy studies.
Figure 5.
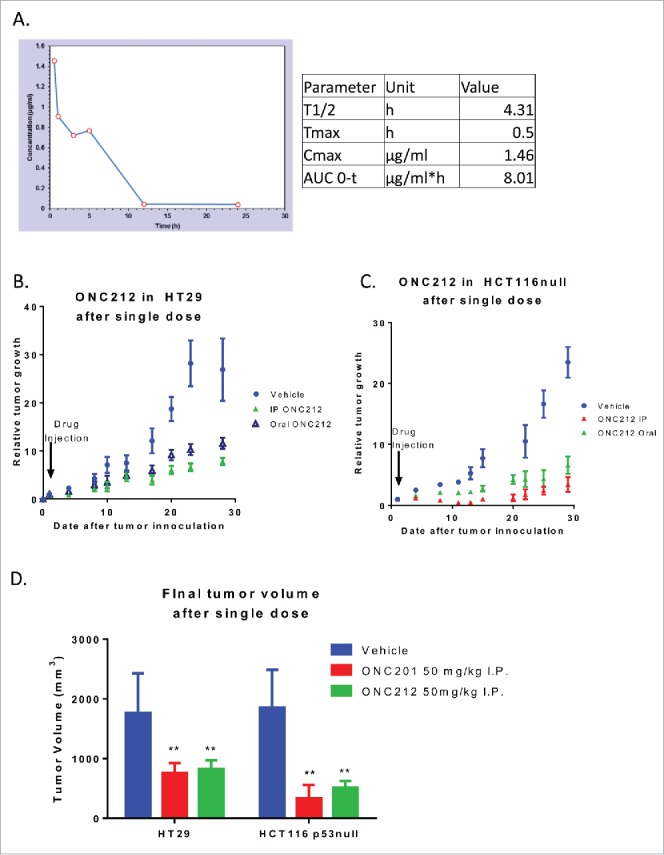
Pharmacokinetics and efficacy of oral ONC212. A) Pharmacokinetic profile through mass spectrometry at a dose of 125 mg/kg with blood extracted into a MeOH pellet through oral administration n = 3. Efficacy of ONC212 at 50 mg/kg in athymic nude mice. Comparison of IP vs oral in B) HT29 and C) HCT116 xenografts overtime D) Final tumor volume comparison of ONC212 in two CRC xenografts. HT29 after 30 days. HCT116 p53-null after 6 weeks. PK n = 3; efficacy n = 6. **p<.01
To determine ONC212's oral efficacy, the impact of oral and intra-peritoneal administration of ONC212 on tumor xenograft growth was evaluated. ONC212 significantly inhibited tumor growth of both HT29 and HCT116p53−/− xenografts, regardless of the route of administration. This suggests that ONC212 has favorable activity when administered via oral gavage (Fig. 5b, Fig. S3b). Furthermore, despite its shorter half-life vs. ONC201, ONC212's efficacy at 30 days after administration was comparable to ONC201. This suggests that ONC212 has a prolonged pharmacodynamic effect despite systemic clearance (Fig. 5d).
ONC212 has broad spectrum anti-cancer efficacy with improved efficacy in skin cancer relative to ONC201
Given that ONC212 was well tolerated in vivo, was orally active, and demonstrated accelerated kinetics of signaling in vitro, it was selected for subsequent efficacy testing in vitro and in vivo. The Genomic of Drug Sensitivity in Cancer (GDSC) collection of cell lines (1,068 human cancer cell lines) was used to test the in vitro efficacy of ONC212. Most solid tumors and hematological malignancies were sensitive to ONC212 in the low nanomolar range (Fig. 2e). Skin cancer was one of the most ONC212-sensitive solid tumor types. Furthermore, it was more sensitive to ONC212 than to ONC201 (Fig. 6a-c). Among the 53 skin cancer cell lines tested, 51 were melanoma cell lines. ONC212 reduced cell viability independent of BRAF V600E mutation status (Fig. 6d-e).
Figure 6.
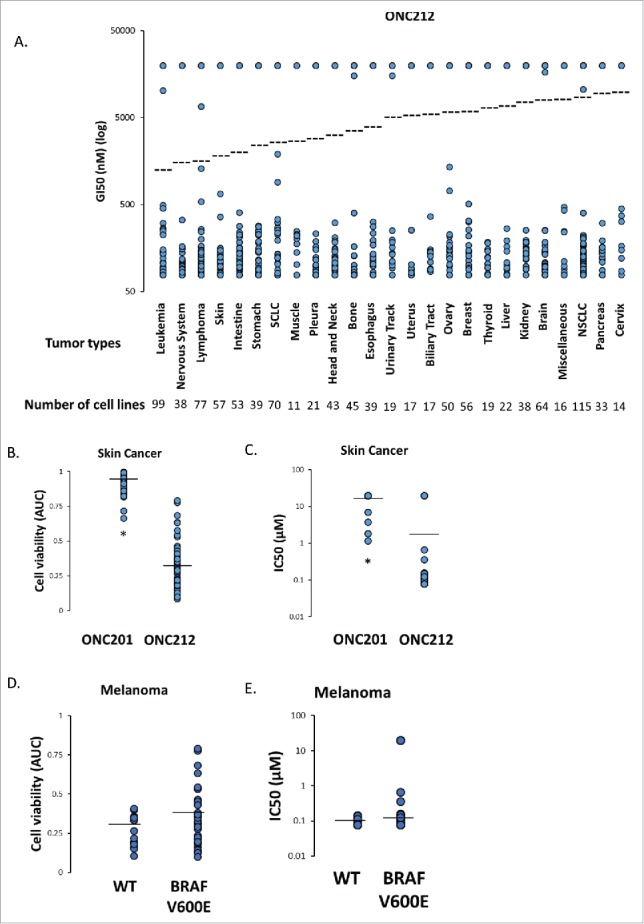
Genomic of Drug Sensitivity in Cancer (GDSC) Screen. A) In vitro sensitivity of 1068 human cancer cell lines to ONC212 (78nM – 20µM, 72h) organized by tumor type. The results are shown as ONC212 GI50 (nM) with representation of all cell lines in each tumor type. Dotted line represents average GI50 for each tumor type. The number of cell lines tested per tumor type are indicated. B) Average cell viability (area under curve, AUC) and C) IC50 with 72 hour ONC201 and ONC212 (0.078-20µM) treatment in a panel of 53 skin cancer cell lines in the GDSC screen. * indicates p < 6.68 × 10–24. D) Average cell viability (area under curve, AUC) and E) IC50 with 72 hour ONC212 (0.078-20µM) treatment in a panel of 15 wild-type (WT) and 32 BRAF V600E melanoma cell lines in the GDSC screen. P-value 0.0595
Oral ONC212 shows potent anti-tumor efficacy in a human melanoma xenograft and hepatocellular model
In vitro experiments showed that both melanoma and hepatocellular carcinoma cell lines were more sensitive to ONC212 than ONC201 (Fig. 2a-e). Similarly, BRAF V600E melanoma MALME, UAC-903 xenografts and hepatocellular carcinoma Hep3B xenografts were more sensitive to weekly dosing of ONC212 than to weekly dosing of ONC201 (Fig. 7a-c, Fig. S4a-b). Immunohistochemical analyses of Ki67 and caspase-3 demonstrated that ONC212 reduced tumor-cell proliferation and induced apoptosis in UACC-903 and MALME xenografts to a greater extent than ONC201 (Fig. 7d, Fig. S4c).
Figure 7.
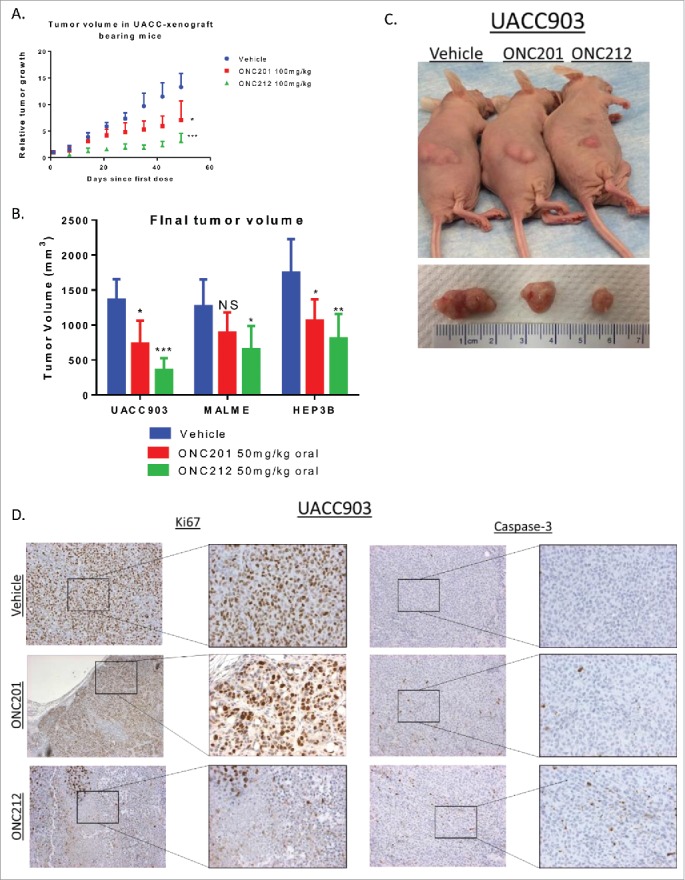
Efficacy of ONC212 in melanoma and liver cancer. A) relative tumor growth and B) Final tumor volume of MALME, UACC-903, and HEP3B xenograft bearing mice treated with Vehicle, ONC201, or ONC212 weekly. Final tumor volumes measured after 5 weeks for UACC903, 5 weeks for MALME, and 30 days for HEP3B as these where end of study. Statistics are compared to vehicle of each tissue type C) Photographic imaging of representative cohorts. D) Ki67 (left) and Caspase-3 (right) staining of selected cohorts at 5 weeks. N = 6 *p<.05; **p<.01 ***p<.005 IHC performed in 3 tumors from each cohort.
Discussion
The chemical structure of ONC201 was modified to produce analogs that preserved the imipridone core structure and possessed potent cytotoxic effects toward tumor cells across various tumor types, including some that are less sensitive to ONC201. Manipulating the R1 benzyl group and introducing halogen substituents yielded analogs with increased potency compared to ONC201. We selected ONC212 and ONC206 for further examination due to their distinct efficacy spectrum and favorable in vitro therapeutic window. We demonstrate that both analogs caused similar TRAIL/DR5 upregulation and activation of the ISR as ONC201, with ONC212 having relatively rapid kinetics vs. ONC201 or ONC206. Although the kinetics of TRAIL/DR5 upregulation are earlier in ONC212 treated cells, apoptosis still occurs at 72 hours, congruent with ONC201 treated cells. Therefore, further assessment of other signaling pathways should be performed to determine if there are unique signaling differences between these imipridones.
Interestingly, ONC206 and ONC201 both inhibited invasion and migration of tumor cells while ONC212 inhibited only invasion. These observations on kinetics of response and invasion suggest that ONC212 possesses distinct anti-cancer properties relative to other imipridones. We selected ONC212 for in vivo evaluation considering the strong potency and differentiated kinetics of signaling. Given the high MTD of ONC212 at 250 mg/kg, the in vivo anti-tumor activity of ONC212 was investigated further. ONC212 exhibited a shorter half-life as compared to ONC201 and potent anti-tumor efficacy in vivo at doses that appeared non-toxic. ONC212 was efficacious at single doses of 50 mg/kg - 100 mg/kg with oral administration, suggesting this route of administration is viable like ONC201. Interestingly, despite its shorter half life, ONC212 possessed similar efficacy in CRC to that of ONC201; demonstrating a potentially prolonged mechanism of action that allows for a sustained anti-tumor effect that warrants further investigation.
Finally, we have observed activity of ONC212 in vivo that shows an advantage in specific tumor types vs. the parent imipridone compound ONC201. Specifically, the more pronounced anti-tumor activity of ONC212 in BRAF V600E melanoma and hepatocellular carcinoma xenografts is an important demonstration of the distinct spectrum of ONC212 anti-cancer activity in cancer. The stronger effect in Ki67+ cells compared to apoptotic cell marker caspase-3 suggests that ONC212's anti-tumor activity may be primarily dependent on its anti-proliferative signaling mechanism. While melanoma and hepatocellular carcinoma emerged as promising ONC212 indications, other solid tumors and hematological malignancies among the >1000 cell line screen remain to be explored for future development.
Materials and methods
Reagents and cell-based assays
All cell lines were obtained from the American Type Culture Collection or discussed previously.3 Cell lines were authenticated by short tandem repeat profiling and were free of mycoplasma contamination. Analogs were synthesized and provided by Provid pharmaceuticals. ONC201 was obtained from Oncoceutics. Cell titer-glo assays (Promega, Madison, WI) were performed following manufacturer's instructions and luminescence imaging was performed on a Xenogen IVIS system (Xenogen, Almadea, CA). Colony assays were performed by seeding 500 cells/well, treating for 3 days at indicated doses, and allowing the colonies to form for approximately 10 days. Media was replenished every 3–4 days. Colonies were gently washed in PBS, fixed with methanol, stained with Coomassie blue, rinsed, and allowed to dry prior to enumeration. GI50 values were determined using PRISM software (Graphpad).
Floating and adherent cells were analyzed on a LSRII cytometer. For surface TRAIL experiments, adherent cells were harvested by brief trypsinization, fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min, incubated overnight with an anti-TRAIL antibody (Abcam, ab2435) at 1:250, washed and incubated with anti-rabbit Alexa Fluor 488 (Invitrogen) for 30 min, and analyzed. Cells were gated on forward and side scatter to eliminate debris and dead cells from the analysis. Surface TRAIL data were expressed as median fluorescence intensity relative to that of control samples unless indicated otherwise. For sub-G1 content and cell cycle profile analyses, all cells were pelleted and ethanol-fixed, followed by staining with propidium iodide (Sigma) in the presence of RNase.
Western blot analysis, qRTPCR, and immunohistochemistry
Western blot analysis was conducted as previously described3 with NuPAGE 4 to 12% bis-tris gel and visualized with ECL Prime Western Blotting Detection Reagent (Amersham) with CytoSMART Live Imaging System (Lonza). For all lysis buffers, fresh protease inhibitor (Roche) was added immediately. All antibodies were purchased from Cell Signaling except anti-DR5 antibody (Abcam ab1675).
RNA was isolated using RNeasy kit (Qiagen) or Quick-RNA Miniprep kit (Zymo Research) according to manufacturers’ instructions. RNA was quantitated using a Nanodrop spectrophotometer. cDNA was synthesized using a SuperScript II RT kit while real-time PCR was performed using a Quantitect SYBR Green PCR mix. Relative amounts of target mRNA were quantitated using the 2ΔΔCt method using GAPDH as internal control. At least three technical replicates per biological replicate were analyzed.
After tissue fixation, the tumor samples were embedded in paraffin and 8 μm sections were cut and mounted on slides. The sections were then processed and analyzed using immunohistochemistry with TRAIL, Ki67, and caspase-3 antibodies similar to the method described previously.3
In vitro tumor migration and invasion assays
Boyden assays were performed using the QCM ECMatrix Cell invasion assay (Millipore, ECM550) and the cultrex cell migration assays (R&D systems, 3465-096-K). Cell migration and invasion were also assessed using the real-time xCelligence system. Invasion assays in the xCelligence system were conducted with Matrigel.13 Wound healing assays were performed with the CytoSelect Wound healing assay kit (Cell BioLabs, CBA-120T).
In vivo studies
All animal experiments were conducted in accordance with the Institutional Animal Care and Use Committee at the Fox Chase Cancer Center. Mouse experiments were free of pathogens including mouse hepatitis virus and c. bovis. For first-in animal studies, ONC212 was introduced in single doses in C57/BL6 mice at 0 mg/kg, 12.5 mg/kg, 25 mg/kg, 50 mg/kg, and 100 mg/kg. Mice were monitored for 48 hours for any symptoms including gait problems, sweating, dizziness, and fatigue. Mice were sacrificed, and toxicology and pathology studies were performed as described below. For maximum tolerated dose studies, ONC212 and ONC206 were administered to C57/BL6 mice in single dose increments starting at 50 mg/kg. Mice were monitored for 5 days and symptoms were monitored before pathology and toxicology analyses were performed. Once an MTD was established, efficacy studies were performed.
For subcutaneous xenograft studies, 6-week-old female athymic nu/nu mice (Taconic Biosciences) were inoculated with 1×106 cells of the HT29-luciferase, HCT116 p53−/−, UACC903, MALME, and HEP3B cell lines in each rear flank, in a 150 μl suspension of 1:1 Matrigel (BD). All subcutaneous tumors were allowed to establish for 1 to 3 weeks after injection. Treatment was initiated once xenografts reached a volume of ∼200 mm3. Mice were monitored every 3 days and tumors volumes were measured using calipers. ONC201 and ONC212 injections were administered subcutaneously at indicated doses in 20:80 DMSO:PBS or orally in 10:70:20 DMSO:PBS:Cremphor El as described by Allen et al.3 Tumor volumes were measured according to the formula (L*W^2)/2.
In vivo pathology and toxicology
Toxicity during the course of ONC201, and ONC212 treatment was judged by body weight decrease of greater than 10%, tumor growth of more than 10% of body weight, or a body condition scoring <2. Serum and plasma samples were collected through orbital bleeding and cardiac puncture before sacrifice, and were immediately stored at 4°C and processed by Antech Diagnostics for CBC and chemistry panels. Results were analyzed by a board-certified toxicologist. Tumor volumes were measured post-mortem through caliper and water density examination. Organ and tumor samples were processed in 10% formalin and fixed in paraffin. Hematoxylin-stained samples were analyzed by a board-certified pathologist to determine whether tumor cells existed on any organs or necrosis occurred in tumors. A board-certified veterinary pathologist also indicated whether or not signs of toxicity were present.
Pharmacokinetics
HPLC analysis was performed by absorbance detection at 239 nm by the Wistar Institute Mass Spectrometry Facility. An acetonitrile (ACN) gradient was carried out for elution as 15–20% ACN for 0–5 minutes, 20–23% for 5–12 minutes, 25% for 12–18 minutes. The standard curve was generated by spiking varying concentrations of TIC10/ONC201 into plasma harvested from untreated non-tumor bearing C57/BL6 mice. For all plasma samples, blood was obtained by terminal cardiac puncture of the left ventricle and collected into EDTA tubes (BD). To prepare a methanolized pellet of treated mouse samples and spiked blood; blood samples from EDTA tubes where centrifuge at 14,000 g for 10 min in a cold room (4–8°C) methanol (cooled to −80°C) was added to the supernatant to make a final 80% (vol/vol) methanol solution. The solution was shaken and incubated for 6–8 h at −80°C. Samples were centrifuged at 14,000 g for 10 min (4–8°C) and then centrifuged in the SpeedVac to lyophilize to a pellet using no heat. The final pellet was solubilized and immediately injected into the HPLC as described in.14 AUC was normalized to an internal serum peak with a retention time of 8.1 minutes. AUC data vs. time was fit with a two-compartment open model, assuming first order elimination from central compartment with the equation AUC = Ae-αt + Be-βt, where t = time, and A and B are the extrapolated concentrations at the initiation of the two phases (distribution and elimination). Half-lives were calculated as t1/2α = 0.693/α and t1/2β = 0.693/β. Other equations used for calculation included CL = dose/AUC0-∞ and Vd = dose/(AUC0-∞X β).
In vivo imaging
For luciferase-expressing cell lines, D-Luciferin from Gold Bio was administered weekly following the manufacturers ’ instructions (60 μL, 50 mg/ml stock) and imaging was performed on a Xenogen IVIS system (Xenogen, Almadea, CA).
Genomics of drug sensitivity in cancer (GDSC) cell line screening
ONC212 (78 nM up to 20 µM) was tested in 1,068 human cancer cell lines. Cell viability was determined at 72 hours post-treatment. Dose responses curves were generated and IC50/area under curve (AUC) was determined.15,16
Crystallization of ONC212 for X-ray structure determination
A 75-mg sample of ONC212•2HCl•1H2O was dissolved in 0.75 mL of ethanol with heating. Approximately 20 microliters of water was added resulting in precipitation of a white solid. The mixture was heated until the solid was dissolved and 0.5 mL of dioxane was added. The was filtered into a vial, concentrated by heating by about 50%, and allowed to stand at room temperature overnight to crystallize. The crystal structure determination is described in the Supplemental Data and the structure is shown in Fig. 1.
Supplementary Material
Disclosure of potential conflicts of interest
Wafik El-Deiry is a co-founder of Oncoceutics Inc., and is fully compliant with NIH and institutional disclosure guidelines. J.E.A., V.V.P., M.S., and W.O. are also shareholders of Oncoceutics Inc.
Acknowledgments
We thank the Fox Chase Cancer Center Animal Facility and Dr. Patrick Carroll of the X-ray facility at the Department of Chemistry at the University of Pennsylvania for X-ray crystallographic studies. This work was presented in part at the 2016 American Association for Cancer Research (AACR) meeting in New Orleans, LA. This work received the AACR-Gerald B. Grindey Memorial Scholar-in-Training Award in 2017 (J.W.). W.S.E-D. is an American Cancer Society Research Professor.
Funding
This work was supported by grants from the NIH (R01 CA173453) and the American Cancer Society to W.S.E-D. and an NIH SBIR grant (R43 CA177002) to G.O. and W.S.E-D.
References
- [1].Ishizawa J, Kojima K, Chachad D, Ruvolo P, Ruvolo V, Jacamo RO, Borthakur G, Mu H, Zeng Z, Tabe Y, et al.. ATF4 induction through an atypical integrated stress response to ONC201 triggers p53-independent apoptosis in hematological malignancies. Sci Signal 2016; 9(415):ra17; PMID:26884599; https://doi.org/ 10.1126/scisignal.aac4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2α kinases. Sci Signal 2016; 9(415):ra18; PMID:26884600; https://doi.org/ 10.1126/scisignal.aac4374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Allen JE, Krigsfeld G, Mayes PA, Patel L, Dicker DT, Patel AS, Dolloff NG, Messaris E, Scata KA, Wang W, et al.. Dual inactivation of Akt and ERK by TIC10 signals Foxo3a nuclear translocation, TRAIL gene induction, and potent antitumor effects. Sci Transl Med 2013; 5(171):171ra17; PMID:23390247; https://doi.org/ 10.1126/scitranslmed.3004828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ashkenazi A. Targeting the extrinsic apoptosis pathway in cancer. Cytokine Growth Factor Rev 2008; 19(3-4):325-31; PMID:18495520; https://doi.org/ 10.1016/j.cytogfr.2008.04.001 [DOI] [PubMed] [Google Scholar]
- [5].Pan G, et al.. The receptor for the cytotoxic ligand TRAIL. Science 1997; 276(5309):111-3; PMID:9082980; https://doi.org/ 10.1126/science.276.5309.111 [DOI] [PubMed] [Google Scholar]
- [6].Prabhu VV, Allen JE, Dicker DT, El-Deiry WS. Small-molecule ONC201/TIC10 targets chemotherapy-resistant colorectal cancer stem-like cells in an Akt/Foxo3a/TRAIL-dependent manner. Cancer Res 2015; 75(7):1423-32; PMID:25712124; https://doi.org/ 10.1158/0008-5472.CAN-13-3451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang Q, Wang H, Ran L, Zhang Z, Jiang R. The preclinical evaluation of TIC10/ONC201 as an anti-pancreatic cancer agent. Biochem Biophys Res Commun 2016; 476(4):260-6; PMID:27233611; https://doi.org/ 10.1016/j.bbrc.2016.05.106 [DOI] [PubMed] [Google Scholar]
- [8].Allen J, Kline CL, Prabhu VV, Wagner J, Ishizawa J, Madhukar N, Lev A, Baumeister M, Zhou L, Lulla A, et al.. Discovery and clinical introduction of first-in-class imipridone ONC201. Oncotarget 2016; 7(45):74380-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, Chan N, Malhotra J, Rodriguez-Rodriguez L, Aisner J, et al.. First-in-human Clinical Trial of Oral ONC201 in Patients with Refractory Solid Tumors. Clin Cancer Res 2017; 23(15): 4163-69; PMID:28331050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wagner J, Kline CL, Pottorf RS, Nallaganchu BR, Olson GL, Dicker DT, Allen JE, El-Deiry WS. The angular structure of ONC201, a TRAIL pathway-inducing compound, determines its potent anti-cancer activity. Oncotarget 2014; 5(24):12728-37; PMID:25587031; https://doi.org/ 10.18632/oncotarget.2890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Takeda K, Hayakawa Y, Smyth MJ, Kayagaki N, Yamaguchi N, Kakuta S, Iwakura Y, Yagita H, Okumura K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat Med 2001; 7(1):94-100; PMID:11135622; https://doi.org/ 10.1038/83416 [DOI] [PubMed] [Google Scholar]
- [12].Grosse-Wilde A, Voloshanenko O, Bailey SL, Longton GM, Schaefer U, Csernok AI, Schütz G, Greiner EF, Kemp CJ, Walczak H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Invest 2008; 118(1):100-10; PMID:18079967; https://doi.org/ 10.1172/JCI33061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eisenberg MC, Kim Y, Li R, Ackerman WE, Kniss DA, Friedman A. Mechanistic modeling of the effects of myoferlin on tumor cell invasion. Proc Natl Acad Sci U S A 2011; 108(50):20078-83; PMID:22135466; https://doi.org/ 10.1073/pnas.1116327108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Yuan M, Breitkopf SB, Yang X, Asara JM. A positive/negative ion-switching, targeted mass spectrometry-based metabolomics platform for bodily fluids, cells, and fresh and fixed tissue. Nat Protoc 2012; 7(5):872-81; PMID:22498707; https://doi.org/ 10.1038/nprot.2012.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al.. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012; 483(7391):570-5; PMID:22460902; https://doi.org/ 10.1038/nature11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al.. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 2013; 41(Database issue):D955-61; PMID:23180760; https://doi.org/ 10.1093/nar/gks1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.