

ABSTRACT
Chondroblastoma is a cartilaginous tumor that typically arises under 25 y of age (80%). Recent studies have identified a somatic and heterozygous mutation at the H3F3B gene in over 90% chondroblastoma cases, leading to a lysine 36 to methionine replacement (H3.3K36M). In human cells, H3F3B gene is one of 2 genes that encode identical H3.3 proteins. It is not known how H3.3K36M mutant proteins promote tumorigenesis. We and others have shown that, the levels of H3K36 di- and tri-methylation (H3K36me2/me3) are reduced dramatically in chondroblastomas and chondrocytes bearing the H3.3K36M mutation. Mechanistically, H3.3K36M mutant proteins inhibit enzymatic activity of some, but not all H3K36 methyltransferases. Chondrocytes harboring the same H3F3B mutation exhibited the cancer cell associated phenotypes. Here, we discuss the potential effects of H3.3K36M mutation on epigenomes including H3K36 and H3K27 methylation and cellular phenotypes. We suggest that H3.3K36M mutant proteins alter epigenomes of specific progenitor cells, which in turn lead to cellular transformation and tumorigenesis.
KEYWORDS: Asf1, Cancer cells, Chondroblastoma, Chromosomes, Epigenetic, H3K36me3, Histone methylation, Tumorigenesis
Introduction
In eukaryotic cells, DNA is assembled into chromatin through the basic repeat unit called nucleosome, which contains one histone (H3-H4)2 tetramer and 2 histone H2A-H2B dimers wrapped around 147 bp of DNA.1,2 Histone proteins are modified extensively by post-translational modification.3,4 These histone modifications, including ubiquitylation, phosphorylation, acetylation and methylation,5-8 are thought to play an important role to regulate a variety of cellular processes such as gene transcription,9,10 DNA replication,11,12 and DNA repair.13-15 However, the approaches to address the function of a histone modification in higher eukaryotic cells are still limited. Unlike lower eukaryotic cells, such as budding yeast that contains only 2 copies of different histone genes,16,17 mammalian cells contain multiple histone genes.18,19 Consequently, it is almost impossible to mutate the modified residue to probe the function of the modification in mammalian cells. Therefore, the most common approach used in higher eukaryotic cells is to mutate or deplete the enzyme that catalyzes the modification. However, most histone modifying enzymes have non-histone substrates. It is then that histone acetyltransferase and histone methyltransferases were renamed as lysine acetyltransferases20-22 and lysine methyltransferases23-25 separately. In addition, in higher eukaryotic cells, there exits multiple enzymes that catalyze the same modification. Therefore, probing the function of a histone modification through depletion and/or knockout the cognate enzyme, an approach that is frequently used, carries caveats.
Recently, histone H3 lysine to methionine mutations have been found in brain26-28 and bone tumors.29 Specifically, one allele of H3F3A gene, one of 2 genes encoding histone H3 variant H3.330,31, is mutated in over 60% diffuse intrinsic pontine gliomas (DIPG) cases, replacing lysine 27 with methionine (H3.3K27M). In addition, one allele of H3F3B gene, the other gene encoding histone H3.3, is mutated in over 90% chondroblastoma cases, leading to changes of lysine 36 to methionine (H3.3K36M). Studies from different laboratories have shown that expressing a histone lysine (K) to methionine (M) mutant transgene results in dramatic loss of histone methylation of the corresponding lysine residues on wild type and endogenous histones.32-35 Moreover, structural and biochemical studies using several K to M mutations indicate that the global reduction of histone methylation is due to inhibition of the corresponding lysine methyltransferases.36,37 All these results indicate that histone K to M mutation is a good tool to address the function of specific histone methylation in cells. In the following sections, we will highlight the lessons we learned on the utilization of H3.3K36M mutation to address the function of H3K36 methylation based on our recent studies of H3.3K36M mutation in chondroblastoma.38
In budding yeast, histone H3K36 methylation is catalyzed by SET2, the only enzyme catalyzing mono-, di- and tri-methylation.39,40 In human cells, multiple enzymes catalyze H3K36 methylation. For instance, while ASH1L, NSD1, and NSD2/MMSET/WHSC1 are the prominent methyltransferases that catalyze mono- and di-methylation of H3K36,41-43 SETD2 is the only characterized enzyme catalyzing tri-methylation of H3K36.44 Genome-wide mapping of H3K36me2 and H3K36me3 using ChIP-seq indicate that H3K36me3 is localized predominantly at gene bodies, whereas H3K36me2 is localized at gene bodies as well as intergenic regions.45,46 In budding yeast, H3K36me3 at gene body is important for recruiting histone deacetylases to remove acetylation of new histone H3 following gene transcription and thereby suppresses cryptic gene transcription.47 In addition, H3K36me2 is regulated co-transcriptionally and is associated with active gene transcription.46 In human cells, splicing enhances H3K36 methylation through recruitment of SETD2.48 Therefore, the localization of H3K36me2/me3 at gene body is associated with active gene transcription. However, the function of H3K36me2 at intergenic regions remains largely unknown. In addition to gene transcription, H3K36 methylation is also found to be involved in alternative splicing,49 DNA replication,50,51 and DNA repair.52 Moreover, H3K36 methylation at gene body is also associated with DNA methylation at gene body.53-56 Therefore, H3K36 methylation likely impacts various cellular processes, and it is not surprising that alteration of the function/expression of histone H3K36 methyltransferases is also associated with different cancers. For instance, overexpression of MMSET has been implicated in multiple myeloma and neuroblastoma.57 Mutations in SETD2 are found to be the causal for renal cell carcinoma and breast cancer.58,59 The causal relationships between these alterations in the enzymes and histone H3K36 methylation in tumorigenesis remain to be determined.
Overexpression of H3.3K36M mutant in 293T cells results in splicing defects
H3K36 methylation and SETD2 play important roles in regulating RNA splicing.48,60 Therefore, we analyzed whether RNA splicing was affected in H3.3K36M knock-in chondrocytes by RNA-seq but have not detected any splicing defects in mutant chondrocytes cells compared with parental wild type T/C28a2 cells (Fig. 1A). To confirm this, we also overexpressed the H3.3K36M mutant transgene in 293T cells which results in a greater reduction of H3K36 methylation (data not shown) possibly due to higher expression of the H3.3K36M mutant transgene. Surprisingly, we detected increased splicing defects by RNA-seq (Fig. 1B). We also analyzed the RNA splicing in 2 wild type bone tissue samples and 3 H3.3K36M mutant chondroblastoma tissue samples. Consistent with the findings in T/C28a2 cells, no major RNA splicing defects were detected in the chondroblastoma tissues harbor the H3.3K36M mutation compared to normal control tissues (Fig. 1C). We have shown that chondrocyte cells engineered with the same genetic mutation as chondroblastoma using CRISPR/CAS9 exhibited defects in homologous recombination.38 However, the mutant chondrocytes do not show defects in RNA splicing and mismatch repair.38 One possible explanation is that different cell lines used (293T cells vs T/C28a2 cells) contribute to the difference in RNA splicing defects. Alternatively, H3.3K36M mutant chondrocytes, while having low levels of H3K36 methylation, still retain considerable amounts of H3K36 methylation, which can support the splicing process. We suggest that different levels of H3K36M mutant proteins expressed (transgenes vs. engineered K36M mutation) may play a bigger role.
Figure 1.
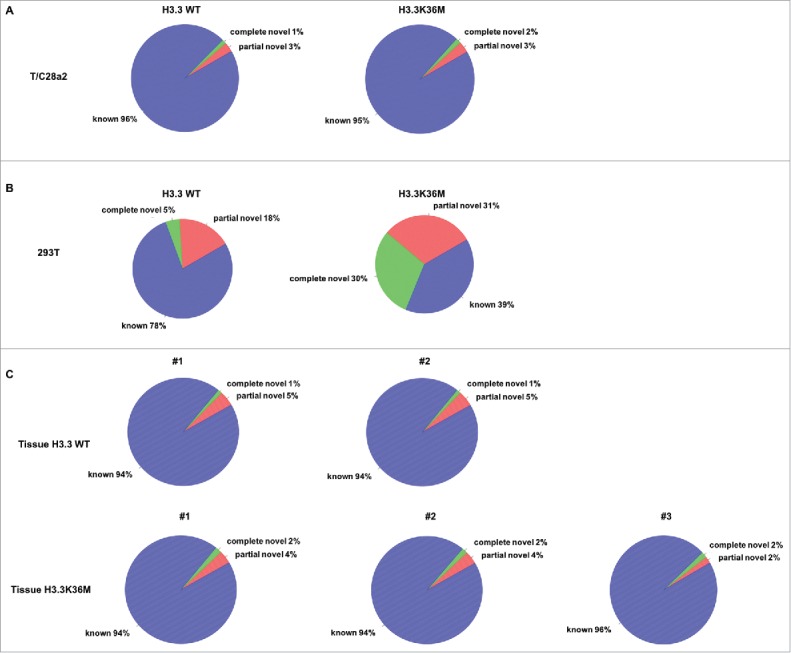
Expression of H3.3K36M mutant protein in distinct cells leads to different effects on RNA splicing. Expression of H3.3K36M mutant protein in 293T cells (B) but not T/C28a2 cells (A) leads to increase in splicing defects. (C) The chondroblastoma tissue samples with H3.3K36M mutation showed the similar RNA splicing pattern as the wild type bone tissues. The annotation of the splice junction in different RNA-seq data sets was indicated in the figure. The genes from Ensembl, GENECODE, UCSC, and RefSeq were combined and used as a reference gene model. Known: the junction is part of the gene model; complete_novel: complete new junction; partial_novel: one of the splice junction (SJ, either 5SJ or 3SJ) is new, while the other splice junction is annotated (known).
Expression of H3.3K36M,R129E mutant results in reduced levels of H3K36 methylation
It is known that histone chaperone HIRA complex deposits H3.3 at the generic region,61 and Asf1a delivers H3.3 to HIRA (possibly Daxx) for nucleosome assembly.62-64 The structure of Asf1-H3 have been identified and several H3 residues have been identified to bind Asf1.65,66 For instance, replacing histone H3 arginine 129 with glutamic acid (H3R129E) destroys the Asf1a-H3 interaction65-67. Therefore, we made H3.3R129E mutant alone or in combination with H3.3K36M mutation and tested how the expression of these mutants affected H3K36me2 and H3K36me3. Expression of H3.3R129E mutant had no apparent effect on H3K36me2/me3 in 293T cells. In contrast, expression of H3.3K36M,R129E double mutant protein resulted in dramatic reduction of H3K36me2 and H3K36me3 levels (Fig. 2A). This result suggests that H3.3K36M mutant proteins can exert their effects on H3K36 methylation before they are assembled into nucleosomes. However, we could not detect the expression of H3.3R129E and H3.3K36M,R129E mutant proteins in whole cell extracts or on chromatin by Western blot analysis, suggesting that H3.3R129E mutant proteins are not stable.
Figure 2.
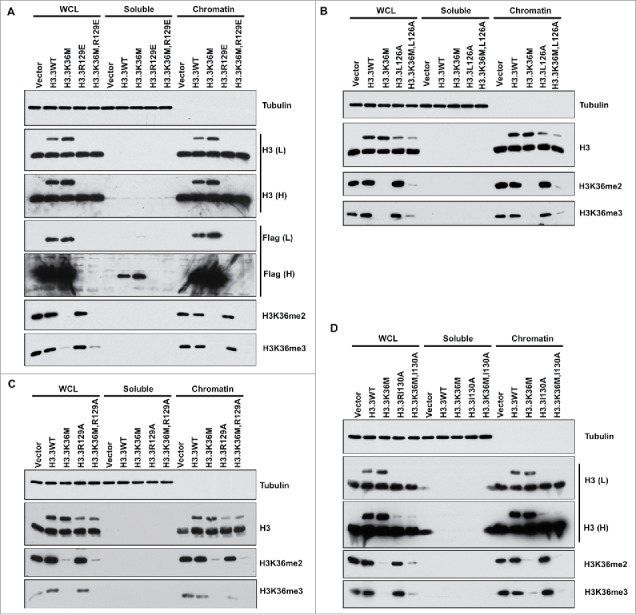
Expression of double mutants containing the H3.3K36M and each Asf1 binding deficient mutant results in reduced H3K36me2/me3 levels in T/C28a2 cells. T/C28a2 cells were infected with virus expressing FLAG-tagged wild type H3.3, or H3.3 mutants with relevant mutations indicated in the figure (A-D). Cells were collected for chromatin fractionation assays to separate proteins into soluble and chromatin fractions. Proteins in whole cell extracts (WCE), chromatin and soluble fractions were analyzed by Western blot.
To resolve this complication for interpreting the result, we first started to analyze other H3 mutants that exhibit defects in Asf1a binding including H3.3L126A (Fig. 2B), H3.3R129A (Fig. 2C), and H3.3I130A (Fig. 2D). A combination of each mutation with H3.3K36M affected H3K36me2 and H3K36me3 levels, whereas single mutation without H3.3K36M had no apparent effects. Based on chromatin fractionation assays, all these mutant proteins could be detected on chromatin, suggesting that these mutant proteins were assembled into nucleosomes. These results indicate that these Asf1a binding deficient mutants were assembled into nucleosomes, therefore, could not be used to address whether the H3.3K36M mutant protein can exert their effects on H3K36 methylation before assembled into nucleosomes.
H3.3R129E mutant is incorporated into chromatin
Asf1 is known to bind H3-H4 before nuclear import of H3-H4.5,66,68,69 Therefore, it is possible that H3.3R129E mutant proteins were not stable. To test this idea, we analyzed whether H3.3R129E mutant alone or in combination with H3.3K36M mutant could be detected by Western blot in cells treated with the proteasome inhibitor MG132. However, proteasome inhibition failed to detect H3.3R129E or H3.3K36M,R129E mutant proteins in cell extracts (data not shown). It has been reported that chaperone-mediated autophagy is involved in degradation of soluble histone in mammalian cells.70 Therefore, in independent experiments, we depleted HSC70 and LAMP2a, 2 proteins controlled chaperone-mediated autophagy, but we still could not detect H3.3R129E mutant proteins in cell extracts and chromatin by Western blot (data not shown). These results indicate that the low levels of H3.3R129E are unlikely due to degradation of the proteins by proteasome and autophagy pathways.
After these trials, we tested whether H3.3R129E and H3.3K36M,R129E mutants could be detected after immunoprecipitating these proteins from soluble extracts and from chromatin (Fig. 3A). In these experiments, we were able to detect H3.3R129E and H3.3K36M,R129E mutant proteins. Furthermore, most H3.3R129E and H3.3K36M,R129E mutant proteins were detected on the chromatin. These results indicate that H3.3R129E mutant proteins that are known to be defective in binding to the histone chaperone Asf1, could still be assembled into nucleosomes. To confirm this result, we performed ChIP-PCR experiments and observed that H3.3R129E and H3.3K36M,R129E mutant proteins were assembled into chromatin (Fig. 3B). In addition, the H3K36me3 levels at the incorporated loci were dramatically decreased (Fig. 3C). Based on these results, we suggest the following 3 ideas. First, the nucleosome assembly of H3.3R129E mutant proteins is very inefficient, and consequently, those that are not assembled into nucleosomes are degraded, contributing to the low levels of H3.3R129E mutant proteins. Second, very low levels of H3.3K36M mutant proteins, such as those that cannot be detected by Western blot in cell extracts, can lead to a dramatic reduction of H3K36me3 in cells, suggesting that while there are 8 H3K36 methyltransferases, their expression levels may be low. Third, these results further strengthen our conclusion that the H3.3K36M mutant proteins likely exert their inhibitory effect on H3K36 methylation after being incorporated into nucleosomes.38
Figure 3.
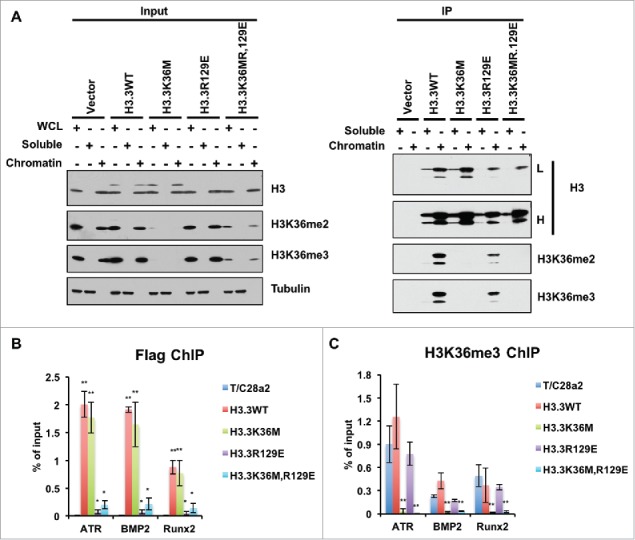
H3.3R129E and H3.3K36MR129E mutant proteins were enriched on chromatin. (A) Soluble histones or mononucleosomes were purified from HEK293T cells with vector (negative control), HEK293T cells expressing FLAG-tagged WT H3.3, H3.3K36M, H3.3R129E, or H3.3K36MR129E mutant. Proteins from Input and immunoprecipitated (IP) samples were analyzed by Western blotting using the indicated antibodies. (B) ChIP-qPCR results of incorporations of different mutant histone H3.3 proteins and reduction of H3K36me3 at different loci. ChIP-qPCR is performed by using T/C28a2 cells after infection of different mutant histone H3.3 expressing virus. Data represents the average and standard deviations of 3 independent experiments. (*P < 0.05, **P < 0.01).
“Cross-talk” between H3K36me2 and H3K27me3 at intergenic regions
It has been reported that H3K36 methylation can inhibit the activity of PCR2 complex.71,72 H3K27me3 increased at intergenic regions in mouse mesenchymal stem cells expressing H3.3K36M mutant transgene, most likely due to the dramatic reduction of H3K36me2.73 We also performed H3K27me3 ChIP-seq in H3.3K36M mutant chondrocytes and their parental line, T/C28a2. To determine whether H3K27me3 levels at intergenic regions are also altered in chondrocytes with H3.3K36M mutation, we calculated the H3K27me3 ChIP-seq reads density at the intergenic regions and observed that H3K27me3 increased dramatically at intergenic regions with reduced H3K36me2 compared with those with H3K36me2 (Fig. 4A). In contrast, we did not detect major changes in H3K27me3 at genic regions surrounding TSS, where H3K37me3 was enriched when all genes were taken into consideration (Fig. 4B). This observation is consistent with our Western blot analysis of H3K27me3 in H3.3K36M mutant chondrocyte cells.38 However, when genes were separated into 3 groups based on their expression levels (high, middle and low) in wild type chondrocyte cells, we observed that H3K27me3 was reduced at promoters of low expressed genes in H3.3K36M mutant lines compared with wild type line (compared Fig. 4C to E). These results are consistent with the idea that loss of H3K36me2 results in redistribution of H3K27me373 in cells expressing H3.3K36M mutant proteins. Based on these results, we suggest that one of the functions of H3K36me2 at intergenic regions is to inhibit PRC2 mediated H3K27 methylation at these regions.
Figure 4.
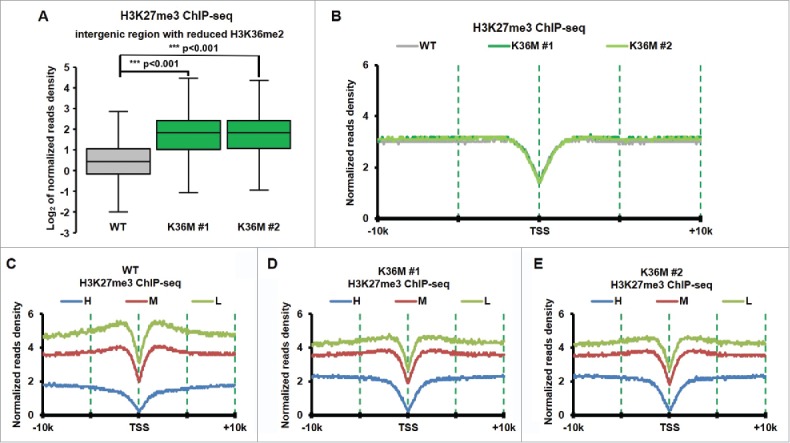
H3K27me3 increased dramatically at intergenic regions with reduced of H3K36me2 in H3.3K36M cell compare with WT cell. (A) Normalized tag distribution profiles of H3K27me3 for intergenic regions with reduced H3K36me2 in 2 H3.3K36M mutant cell lines compare with WT cells. The reads are normalized to reads per kilobase per 10 million mapped reads. (B) The normalized reads density of H3K27me3 ChIP-Seq was plotted from 10 kb upstream to 10Kb downstream from TSS sites in WT and H3.3K36M cells. The reads are normalized to reads per kilobase per 10 million mapped reads. (C-E) The same calculation as in B, but the whole human genes were split into equal number of 3 groups according to their expression levels. H: high expression, M: medium expression, and L: Low expression.
Discussion/perspective
These results presented here likely have the following implications. It is possible that the distinct expression levels of H3.3K36M mutant proteins will impact the H3K36 methylation levels and thereby cellular phenotypes differently. This is not surprising as H3.3K36M mutant proteins likely reduce H3K36 methylation through a mechanism of competitive inhibition, one would expect that the relative amount of H3.3K36M mutant proteins and H3K36 methyltransferases in cells will determine the reduced levels of H3K36 methylation. Supporting this idea, it has been shown recently that overexpression of Clr4, which is the H3K9 methyltransferase in S. pombe, can relieve the silencing defects of H3K9M mutant proteins.74 Therefore, the expression levels of a histone lysine to methionine mutant protein as well as its targeted methyltransferases will have a profound impact on histone methylation and cellular phenotypes.
We made a surprising finding that expression of H3.3K36M,R129E mutant can still lead to the reduction of H3K36 methylation despite the fact that the mutant proteins were not detectable in whole cell extracts by Western blot. Even more surprising is the observation that H3.3R129E and H3.3K36M,R129E mutant proteins are still assembled into chromatin despite the fact that they are defective in binding to Asf1a/b. These results indicate that H3.3R129E and H3.3K36M,R129E mutant proteins are not stable, most likely due to the fact that they are defective in nucleosome assembly. Therefore, most of proteins that cannot be assembled into nucleosomes would be degraded. It's known that proteasome-mediated protein degradation and the chaperone-mediated autophagy are involved in the elimination of unstable protein.70 However, inhibition of these 2 pathways didn't increase the levels of the H3.3R129E or H3.3K36M,R129E mutant proteins. In the future, it would be interesting to determine why the mutant proteins are not stable.
While the primary effects of H3.3K36M mutant proteins are likely on H3K36 methylation, it is clear that H3K27me3 are also altered on chromatin.73 More specifically, H3K27me3 levels at intergenic regions increase in cells expressing H3.3K36M mutant chondrocytes. Therefore, it is possible that one function of H3K36me2 at intergenic regions is to suppress H3K27 methylation at these regions. In addition, it has been suggested that specific patterns of histone modifications have context-dependent effects.6 Therefore, the cellular phenotypes displayed in cells expressing H3.3K36M mutant proteins reflect changes not only in H3K36 methylation, but also other induced changed on histone modifications including H3K27me3. Considering the fact that the chondroblastomas typically arise under 25 y of age (80%),75-77 we suggest that H3.3K36M mutant proteins reprogram epigenomes of specific progenitor cells, which in turn leads to cellular transformation and tumorigenesis.
Methods and materials
Preparation of chromatin and soluble fractions
Cell fractions were prepared as described before.78 Cells were incubated in fraction buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1 mM DTT) for 5 mins. The whole cell lysate was collected by directly add Laemmli buffer to cell mix. Other fractions were separated by centrifugation at 1300 g, 4 °C, 10 min. The nuclear pellet was washed with fraction buffer and serve as the chromatin fraction. The supernatant was treated as soluble fraction.
Chromatin immunoprecipitation-deep sequencing (ChIP-seq) and ChIP-qPCR
ChIP was performed as described before.38 In brief, cells were fixed with 1% for 10 min and then quenched with 125 mM glycine for 5 min at room temperature. The chromatin was prepared with MNase digestion and sonication. 2 μg of rabbit polyclonal anti-H3K36me3 antibody (Active Motif, Cat. #61101, lot # 32412003), 1.5 μg of rabbit monoclonal anti-H3K36me2 antibody (Cell Signaling Technology, Cat. #2901, lot# 5), 2 μg of rabbit monoclonal anti-H3K27me3 antibody (Cell Signaling Technology, Cat. #9733), or 2 ug of mouse monoclonal anti-Flag antibody (Sigma, clone M2) was used to incubate with chromatin overnight at 4°C. 30 μL protein G-magnetic beads (Life Technologies) were then added for incubation 3 hours. After extensively washed, the bound chromatin was eluted and reverse-crosslinked at 65°C overnight. DNAs were purified using Min-Elute PCR purification kit (Qiagen) after the incubation with RNase A and proteinase K.
Immunoprecipitation (IP)
Cells fractions were prepared as described before. The chromatin fraction was digested by MNase. Different fractions were diluted into IP buffer (50 mM Tris-HCl, pH8, 10 mM EDTA, 100 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate). Samples were then incubated with 30 μL of prewashed anti-FLAG M2 agarose beads (Sigma) overnight at 4°C. The beads were extensively washed with washing buffer (50 mM HEPES-KOH, pH 7.4, 200 mM NaCl, 0.5% Triton X-100, 10% glycerol, 10 mM EDTA). Beads were then boiled with 60 μL Laemmli buffer and analyzed by Western blotting.
RNA-seq data analysis
RNA-seq sequence reads were aligned to the human genome hg19 and to gene annotations from Refseq gene using TopHat v2.0579 RSeQC80 was used to analysis the RNA splicing.
H3K27me3 ChIP-seq data analysis
Paired-end reads from H3K27me3 ChIP-seq were aligned to the human genome (hg19) using the Bowtie2 software.81 The consistent pair reads were only used for the further analysis. The reads density scan around TSS and count reads in intergenic was performed by in-house Perl programs.
Antibodies
For Western blotting analysis, antibodies against H3K36me2 (Cat. #2901, lot 5) were purchased from Cell signaling. H3K36me3 (Cat. #61101, lot 32412003) were purchased from Active motif. Antibodies against α-Tubulin (Cat. #T9026) and FLAG (Cat. #F1804) were purchased from Sigma. Antibodies against histone H3 were described previously.38
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by NIH grant CA157489.
References
- [1].Jenuwein T, Allis CD. Translating the Histone Code. Science (80- ) 2001; 293:1074-80; http://dx.doi.org/ 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- [2].Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-5; PMID:10638745; http://dx.doi.org/ 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- [3].Zhang Y, Reinberg D. Transcription regulation by histone methylation: Interplay between different covalent modifications of the core histone tails. Genes Dev 2001; 15:2343-60; PMID:11562345; http://dx.doi.org/ 10.1101/gad.927301 [DOI] [PubMed] [Google Scholar]
- [4].Deal RB, Henikoff S. Histone variants and modifications in plant gene regulation. Curr. Opin. Plant Biol 2011; 14:116-22; PMID:21159547; http://dx.doi.org/ 10.1016/j.pbi.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Han J, Zhang H, Zhang H, Wang Z, Zhou H, Zhang Z. XA Cul4 E3 ubiquitin ligase regulates histone hand-off during nucleosome assembly. Cell 2013; 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Lee JS, Smith E, Shilatifard A. The Language of Histone Crosstalk. Cell 2010; 142:682-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Burgess RJ, Zhang Z. Histones, histone chaperones and nucleosome assembly. Protein Cell 2010; 1:607-12; PMID:21203931; http://dx.doi.org/ 10.1007/s13238-010-0086-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011; 146:1016-28; PMID:21925322; http://dx.doi.org/ 10.1016/j.cell.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997; 389:349-52; PMID:9311776; http://dx.doi.org/ 10.1038/38664 [DOI] [PubMed] [Google Scholar]
- [10].Weber CM, Henikoff S. Histone variants: dynamic punctuation in transcription. Genes Dev 2014; 28:672-82; PMID:24696452; http://dx.doi.org/ 10.1101/gad.238873.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Abmayr SM, Workman JL. Holding on through DNA replication: Histone modification or modifier? Cell 2012; 150:875-7; PMID:22939615; http://dx.doi.org/ 10.1016/j.cell.2012.08.006 [DOI] [PubMed] [Google Scholar]
- [12].Han J, Zhou H, Horazdovsky B, Zhang K, Xu R, Zhang Z. Rtt109 acetylates histone H3 lysine 56 and functions in DNA replication. Sci (New York, NY) 2007; 315:653-5; http://dx.doi.org/ 10.1126/science.1133234 [DOI] [PubMed] [Google Scholar]
- [13].van Attikum H, Gasser SM. The histone code at DNA breaks: a guide to repair? Nat Rev Mol Cell Biol 2005; 6:757-65; PMID:16167054; http://dx.doi.org/ 10.1038/nrm1737 [DOI] [PubMed] [Google Scholar]
- [14].Jiang Y, Qian X, Shen J, Wang Y, Li X, Liu R, Xia Y, Chen Q, Peng G, Lin S-Y, et al.. Local generation of fumarate promotes DNA repair through inhibition of histone H3 demethylation. Nat Cell Biol 2015; 17:1158-68; PMID:26237645; http://dx.doi.org/ 10.1038/ncb3209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lowndes NF, Toh GWL. DNA repair: The importance of phosphorylating histone H2AX. Curr. Biol 2005; 15. [DOI] [PubMed] [Google Scholar]
- [16].Eriksson PR, Ganguli D, Nagarajavel V, Clark DJ. Regulation of histone gene expression in budding yeast. Genetics 2012; 191:7-20; http://dx.doi.org/ 10.1534/genetics.112.140145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science (80- ) 2007; 315:1405-8; http://dx.doi.org/ 10.1126/science.1134053 [DOI] [PubMed] [Google Scholar]
- [18].Bernstein BE, Meissner A, Lander ES. The Mammalian Epigenome. Cell 2007; 128:669-81. [DOI] [PubMed] [Google Scholar]
- [19].Etchegaray J, Lee C, a Wade P, Reppert SM. Rhythmic histone acetylation underlies transcription in the mammalian circadian clock. Nature 2003; 421:177-82; PMID:12483227; http://dx.doi.org/ 10.1038/nature01314 [DOI] [PubMed] [Google Scholar]
- [20].Carrozza MJ, Utley RT, Workman JL, Côté J. The diverse functions of histone acetyltransferase complexes. Trends Genet 2003; 19:321-9; PMID:12801725; http://dx.doi.org/ 10.1016/S0168-9525(03)00115-X [DOI] [PubMed] [Google Scholar]
- [21].Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal a K, Zhou MM. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999; 399:491-6; PMID:10365964; http://dx.doi.org/ 10.1038/20974 [DOI] [PubMed] [Google Scholar]
- [22].Lee KK, Workman JL. Histone acetyltransferase complexes: one size doesn't fit all. Nat Rev Mol Cell Biol 2007; 8:284-95; PMID:17380162; http://dx.doi.org/ 10.1038/nrm2145 [DOI] [PubMed] [Google Scholar]
- [23].Rea S, Eisenhaber F, O'Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, et al.. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 2000; 406:593-9; PMID:10949293; http://dx.doi.org/ 10.1038/35020506 [DOI] [PubMed] [Google Scholar]
- [24].Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD. Histone Methyltransferases Direct Different Degrees of Methylation to Define Distinct Chromatin Domains. Mol Cell 2003; 12:1591-8; PMID:14690610; http://dx.doi.org/ 10.1016/S1097-2765(03)00479-9 [DOI] [PubMed] [Google Scholar]
- [25].Trievel RC. Structure and function of histone methyltransferases. Crit Rev Eukaryot Gene Expr 2004; 14:147-69; PMID:15248813; http://dx.doi.org/ 10.1615/CritRevEukaryotGeneExpr.v14.i3.10 [DOI] [PubMed] [Google Scholar]
- [26].Schwartzentruber J, Korshunov A, Liu X-Y, Jones DTW, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang D-AK, Tönjes M, et al.. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012; 482:226-31; PMID:22286061; http://dx.doi.org/ 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- [27].Wu G, Broniscer A, a McEachron T, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al.. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet 2012; 44:251-3; PMID:22286216; http://dx.doi.org/ 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sturm D, Witt H, Hovestadt V, Khuong-Quang DA, Jones DTW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, et al.. Hotspot Mutations in H3F3A and IDH1 Define Distinct Epigenetic and Biological Subgroups of Glioblastoma. Cancer Cell 2012; 22:425-37; PMID:23079654; http://dx.doi.org/ 10.1016/j.ccr.2012.08.024 [DOI] [PubMed] [Google Scholar]
- [29].Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, Wedge DC, Cooke SL, Gundem G, Davies H, et al.. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet 2013; 45:1479-82; PMID:24162739; http://dx.doi.org/ 10.1038/ng.2814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Szenker E, Ray-Gallet D, Almouzni G. The double face of the histone variant H3.3. Cell Res 2011; 21:421-34; PMID:21263457; http://dx.doi.org/ 10.1038/cr.2011.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Talbert PB, Henikoff S. Histone variants-ancient wrap artists of the epigenome. Nat Rev Mol Cell Biol 2010; 11:264-75; PMID:20197778; http://dx.doi.org/ 10.1038/nrm2861 [DOI] [PubMed] [Google Scholar]
- [32].Bender S, Tang Y, Lindroth AM, Hovestadt V, Jones DTW, Kool M, Zapatka M, Northcott P a, Sturm D, Wang W, et al.. Reduced H3K27me3 and DNA Hypomethylation Are Major Drivers of Gene Expression in K27M Mutant Pediatric High-Grade Gliomas. Cancer Cell 2013; 24(5):660-72. [DOI] [PubMed] [Google Scholar]
- [33].Lewis PW, Muller MM, Koletsky MS, Cordero F, Lin S, Banaszynski LA, Garcia BA, Muir TW, Becher OJ, Allis CD. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science (80- ) 2013; 340:857-61; http://dx.doi.org/ 10.1126/science.1232245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chan KM, Han J, Fang D, Gan H, Zhang Z. A lesson learned from the H3.3K27M mutation found in pediatric glioma A new approach to the study of the function of histone modifications in vivo? Cell Cycle 2013; 12:2546-52; PMID:23907119; http://dx.doi.org/ 10.4161/cc.25625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chan KM, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, David James C, Jenkins R, et al.. The histone H3.3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Dev 2013; 27:985-90; PMID:23603901; http://dx.doi.org/ 10.1101/gad.217778.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 2015; 350:291; http://dx.doi.org/ 10.1126/science.aab2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al.. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun 2016; 7:11316; PMID:27121947; http://dx.doi.org/ 10.1038/ncomms11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, et al.. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science (80- ) 2016; 352(6291):1344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li J, Moazed D, Gygi SP. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem 2002; 277:49383-8; PMID:12381723; http://dx.doi.org/ 10.1074/jbc.M209294200 [DOI] [PubMed] [Google Scholar]
- [40].Strahl BD, Grant PA, Briggs SD, Sun Z-W, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, et al.. Set2 Is a Nucleosomal Histone H3-Selective Methyltransferase That Mediates Transcriptional Repression. Mol Cell Biol 2002; 22:1298-306; PMID:11839797; http://dx.doi.org/ 10.1128/MCB.22.5.1298-1306.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rayasam GV, Wendling O, Angrand PO, Mark M, Niederreither K, Song L, Lerouge T, Hager GL, Chambon P, Losson R. NSD1 is essential for early post-implantation development and has a catalytically active SET domain. EMBO J 2003; 22:3153-63; PMID:12805229; http://dx.doi.org/ 10.1093/emboj/cdg288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Li Y, Trojer P, Xu CF, Cheung P, Kuo A, Drury WJ, Qiao Q, Neubert TA, Xu RM, Gozani O, et al.. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem 2009; 284:34283-95; PMID:19808676; http://dx.doi.org/ 10.1074/jbc.M109.034462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].An S, Yeo KJ, Jeon YH, Song JJ. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J Biol Chem 2011; 286:8369-74; PMID:21239497; http://dx.doi.org/ 10.1074/jbc.M110.203380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Edmunds JW, Mahadevan LC, Clayton AL. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J 2008; 27:406-20; PMID:18157086; http://dx.doi.org/ 10.1038/sj.emboj.7601967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kolasinska-zwierz P, Down T, Latorre I, Liu T, Liu XS, Ahringer J. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat Genet 2009; 41:376-81; PMID:19182803; http://dx.doi.org/ 10.1038/ng.322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rao B, Shibata Y, Strahl BD, Lieb JD. Dimethylation of histone H3 at lysine 36 demarcates regulatory and nonregulatory chromatin genome-wide. Mol Cell Biol 2005; 25:9447-59; PMID:16227595; http://dx.doi.org/ 10.1128/MCB.25.21.9447-9459.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Sen P, Dang W, Donahue G, Dai J, Dorsey J, Cao X, Liu W, Cao K, Perry R, Lee JY, et al.. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev 2015; 29:1362-76; PMID:26159996; http://dx.doi.org/ 10.1101/gad.263707.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].de Almeida SF, Grosso AR, Koch F, Fenouil R, Carvalho S, Andrade J, Levezinho H, Gut M, Eick D, Gut I, et al.. Splicing enhances recruitment of methyltransferase HYPB/Setd2 and methylation of histone H3 Lys36. Nat Struct Mol Biol 2011; 18:977-83; PMID:21792193; http://dx.doi.org/ 10.1038/nsmb.2123 [DOI] [PubMed] [Google Scholar]
- [49].Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science 2010; 327:996-1000; PMID:20133523; http://dx.doi.org/ 10.1126/science.1184208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pryde F, Jain D, Kerr A, Curley R, Mariotti FR, Vogelauer M. H3 K36 methylation helps determine the timing of Cdc45 association with replication origins. PLoS One 2009; 4(6):e5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Kim HS, Rhee DK, Jang YK. Methylations of histone H3 lysine 9 and lysine 36 are functionally linked to DNA replication checkpoint control in fission yeast. Biochem Biophys Res Commun 2008; 368:419-25; PMID:18252195; http://dx.doi.org/ 10.1016/j.bbrc.2008.01.104 [DOI] [PubMed] [Google Scholar]
- [52].Wagner EJ, Carpenter PB. Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 2012; 13:115-26; PMID:22266761; http://dx.doi.org/ 10.1038/nrm3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Li B, Carey M, Workman JL. The Role of Chromatin during Transcription. Cell 2007; 128:707-19. [DOI] [PubMed] [Google Scholar]
- [54].Dhayalan A, Rajavelu A, Rathert P, Tamas R, Jurkowska RZ, Ragozin S, Jeltsch A. The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J Biol Chem 2010; 285:26114-20; PMID:20547484; http://dx.doi.org/ 10.1074/jbc.M109.089433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lucio-Eterovic AK, Singh MM, Gardner JE, Veerappan CS, Rice JC, Carpenter PB. Role for the nuclear receptor-binding SET domain protein 1 (NSD1) methyltransferase in coordinating lysine 36 methylation at histone 3 with RNA polymerase II function. Proc Natl Acad Sci U S A 2010; 107:16952-7; PMID:20837538; http://dx.doi.org/ 10.1073/pnas.1002653107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Fnu S, Williamson EA, De Haro LP, Brenneman M, Wray J, Shaheen M, Radhakrishnan K, Lee S-H, Nickoloff JA, Hromas R. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci 2011; 108:540-5; PMID:21187428; http://dx.doi.org/ 10.1073/pnas.1013571108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Hudlebusch HR, Santoni-Rugiu E, Simon R, Ralfkiær E, Rossing HH, Johansen JV, Jørgensen M, Sauter G, Helin K. The histone methyltransferase and putative oncoprotein MMSET is overexpressed in a large variety of human tumors. Clin Cancer Res 2011; 17:2919-33; PMID:21385930; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-1302 [DOI] [PubMed] [Google Scholar]
- [58].Duns G, van den Berg E, van Duivenbode I, Osinga J, Hollema H, Hofstra RMW, Kok K. Histone methyltransferase gene SETD2 is a novel tumor suppressor gene in clear cell renal cell carcinoma. Cancer Res 2010; 70:4287-91; PMID:20501857; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0120 [DOI] [PubMed] [Google Scholar]
- [59].Newbold RF, Mokbel K. Evidence for a tumour suppressor function of SETD2 in human breast cancer: A new hypothesis. Anticancer Res 2010; 30:3309-11; PMID:20944102 [PubMed] [Google Scholar]
- [60].Kim S, Kim H, Fong N, Erickson B, Bentley DL. Pre-mRNA splicing is a determinant of histone H3K36 methylation. Proc Natl Acad Sci U S A 2011; 108:13564-9; PMID:21807997; http://dx.doi.org/ 10.1073/pnas.1109475108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Goldberg AD, Banaszynski LA, Noh KM, Lewis PW, Elsaesser SJ, Stadler S, Dewell S, Law M, Guo X, Li X, et al.. Distinct Factors Control Histone Variant H3.3 Localization at Specific Genomic Regions. Cell 2010; 140:678-91; PMID:20211137; http://dx.doi.org/ 10.1016/j.cell.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tang Y, Poustovoitov M V, Zhao K, Garfinkel M, Canutescu A, Dunbrack R, Adams PD, Marmorstein R. Structure of a human ASF1a-HIRA complex and insights into specificity of histone chaperone complex assembly. Nat Struct Mol Biol 2006; 13:921-9; PMID:16980972; http://dx.doi.org/ 10.1038/nsmb1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Yamane K, Mizuguchi T, Cui B, Zofall M, Noma KI, Grewal SIS. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol Cell 2011; 41:56-66; PMID:21211723; http://dx.doi.org/ 10.1016/j.molcel.2010.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Elsässer SJ, Huang H, Lewis PW, Chin JW, Allis CD, Patel DJ. DAXX envelops a histone H3.3-H4 dimer for H3.3-specific recognition. Nature 2012; 491:560-5; PMID:23075851; http://dx.doi.org/ 10.1038/nature11608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Agez M, Chen J, Guerois R, van Heijenoort C, Thuret JY, Mann C, Ochsenbein F. Structure of the Histone Chaperone Asf1 Bound to the Histone H3 C-Terminal Helix and Functional Insights. Structure 2007; 15:191-9; PMID:17292837; http://dx.doi.org/ 10.1016/j.str.2007.01.002 [DOI] [PubMed] [Google Scholar]
- [66].English CM, Adkins MW, Carson JJ, Churchill MEA, Tyler JK. Structural Basis for the Histone Chaperone Activity of Asf1. Cell 2006; 127:495-508; PMID:17081973; http://dx.doi.org/ 10.1016/j.cell.2006.08.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Duarte LF, Young ARJ, Wang Z, Wu H-A, Panda T, Kou Y, Kapoor A, Hasson D, Mills NR, Ma'ayan A, et al.. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat Commun 2014; 5:5210; PMID:25394905; http://dx.doi.org/ 10.1038/ncomms6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Mello JA, Silljé HHW, Roche DMJ, Kirschner DB, Nigg EA, Almouzni G. Human Asf1 and CAF-1 interact and synergize in a repair-coupled nucleosome assembly pathway. EMBO Rep 2002; 3:329-34; PMID:11897662; http://dx.doi.org/ 10.1093/embo-reports/kvf068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bao Y, Shen X. Asf1, a Loveseat for a Histone Couple. Cell 2006; 127:458-60. [DOI] [PubMed] [Google Scholar]
- [70].Cook AJL, Gurard-Levin ZA, Vassias I, Almouzni G. A Specific Function for the Histone Chaperone NASP to Fine-Tune a Reservoir of Soluble H3-H4 in the Histone Supply Chain. Mol Cell 2011; 44:918-27; PMID:22195965; http://dx.doi.org/ 10.1016/j.molcel.2011.11.021 [DOI] [PubMed] [Google Scholar]
- [71].Schmitges FW, Prusty AB, Faty M, Stützer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al.. Histone Methylation by PRC2 Is Inhibited by Active Chromatin Marks. Mol Cell 2011; 42:330-41; PMID:21549310; http://dx.doi.org/ 10.1016/j.molcel.2011.03.025 [DOI] [PubMed] [Google Scholar]
- [72].Yuan W, Xu M, Huang C, Liu N, Chen S, Zhu B. H3K36 methylation antagonizes PRC2-mediated H3K27 methylation. J Biol Chem 2011; 286:7983-9; PMID:21239496; http://dx.doi.org/ 10.1074/jbc.M110.194027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lu C, Jain SU, Hoelper D, Bechet D, Molden RC, Ran L, Murphy D, Venneti S, Hameed M, Pawel BR, et al.. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science (80- ) 2016; 352:844-9; http://dx.doi.org/ 10.1126/science.aac7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Shan C-M, Wang J, Xu K, Chen H, Yue J-X, Andrews S, Moresco JJ, Yates JR III, Nagy PL, Tong L, et al.. A histone H3K9M mutation traps histone methyltransferase Clr4 to prevent heterochromatin spreading. Elife 2016; 5:e17903; PMID:27648579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].De Mattos CBR, Angsanuntsukh C, Arkader A, Dormans JP. Chondroblastoma and chondromyxoid fibroma. J Am Acad Orthop Surg 2013; 21:225-33; PMID:23545728; http://dx.doi.org/ 10.5435/JAAOS-21-04-225 [DOI] [PubMed] [Google Scholar]
- [76].Turcotte RE, Kurt AM, Sim FH, Unni KK, McLeod RA. Chondroblastoma. Hum Pathol 1993; 24:944-9; PMID:8253461; http://dx.doi.org/ 10.1016/0046-8177(93)90107-R [DOI] [PubMed] [Google Scholar]
- [77].Romeo S, Hogendoorn PCW, Dei Tos AP. Benign cartilaginous tumors of bone: from morphology to somatic and germ-line genetics. Adv Anat Pathol 2009; 16:307-15; PMID:19700940; http://dx.doi.org/ 10.1097/PAP.0b013e3181b506a1 [DOI] [PubMed] [Google Scholar]
- [78].Chan KM, Han J, Fang D, Gan H, Zhang Z. A lesson learned from the H3.3K27M mutation found in pediatric glioma. Cell Cycle 2013; 12:2546-52; PMID:23907119; http://dx.doi.org/ 10.4161/cc.25625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28:511-5; PMID:20436464; http://dx.doi.org/ 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Benjamini Y, Speed TP. RSeQC: Quality Control of RNA-seq experiments. Bioinformatics 2012; 40:e72. [DOI] [PubMed] [Google Scholar]
- [81].Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10:R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]