

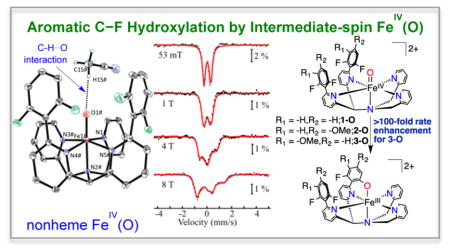
Abstract
The synthesis and reactivity of a series of mononuclear nonheme iron complexes that carry out intramolecular aromatic C–F hydroxylation reactions is reported. The key intermediate prior to C–F hydroxylation, [FeIV(O)-(N4Py2Ar1)](BF4)2 (1-O, Ar1 = −2,6-difluorophenyl), was characterized by single-crystal X-ray diffraction. The crystal structure revealed a nonbonding C–H···O=Fe interaction with a CH3CN molecule. Variable-field Mössbauer spectroscopy of 1-O indicates an intermediate-spin (S = 1) ground state. The Mössbauer parameters for 1-O include an unusually small quadrupole splitting for a triplet FeIV(O) and are reproduced well by density functional theory calculations. With the aim of investigating the initial step for C–F hydroxylation, two new ligands were synthesized, N4Py2Ar2 (L2, Ar2 = −2,6-difluoro-4-methoxyphenyl) and N4Py2Ar3 (L3, Ar3 = −2,6-difluoro-3-methoxyphenyl), with –OMe substituents in the meta or ortho/para positions with respect to the C–F bonds. FeII complexes [Fe(N4Py2Ar2)(CH3CN)]-(ClO4)2 (2) and [Fe(N4Py2Ar3)(CH3CN)](ClO4)2 (3) reacted with isopropyl 2-iodoxybenzoate to give the C–F hydroxylated FeIII–OAr products. The FeIV(O) intermediates 2-O and 3-O were trapped at low temperature and characterized. Complex 2-O displayed a C–F hydroxylation rate similar to that of 1-O. In contrast, the kinetics (via stopped-flow UV–vis) for complex 3-O displayed a significant rate enhancement for C–F hydroxylation. Eyring analysis revealed the activation barriers for the C–F hydroxylation reaction for the three complexes, consistent with the observed difference in reactivity. A terminal FeII(OH) complex (4) was prepared independently to investigate the possibility of a nucleophilic aromatic substitution pathway, but the stability of 4 rules out this mechanism. Taken together the data fully support an electrophilic C–F hydroxylation mechanism.
Graphical Abstract
INTRODUCTION
The aromatic amino acid hydroxylases phenylalanine hydroxylase (PheH), tryptophan hydroxylase (TrpH), and tyrosine hydroxylase (TyrH) comprise a class of nonheme iron oxygenases that are responsible for the hydroxylation of aromatic rings on the amino acid side chain. The proposed mechanism of the catalytic cycle involves formation of a high-valent FeIV(O) (ferryl) intermediate that carries out an electrophilic attack on the aromatic ring as the initial step in the hydroxylation reaction.1–3 Spectroscopic evidence for the intermediacy of an FeIV(O) species is well-documented for TyrH and PheH.4,5 The analogous hydroxylation of aromatic substrates by synthetic nonheme iron complexes is known, but the nature of the active intermediate(s) has not been established. Both FeIV(O) and FeV(O) species were proposed as active oxidants in the absence of direct spectroscopic evidence.6–20
The spin state of FeIV(O) species has been promoted as one of the key factors that determines reactivity.21–28 Nonheme iron enzymes, including the former hydroxylases, access high-spin (S = 2, quintet) FeIV(O) intermediates,29,30 whereas most synthetic FeIV(O) complexes exhibit intermediate-spin (S = 1, triplet) ground states. No intermediate-spin FeIV(O) complexes were shown to promote aromatic hydroxylation, with the exception of a reaction with anthracene (but not with benzene or naphthalene) to give anthraquinone product.7 Only recently was aromatic hydroxylation by a synthetic high-spin FeIV(O) complex reported.31 The generally enhanced reactivity of nonheme Fe enzymes as compared to that of the synthetic systems has been ascribed, in part, to the difference in spin ground state for the ferryl intermediate, and the observations regarding aromatic hydroxylation appear to follow this trend. The origin of the enhanced reactivity of a quintet versus triplet FeIV(O) species has been debated and was examined mainly by computational studies. Calculations suggested that part of the reason for the low reactivity of triplet FeIV(O) was because of steric clash between the incoming aromatic substrate and the equatorial ligands, which blocked access to the key π* acceptor orbitals on the FeIV(O) unit.6,7,25,32
Recently, we provided experimental evidence showing that FeIV(O) complexes are capable of aromatic hydroxylation provided that the aromatic substrate can be oriented properly in the second coordination sphere.15,33 In one case, we directly characterized the FeIV(O) intermediate [FeIV(O)(N4Py2Ar1)]2+ (Ar1 = −2,6-difluorophenyl), and observed the intramolecular arene hydroxylation reaction for this system by UV–vis, Mössbauer, and cold-spray ionization mass spectrometry (CSIMS). This study provided only the second example of a well-characterized FeIV(O) complex that could mediate arene hydroxylation. It also represented the first example of an aromatic C–F hydroxylation mediated by a nonheme iron complex. The tethered aromatic ring was fluorinated at the site of oxidation.33 Density functional theory (DFT) calculations suggested that this FeIV(O) complex not only had a triplet ground state but also indicated that a quintet excited state may be close in energy. The presumed 6-coordinate geometry of this complex was consistent with a triplet state, although there were recent reports of quintet FeIV(O) accommodated by a weak 6-coordinate ligand field.34,35 The low-field Mössbauer parameters of the FeIV(O) complex exhibited the anticipated low isomer shift (δ = 0.03 mm/s) for an FeIV(O) species, but the magnitude of the quadrupole splitting (ΔEQ = 0.54 mm/s) was noticeably smaller than that of other intermediate-spin FeIV(O).36 Thus, the spin ground state assignment for [FeIV(O)(N4Py2Ar1)]2+ was ambiguous. The spin-state assignment for the active FeIV(O) intermediate is critical to understand whether intermediate spin (S = 1) nonheme FeIV(O) complexes are inherently capable of performing these oxidation reactions.
The mechanism of C–F hydroxylation for [FeIV(O)-(N4Py2Ar1)]2+ was suggested to involve electrophilic attack of FeIV(O) on the nearby aryl group. However, the difluorosubstituted phenyl groups are electron-poor, making electrophilic attack challenging. Fluoro-substituted phenyl rings are known to undergo nucleophilic substitution, including with a metal-bound hydroxide.37,38 An alternative mechanism would involve nucleophilic aromatic substitution by a putative FeIII(OH) intermediate, which could form via H atom transfer to FeIV(O) from solvent or perhaps from a comproportionation reaction between FeIV(O) and the starting FeII complex.
In the current work we determine the spin state of the FeIV(O) intermediate and identify the nature of the initial step in the mechanism of the C–F hydroxylation by a combination of ligand design, reactivity studies, and spectroscopic methods. The FeII complexes of two new ligands N4Py2Ar2 (Ar2 = −2,6-difluoro-4-methoxyphenyl) and N4Py2Ar3 (Ar3 = −2,6-difluoro-3-methoxyphenyl) were synthesized. These ligands were designed to include electron-donating methoxy substituents on the phenyl rings in either the meta or ortho/para positions in relation to the fluorine groups. Both of the new FeII complexes undergo intramolecular aromatic C–F hydroxylation upon reaction with the appropriate oxidant. The relative reactivity of these complexes, together with the synthesis of a new terminal iron(II)–hydroxide complex, provide critical insight into the mechanism of C–F hydroxylation. The geometric and electronic structure of the key metastable FeIV(O) species was determined by variable-field Mössbauer spectroscopy, as well as by single-crystal X-ray crystallography.
RESULTS AND DISCUSSION
Structural Characterization of the Ferryl Intermediate [FeIV(O)(N4Py2Ar1)](BF4)2 (1-O)
In an earlier report, it was shown that the FeII precursor [FeII(N4Py2Ar1)(CH3CN)](BF4)2 (1, Ar1 = 2,6-difluorophenyl) reacted with O atom transfer agents to give the ferryl complex 1-O, which could be trapped at −20 °C and characterized by UV–vis, low-field Mössbauer spectroscopy, and CSIMS.33 Upon warming, this complex readily converted to the arene hydroxylated product 1-OAr as shown in Scheme 1, and time-dependent studies confirmed 1-O was the reactive intermediate in this rare C–F hydroxylation reaction. Given the novel reactivity of this FeIV(O) complex, we were motivated to obtain the complete structural and electronic characterization of this complex by X-ray crystallography and variable-field Mössbauer spectroscopy. Although a number of synthetic nonheme FeIV(O) complexes have been reported, only a few have been crystallographically characterized to date.39–45
Scheme 1.

Ligand N4Py2Ar1 (L1) and the [(L1)FeIV(O)]2+- Mediated Aromatic C–F Hydroxylation
The [FeIV(O)(N4Py2Ar1)]2+ complex (1-O) was generated from the reaction of [FeII(N4Py2Ar1)(CH3CN)]2+ (1) and isopropyl 2-iodoxybenzoate (IBX-ester) at −20 °C. Yellow crystals of 1-O were obtained by layering of Et2O into the CH3CN reaction mixture at −70 °C. The crystal structure of 1-O is given in Figure 1 and shows a 6-coordinate iron complex with a terminal oxo ligand in the open site. The short Fe–O distance of 1.6600(16) Å is characteristic of an iron(IV)–oxo complex.39–45 The Fe–N distances (1.9730(18)–2.0511(17) Å) are also in the expected range for an FeIV(O) species. As depicted in Figure 1, a single CH3CN molecule is found in close proximity to the FeIV(O) unit. The H···O distance (2.54(3) Å) and C–H···O angle (170(3)°) indicate a non-bonding interaction between the methyl C–H of the solvent molecule and the terminal oxo ligand. Similar nonbonding C–H···O interactions have been observed in other structurally characterized FeIV(O) complexes, but the C–H–O angles in these structures are significantly bent (100.4–131.6°).39,42,43 The experimentally determined C–H–O angle in 1-O is 170(3)° and is the closest to linearity among the synthetic FeIV(O) complexes. It is intriguing to consider that this interaction resembles the early stage of an H atom transfer from the C–H bond to the FeIV(O) unit.
Figure 1.

Displacement ellipsoid plot (30% probability) of the cation of 1-O at 110(2) K, depicting the C–H···O interaction in the asymmetric unit. All H atoms except for those in the CH3CN molecule were omitted for clarity. Selected bond distances: Fe1–N1 1.9730(18), Fe1–N2 2.0511(17), Fe1–N3 1.9771(18), Fe1–N4 2.0408(17), Fe1–N5 2.0269(17), Fe1–O1 1.6600(16).
The spin state of FeIV(O) complex 1-O cannot be assigned from a simple analysis of the bond lengths in the crystal structure. As seen by comparison of the high- and intermediate-spin FeIV(O) species in Table 1, there is no clear experimentally observed trend seen for bond lengths versus observed spin ground state.46 However, it should be noted that the S = 2 complexes in Table 1 are 5-coordinate species, whereas the S = 1 complexes are 6-coordinate. The enhanced reactivity of 1-O toward arene hydroxylation suggested that this complex could be an unusual 6-coordinate high-spin FeIV(O). The only other characterized FeIV(O) complex that mediates arene hydroxylation is a high-spin S = 2 species.31 In addition, low-field Mössbauer data showed that 1-O exhibits an isomer shift consistent with either intermediate- or high-spin FeIV(O) but a quadrupole splitting closer to the high-spin complexes (Table 1). It is known that the addition of bulky aryl substituents ortho to pyridyl N donors results in the conversion of low-spin iron(II) to high-spin iron(II) complexes.15 We thus sought to analyze the spin state of 1-O by variable-field Mössbauer spectroscopy.
Table 1.
Select Bond Distances (Å), Mössbauer Parameters (mm/s), and Spin State Assignments for Nonheme FeIV(O) Complexes Characterized by XRD
| FeIV(O) | δ, ΔEQ | Fe–N distances
|
Fe–O distance | spin state | |
|---|---|---|---|---|---|
| avg Fe–Neq | Fe–Nax | ||||
| [FeIV(O)(N4Py)]2+ | −0.04, 0.93 | 1.957 | 2.033(8) | 1.639(5) | 1 (ref 40) |
| [FeIV(O)(TMC-py)]2+ | 0.18, 1.08 | 2.083 | 2.118(3) | 1.667(3) | 1 (ref 44) |
| [FeIV(O)(TMC)(CH3CN)]2+ | 0.17, 1.24 | 2.091 | 2.058(3) | 1.646(3) | 1 (ref 43) |
| [FeIV(O)(d36-TMG3tren)]2+ | 0.09, −0.29 | 2.005 | 2.112(3) | 1.661(2) | 2 (ref 39) |
| [FeIV(Osyn)(TMC)(OTf)]+ | 0.16, 1.55 | 2.068 | 1.625(4) | 1 (ref 45) | |
| [FeIV(O)H3buea]− | 0.02, 0.43 | 1.983 | 2.064(1) | 1.680(1) | 2 (ref 41) |
| [(LNHC)FeIV(O)(MeCN)]2+ | −0.13, 3.08 | 1.661 (3) | 1 (ref 42) | ||
| [FeIV(O)(N4Py2Ar1)]2+ | 0.03, 0.54 | 2.004 | 2.051(2) | 1.660(2) | ? (this work) |
Mössbauer Spectroscopy
The electronic structure of [FeIV(O)(N4Py2Ar1)](BF4)2 (1-O) was examined by variable-field Mössbauer spectroscopy. An 57Fe-enriched sample of 1-O was prepared from combining 57FeII(BF4)2 and free ligand in CH3CN, followed by addition of the oxidant at −20 °C, as previously described.33 Analysis of the spectra (Figure 2, left panel, black vertical bars) reveals that ~60% of the total intensity of the spectrum is attributable to 1-O. The remaining ~40% of the total intensity emanates from one or more ferric complexes and can be reasonably well approximated by the experimental spectra of a duplicate sample that was allowed to decay for 40 min at room temperature (green lines). Removal of the contribution of the ferric decay product results in the reference spectra of 1-O (Figure 2, right panel, black vertical bars). The 4.2 K/53 mT Mössbauer spectrum of 1-O displays a quadrupole doublet with parameters (δ = 0.03 mm/s and ΔEQ = 0.54 mm/s) identical to those previously reported.33 While the isomer shift is typical of ferryl complexes, the absolute magnitude of the quadrupole splitting parameters is smaller than those observed for other ferryl complexes (ΔEQ ≈ 1.2 mm/s).36 However, spectroscopic parameters calculated using density functional theory (DFT) methods on geometry-optimized structures of 1-O and the extensively characterized intermediate-spin ferryl complex with 1,4,8,11-tetramethylcyclam ligand [(TMC)FeIV(O)(CH3CN)]2+ are in good agreement with the experimentally observed values (Table S1). The variable-field Mössbauer spectra of 1-O are similar to those previously reported for intermediate-spin FeIV(O) complexes and establish that 1-O has a S = 1 ground state. Because the spin Hamiltonian parameters required for analysis of the fielddependent spectra are correlated,47 we fixed the values of the A-tensor to those calculated for 1-O and allowed the remaining parameters to vary. This approach yields zero-field splitting (ZFS) parameters D ≈ 23 cm−1 and a small rhombicity (E/D), i.e., values that are typical of intermediate-spin ferryl complexes. The nearly axial ZFS tensor with a positive D produces a well-isolated ground state, which has a moderate spin expectation value in the xy plane, 〈Sx〉 ≈ 〈Sy〉 but only a vanishingly small 〈Sz〉.
Figure 2.

4.2 K Mössbauer spectra of 1-O. Left panel: Spectra recorded in externally applied magnetic field with field strength indicated in the figure (black vertical lines). The solid green lines are the experimental spectra of a duplicate sample of 1 + IBX-ester reacted at 23 °C for 40 min, scaled to 38% of intensity. Right panel: Reference spectra of 1-O generated by removal of the contribution of the decay products (black vertical lines). The solid red lines are simulations for S = 1 spin Hamiltonian using the following parameters: D = 23 cm−1, E/D = 0.08, giso = 2.0, δ = 0.03 mm/s, ΔEQ = +0.54 mm/s, η = 0, and A = (−29.1, −27.8, −6.5) MHz.
New Aryl-Substituted FeII Complexes: Mechanistic Implications
The C–F hydroxylation reaction observed for 1-O was unprecedented and suggested that an intermediate-spin FeIV(O) species could function as a powerful oxidant provided that the substrate was oriented properly in the second coordination sphere. However, fluorinated aromatic rings are strongly resistant to electrophilic attack but are susceptible to nucleophilic aromatic substitution. There is also precedent for nucleophilic substitution of aryl C–F bonds by a metal-bound hydroxide intermediate.37 A similar nucleophilic pathway conceivably could occur if an intermediate Fe(OH) species was generated during the C–F hydroxylation reaction for 1. In our previous report, we postulated that the C–F hydroxylation reaction was likely initiated by electrophilic attack of FeIV(O) on the difluorophenyl ring, although direct experimental evidence was lacking for this mechanism. Subsequent to our initial study, another example of nonheme-iron-mediated intramolecular C–F hydroxylation was reported.20 The possibility of either an electrophilic or nucleophilic mechanism was invoked for the observed reactivity. However, no direct evidence for either mechanism was provided.
To probe the nature of the initial step of the C–F hydroxylation mechanism, the N4Py2Ar1 ligand was modified with electron-donating methoxy substituents on the difluorophenyl rings. The new ligands N4Py2Ar2 (L2) and N4Py2Ar3 (L3) were prepared following a route similar to that for N4Py2Ar1 (Scheme 2).33 The key step involved 2,6-difluoro-4-methoxyphenylboronic acid (for L2) or 2,6-difluoro-3-methoxyphenylboronic acid (for L3) starting materials in Suzuki–Miyaura coupling reactions. Employing XPhos-Pd-G2 as the precatalyst, the resulting C–C coupling products were obtained in reasonable yields (49–65%). The completed ligands L2 and L3 were synthesized according to Scheme 2 and isolated as pure solids after chromatography on basic alumina.
Scheme 2.

Synthetic Route for the Preparation of L2 and L3
Synthesis and Structure of a meta-OMe Substituted FeII Complex
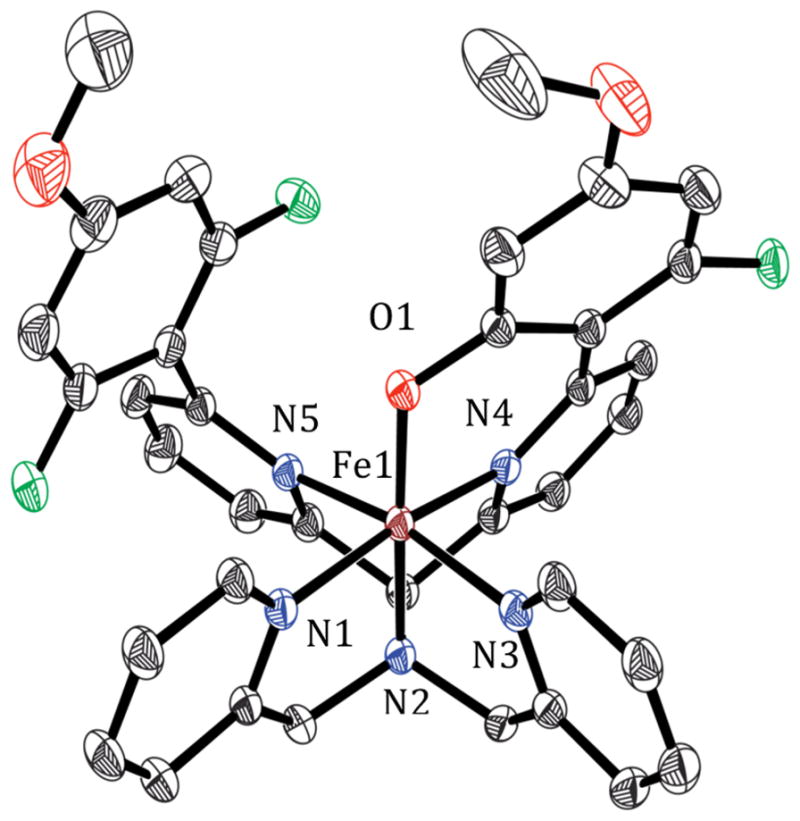
The reaction of N4Py2Ar2 (L2), with an –OMe group positioned meta to the fluorine substituents, and Fe(ClO4)2 in CH3CN followed by vapor diffusion of Et2O yielded crystals of [FeII(N4Py2Ar2)(CH3CN)](ClO4)2 (2). X-ray diffraction analysis (at 110(2) K) of 2 revealed a six-coordinate iron complex with a CH3CN molecule in the open site (Figure 3). The Fe–NPy bond lengths (1.949(2)–2.063(2) Å) seen in the crystal structure at 110(2) K are indicative of a low-spin (ls)-Fe2+ complex.48 The Fe–NPy bonds carrying the fluorinated aryl rings are longer (2.063(2) and 2.030(2) Å) than the other Fe–NPy bonds (1.961(2) and 1.949(2) Å) because of steric interactions caused by the aryl substituents.15,33 Although the crystal structure indicates an ls-FeII species, the 1H NMR spectrum for 2 in CD3CN at 297 K showed paramagnetically shifted peaks (100.3 to −1.43 ppm). A solution-state Evans method measurement for 2 in CD3CN (at 297 K) gave a magnetic moment value of μeff = 4.0 μB, consistent with high-spin (hs)-Fe2+ (S = 2, μeff(calcd) = 4.9 μB). Mössbauer spectra (4.2 K) of 2 ([57Fe(N4Py2Ar2)(CH3CN)](ClO4)2) in frozen solution revealed a mixture of ls- and hs-Fe2+ complexes (Figure S20). These data are consistent with population of both hs- and ls-FeII for 2 in the solution state, which is also temperature-dependent.
Figure 3.

Displacement ellipsoid plot (50% probability) of the cation of 2 at 110(2) K. The H atoms were omitted for clarity. Selected bond distances (in Å): Fe1–N1 1.9614(19), Fe1–N2 1.9599(19), Fe1–N3 1.949(2), Fe1–N4 2.0626(19), Fe1–N5 2.030(2), Fe1–N6 1.927(2).
C–F Hydroxylation with 2
The reaction of 2 at 23 °C with the O atom transfer reagent IBX-ester in CH3CN (Scheme 3) resulted in immediate decay (within 3 min) of the FeII peaks (460 and 370 nm) and formation of a broad, weak band at 750 nm. This band slowly converted into a relatively intense peak at 780 nm (Figure 4) over the next ~40 min. The weak band at 750 nm is characteristic of nonheme FeIV(O) complexes and is similar to the UV–vis feature seen for 1-O. The final spectrum with a peak at 780 nm is close to that previously reported for FeIII–OAr complex [FeIII(N4PyAr1,Ar1O)]2+.33 Analysis of the final green solution by electrospray ionization mass spectrometry (ESIMS) revealed a parent ion at m/z 352.0784, indicating formation of the C–F hydroxylated complex [FeIII(N4PyAr2,Ar2O)]2+ (calcd m/z 352.0781). A peak for a chloride-associated ion, [FeIII(N4PyAr2,Ar2O)Cl]+ (m/z 739.1276), was also observed, where the ESIMS instrument likely provides the adventitious chloride.
Scheme 3.

Conversion of 2 to 2-OAr
Figure 4.

UV–vis spectral changes for the reaction of 2 (0.47 mM; 3.0 mL) + IBX-ester (1 equiv) in CH3CN at 23 °C (left) and plot of A780 versus time and best fit (red line, right).
Conclusive evidence of the occurrence of C–F hydroxylation and the formation of the phenolate-bound iron complex was obtained by X-ray crystallography (Figure 5). Green crystals of the FeIII–OAr complex 2-OAr were obtained by vapor diffusion of Et2O into CH3CN. The structure revealed a 6-coordinate FeIII complex with the expected phenolate coordination resulting from hydroxylation of one of the C–F bonds. The Fe–N (1.921(2)–2.018(2) Å) and Fe–O (1.814(2) Å) bond lengths are consistent with ls-FeIII, which was further supported by the observation of a rhombic S = 1/2 signal [g = (2.42, 2.12, 1.90)] in the X-band EPR spectrum (Figure S18). The signals with effective g-values of 6.37 and 4.24 in the EPR spectrum indicated the presence of a hs-Fe3+ species, which may arise from the [FeIII(F)(N4Py2Ar2)]2+ side-product generated during the reaction. The formation of an FeIII(F) species was observed in the previous example of FeIV(O)-mediated aromatic C–F hydroxylation.33 Manual isolation of the crystals of 2-OAr followed by molar absorptivity measurement of 2-OAr revealed 70% yield for the C–F hydroxylated product at 23 °C.
Figure 5.
Displacement ellipsoid plot (30% probability) of the cation of 2-OAr at 110(2) K. H atoms were omitted for clarity. Selected bond distances (in Å): Fe1–N1 1.963(2), Fe1–N2 1.969(3), Fe1–N3 1.966(3), Fe1–N4 1.921(2), Fe1–N5 2.018(2), Fe1–O1 1.814(2).
Trapping the FeIV(O) Intermediate (2-O)
Performing the reaction of 2 + IBX-ester at −20 °C (Scheme 3) and following by UV–vis revealed a weak absorption band at λmax = 750 nm consistent with formation of [FeIV(O)(N4Py2Ar2)]+ (2-O, Figure 6), similar to that observed for 1-O.33 This band is stable for at least 4 h at −20 °C. CSIMS of the solution at −20 °C revealed the parent ion peak at 361.5774, supporting the formation of [FeIV(O)(N4Py2Ar2)]2+ (calcd m/z 361.5773), and the isotope distribution pattern indicated the presence of another complex with formula consistent with FeIII(OH) in approximately a 2:1 FeIV(O)/FeIII(OH) ratio (Figure S9). Evidence for the formation of the ferryl species was obtained from Mössbauer spectroscopy (Figure S20). The 4.2 K/53 mT Mössbauer spectrum revealed that 2-O exhibits a quadrupole doublet with parameters (δ = 0.03 mm/s, ΔEQ = 0.48 mm/s) almost identical to 1-O. The 4.2 K/8 T spectrum further revealed that 2-O has a S = 1 ground state (Figure S20).
Figure 6.
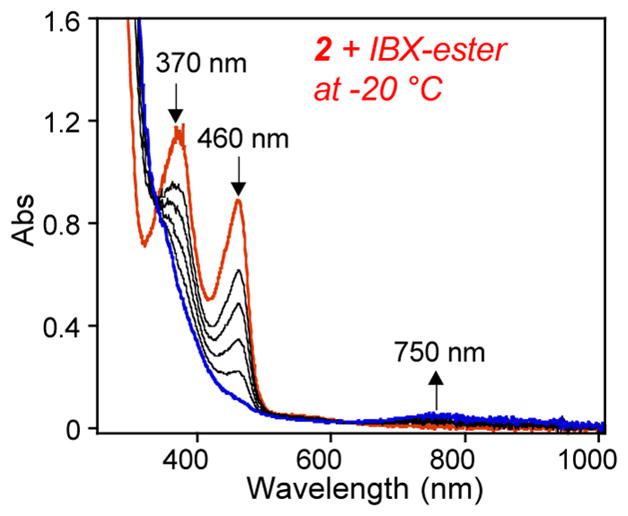
Reaction of 2 (1.04 mM) with IBX-ester (1.5 equiv) at −20 °C, monitored by UV–vis spectroscopy.
Kinetic Analysis for 2
The observance of the FeIV(O) intermediate 2-O by UV–vis allowed us to measure the rate of C–F hydroxylation at room temperature (23 °C). The absorbance at 780 nm was plotted against time, assigning t = 0 following the time required to form FeIV(O) (750 nm). Fitting of these data gave a first-order rate constant of k2 = 6.7(±0.1) × 10−2 min−1. The rate constant is independent of the concentration of the iron complex (Figure S13), consistent with the intramolecular nature of the reaction. Performing the reaction in the presence of excess IBX-ester (5 equiv) also did not have an effect on the rate of C–F hydroxylation (rate constant 7.5(±0.1) × 10−2 min−1). For the C–F hydroxylation reaction mediated by the FeIV(O) complex 1-O, a first-order rate constant of k1 = 5.13 (±0.06) × 10−2 min−1 was reported.33 (the rate constant with 5 equiv of IBX-ester was found to be 8.2 × 10−2 (±0.1) min−1). Thus, the presence of the methoxy groups in the meta position with respect to the fluorine substituents in 2 has little effect on the rate of the C–F hydroxylation reaction.
Synthesis of an ortho-/para-OMe-Substituted FeII Complex
Observing little effect of the meta-methoxy substituent on the rate of C–F hydroxylation, we turned our attention toward preparing an FeII complex of N4Py2Ar3 (L3), with a methoxy substituent ortho/para to the C–F bonds. [Fe(N4Py2Ar3)(CH3CN)](ClO4)2 (3) was prepared similarly to complex 2, from the reaction of L2 with Fe(ClO4)2 in CH3CN. Complex 3 was characterized by UV–vis (λmax = 373, 460 nm), 1H NMR spectroscopy (paramagnetic spectrum in CD3CN at 24 °C), and ESIMS (parent ion at m/z 353.5798, calcd m/z 353.5798 for [FeII(N4Py2Ar3)]2+). The Mössbauer spectrum (4.2 K) for a frozen solution of 57Fe-labeled 3 showed the presence of both hs- and ls-Fe2+ complexes (Figure S21).
C–F Hydroxylation with 3
The reaction of 3 with IBX-ester at 23 °C (Scheme 4) resulted in an immediate change (~2 min) in color from yellow to green with the appearance of a significantly red-shifted, stable peak at 915 nm (ε = 1010 M−1 cm−1; based on total Fe) (Figure 7). ESIMS analysis of the final green solution revealed a parent ion at m/z 739.1238, corresponding to the C–F hydroxylated product with one Cl− associated [(FeIII(N4PyAr3,Ar3O))(Cl)]+ (calcd m/z 739.1260). X-band EPR spectra (Figure S19) confirmed the presence of ls-FeIII [g = (2.37, 2.12, and 1.91)] similar to that of 2-OAr and other previously reported phenolate-bound FeIII complexes. 15,33 A hs-Fe3+ signal was also observed in the EPR spectrum, which is likely due to the formation of the ([FeIII(N4Py2Ar3)(F)]2+ complex. A peak in the ESIMS at m/z 761.1274 supported this assignment (calcd m/z for [FeIII(N4Py2Ar3)(F)(Cl)]+ 761.1279, Figure S11). The observed UV–vis band at 915 nm for 3-OAr ([FeIII(N4PyAr3,Ar3O)]2+) is considerably red-shifted compared to that seen for either 1-OAr (no OMe substitution) or 2-OAr (meta-substituted –OMe).33 A similar red-shift in a phenolate-to-FeIII charge-transfer band was observed for an FeIII-TPA derivative upon substitution of an electron-donating methyl group para to the phenolate donor.9 In contrast to the reaction of 2 + IBX-ester, no evidence for an intermediate FeIV(O) species was observed by UV–vis at 23 °C.
Scheme 4.

Conversion of 3 to 3-OAr
Figure 7.
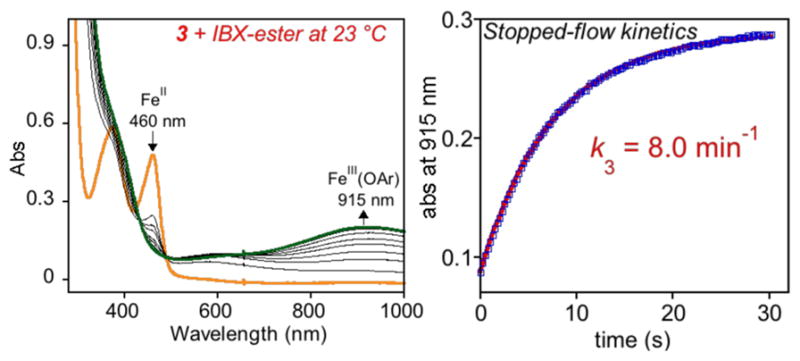
UV–vis spectral changes for 3 (0.2 mM) + IBX-ester (1.1 equiv) in CH3CN at 23 °C (left). Plot of absorbance at 915 nm versus time obtained from stopped-flow UV–vis spectroscopy and the best fit (red line, right).
Trapping the FeIV(O) Intermediate (3-O)
To trap the FeIV(O) intermediate, the reaction of 3 + IBX-ester was performed at −20 °C. UV–vis analysis showed formation of the 755 nm band associated with FeIV(O) (3-O), but unlike meta-substituted 2-O, this species was not stable at −20 °C, converting to the C–F hydroxylated product 3-OAr. Lowering the temperature further to −35 °C resulted in stabilization of the 755 nm band (Figure 8). 1H NMR spectroscopy of the reaction mixture revealed a paramagnetic spectrum with distinct, relatively sharp peaks at 49, −10, and −25 ppm. The 1H NMR spectra for 1-O and 2-O at −20 °C also revealed similar paramagnetically shifted peaks (Figure S6), supporting the assignment of the ferryl complex 3-O. The presence of the FeIV(O) complex at −35 °C was further demonstrated by CSIMS, which revealed a parent ion peak at m/z 362.0802 (calcd m/z for [FeIV(O)(N4Py2Ar3)]2+ 361.5773). Analysis of the isotope distribution pattern indicated almost an equal proportion of [FeIV(O)(N4Py2Ar3)]2+ (3-O) and [FeIII(OH)(N4Py2Ar3)]2+ species (Figure S12).
Figure 8.
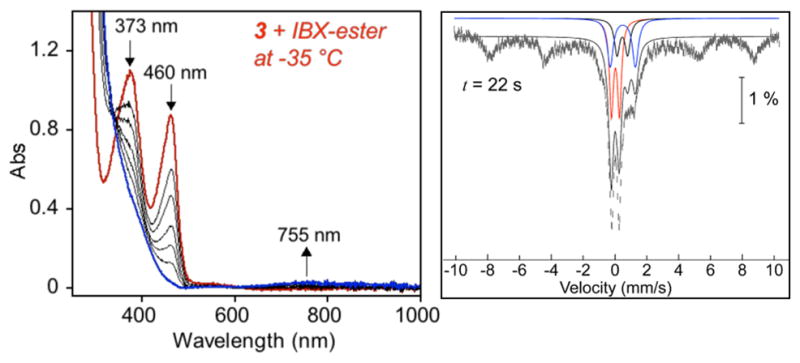
UV–vis spectral changes for the reaction of 3 (0.99 mM) with IBX-ester at −35 °C in CH3CN (left). Freeze-quench Mössbauer spectrum for 3 (5 mM) + IBX-ester at −5 °C in CH3CN/2-methylbutane after 22 s (right). The species are ls-FeII 3, thick solid black; hs-FeII 3, dashed black; FeII intermediate, blue; FeIVO intermediate, red; sum of fitted species, thin black line; Fe(III) products, unfitted.
Attempted Mössbauer characterization of 3-O from the reaction of [57FeII(N4Py2Ar3)(CH3CN)](ClO4)2 + IBX-ester at −35 °C revealed mostly an FeIII signal and thus was not successful. The difficulties in trapping 3-O by Mössbauer spectroscopy are possibly due to a comproportionation reaction between the initially formed FeIV(O) and unreacted FeII. The relatively high concentration of FeII required for Mössbauer analysis and slow kinetics for the reaction between 3 + IBX-ester at −35 °C both would facilitate the intermolecular reaction between the FeII and FeIV species. The lower yield for 3-O at low reaction temperature was also reflected in the lower yield for 3-OAr that arises when the FeIV(O) species generated at −35 °C was warmed to room temperature (21 °C), compared to the yield that was obtained when 3 + IBX-ester was performed at 21 °C (Figure S23). To minimize the possibility of FeIV(O) decay, a rapid-freeze-quench technique was applied for the reaction between 57Fe-labeled 3 + IBX-ester at −5 °C, and the reaction was probed by time-dependent Mössbauer spectroscopy (Figure S21). Analysis of the frozen reaction mixture at early time intervals reveals hs- and ls-FeII signals from starting material and a new intermediate with parameters consistent with a low- or intermediate-spin FeII species (formed within 18 s). These species begin to decay before the appearance of the FeIV(O) species (formed by ~22 s, and decay by ~29 s). The new, putative FeII intermediate was not characterized, although a reasonable possibility for its identity is an FeII(IBX-ester) precursor complex on the way to the FeIV(O) species. The ferryl complex 3-O exhibited a quadrupole doublet with isomer shift of 0.03 mm/s and quadrupole splitting of 0.50 mm/s (Figure 8), similar to those observed for complexes 1-O and 2-O, and suggesting that it has a similar electronic structure.
Kinetic Analysis for 3
Stopped-flow UV–vis spectroscopy was used to detect the FeIV(O) intermediate and obtain the rate constant for C–F hydroxylation in the reaction of 3 with IBX-ester at room temperature (22 °C). Mixing of 3 with IBX-ester in CH3CN led to the formation of the FeIV(O) (3-O) species (λmax = 755 nm) within 0.75 s, which then converted to the aryl hydroxylated complex 3-OAr (λmax = 915 nm) over ~35 s. Fitting of the data for the conversion of 3-O to 3-OAr led to a first-order rate constant of k3 = 8.0(±0.04) min−1 (Figure 7). This value is significantly higher than the rate constant obtained for 1-O (~160-fold increase), as well as for the meta-methoxysubstituted 2-O (~120 fold increase). The presence of the electron-donating –OMe group in the ortho/para position in relation to the C–F bonds results in a large increase in reaction rate.
Possible Mechanisms
Two possible pathways for the initial attack of the iron-bound O atom on the difluorophenyl ring can be envisioned and are shown in Scheme 5. The mechanism in Scheme 5A involves electrophilic attack on the arene ring by FeIV(O). Scheme 5B involves nucleophilic attack by a one-electron reduced iron(III)-hydroxide intermediate. It is important to consider this pathway as significant amounts of FeIII(OH) were seen during the formation of the ferryl complexes, particularly 2-O and 3-O. The thermal stabilities and relative rates of C–F hydroxylation for 1-O, 2-O, and 3-O provide insight into the mechanism of this initial step. Our mechanistic analysis rests on the assumption that the reaction proceeds by the same mechanism involving the ferryl intermediates 1-O, 2-O, and 3-O, which have been trapped and shown to exhibit similar spectroscopic parameters. These data in turn suggest that their electronic structures are similar, as can be expected based on the similarity of the ligands.
Scheme 5.

Possible Mechanistic Pathways for FeIV(O)- Mediated Aromatic C–F Hydroxylation
Addition of an electron-donating –OMe group to the ortho/para position relative to the C–F bonds can be expected to increase the rate of the electrophilic pathway because of its ability to stabilize the electron-deficient radical intermediate through resonance delocalization. A decrease in the rate of the nucleophilic pathway is also expected due to the same reasoning. In contrast, substitution at the meta position should have little influence on the rate of either pathway because it is not in conjugation with either radical or anionic intermediates (Scheme 5). The lower thermal stability of FeIV(O) and enhanced reactivity of C–F hydroxylation for the o-/p-OMesubstituted complex 3, as compared to the unsubstituted 1 and m-OMe-substituted 2, clearly point to the electrophilic mechanism, shown in pathway A. The lack of an effect for the m-OMe-substitution versus unsubstituted 1 is also consistent with this conclusion. We propose that the radical intermediate in Scheme 5A likely gets reduced by one electron, which can subsequently release fluoride to form the final C–F hydroxylated product. The source of the reducing equivalent has not been definitively characterized.
Activation Parameters
Temperature-dependent studies on the rate-constants (k1, k2, and k3) were performed on complexes 1–3 to obtain the activation parameters for the hydroxylation reactions. The plots of ln(k/T) versus 1/T (Eyring plot) for the conversion of FeIV(O) to FeIII(OAr) complex (20–45 °C for 1 and 2, 22 to −15 °C for 3) revealed a linear dependence as shown in Figure S17. Activation parameters (ΔH‡ and ΔS‡) were obtained and are given in Table 2. For the complexes 1 and 2, although the enthalpic (ΔH‡) and entropic contributions (ΔS‡) of the activation energies show some differences, the free energies of activation (ΔG‡) at 298 K are nearly identical. This similarity is expected given the similar reaction rates. The observed entropic contributions to the activation barriers are smaller here compared to bimolecular reactions involving FeIV(O) complexes7,49,50 and are indicative of an intramolecular mechanism. Moreover, ΔH‡ and ΔG‡ for the conversion of 1-O to 1-OAr match very well with those values obtained from previous DFT calculations where an electrophilic pathway was proposed (Table 2).33 Analysis of the Eyring plot for 3-O to 3-OAr revealed that the enthalpic and Gibbs free energy of activation for the o,p-OMe-substituted complex is smaller than the other two complexes (1-O and 2-O) by at least 5 kcal/mol. The smaller activation barrier for 3-O is consistent with the observed reactivity pattern, where 3-O undergoes the C–F hydroxylation reaction at a faster rate compared to the rates observed for 1-O and 2-O. These data further support the assignment of an intramolecular electrophilic pathway for C–F hydroxylation.
Table 2.
Activation Parameters for the FeIV(O)-Mediated Arene Hydroxylation Reactions for 1-O to 1-OAr and 2 to 2-OAr
| Parameters | 1-O to 1-Oar
|
2-O to 2-Oar | 3-O to 3-OAr | |
|---|---|---|---|---|
| exptl. | DFTa | |||
| ΔH‡b | 17.4(0.4) | 16.9 | 20.5(1.4) | 12.0(0.9) |
| ΔS‡c | −12.7(1.4) | −4.9d | −3.1(4.6) | −14.1(3.2) |
| ΔG‡b,e | 21.2(0.6) | 19.65 | 21.4(2.0) | 16.2(1.3) |
The values in the parentheses are the respective errors.
Taken from ref 33.
In kcal mol−1.
In cal mol−1 K−1.
Obtained from ΔG‡ = ΔH‡ − TΔS‡ at 298 K.
At 298 K.
Synthesis of a Terminal FeII(OH) Complex
A final experiment to eliminate the possibility of nucleophilic attack as the initial step in C–F hydroxylation (Scheme 5B) involved the synthesis of an FeII(OH) complex. The complex [FeII(OH)- (N4Py2Ar1)](ClO4) (4) was prepared from the reaction of the FeII(CH3CN) complex and NaOH in CH2Cl2 (Scheme 6). Dark red crystals of 4 were obtained from the layering of pentane with CH2Cl2. X-ray diffraction analysis of crystals of 4 revealed the presence of a terminal –OH group in the open site of the 6-coordinate FeII complex (Figure 9). The H atom attached to the hydroxide was found from difference Fourier maps. The Fe–N bond lengths (2.167(4)–2.304(4) Å and 2.175(4)–2.300(4) Å for two crystallographically independent cations) are indicative of a hs-FeII center in 4. The Fe–O bond distance (1.907(3) and 1.914(3) Å) is consistent with that of other structurally characterized FeII(OH) complexes.51–53
Scheme 6.

Synthesis of [FeII(OH)(N4Py2Ar1)](ClO4) (4)
Figure 9.
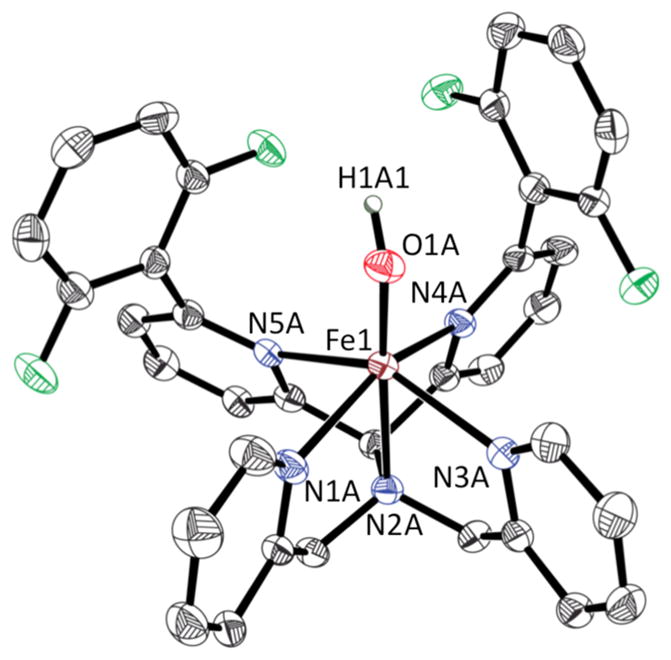
Displacement ellipsoid plot (50% probability) of one of the two crystallographically independent cations of 4 at 110(2) K. All H atoms were omitted for clarity (except for H1A1). Selected bond distances (in Å) for one of the crystallographically independent units: Fe1–N1A 2.175(4), Fe1–N2A 2.269(4), Fe1–N3A 2.223(4), Fe1– N4A 2.251(4), Fe1–N5A 2.300(4), Fe1–O1A 1.914(3).
The FeII(OH) complex 4 is stable both in the solid state as well as in solution upon redissolving the crystalline solid. No evidence for C–F hydroxylation was observed even upon prolonged standing in CH2Cl2. Treatment of the free ligand N4Py2Ar1 (L1) with excess NaOH also does not lead to the substitution of the C–F group. Attempts to prepare analogous FeIII(OH) were unsuccessful. However, given that a putative FeIII(OH) intermediate (Scheme 5B) should be less nucleophilic than either 4 or free OH−, it can be concluded that the nucleophilic pathway is not relevant here.
Summary and Conclusions
FeIV(O) complex 1-O formed during the intramolecular C–F hydroxylation reaction was isolated at low temperature and characterized by singlecrystal XRD. The crystal structure revealed the expected FeIV(O) structure along with a nonbonding C–H···O interaction between FeIV(O) and CH3CN. The spin ground state of this complex could not be determined by previous low-field Mössbauer measurements. Variable-field Mössbauer spectroscopy, together with DFT calculations, was successful in providing a definitive assignment of an intermediate-spin (S = 1) ground state for 1-O.
The hydroxylation of C–F bonds by a nonheme iron(IV)– oxo complex was unprecedented prior to our initial report,33 and we wanted to establish the role of the FeIV(O) complex in this process. There are two general mechanisms that can be envisioned, the first being electrophilic attack of FeIV(O) on the fluorinated aromatic ring and the second being nucleophilic aromatic substitution by an FeIV(O)-derived nucleophile. We reasoned that if the mechanism followed electrophilic attack, it would be enhanced by the addition of electrondonating substituents to the aromatic ring in the appropriate positions. With this hypothesis in hand, new FeII complexes 2 and 3 were synthesized, which incorporated electron-donating –OMe substituents in meta (2) or ortho/para (3) positions, respectively. Both complexes underwent C–F hydroxylation as seen for 1, and an FeIV(O) intermediate in both cases could be trapped at low temperature. The meta-substituted FeIV(O) complex 2-O exhibited similar reaction rates to that of 1-O, but the rate of C–F hydroxylation for ortho-/para-substituted 3-O was significantly higher (>100-fold) than that of either 1-O or 2-O. The reactivity pattern for 1-O, 2-O, and 3-O provides strong evidence that C–F hydroxylation occurs by electrophilic attack of the FeIV(O) on the arene ring. In addition, the synthesis and crystallographic characterization of iron(II)– hydroxide complex [FeII(OH)(N4Py2Ar1)]+ allowed us to examine the propensity for nucleophilic aromatic substitution of the internal C–F bonds. This pathway was ruled out because of the high stability of [FeII(OH)(N4Py2Ar1)]+ and a lack of any evidence for substitution of the C–F groups by hydroxide.
Aromatic hydroxylation is an important biochemical process and is mediated by the nonheme iron aromatic amino acid hydroxylases (TyrH, PheH, and TrpH). Spectroscopic and biochemical studies have pointed to a high-spin (S = 2) FeIV(O) as the reactive oxidant, which performs the key electrophilic attack on the aromatic substrate. Although nonheme iron model complexes were known to mediate aromatic hydroxylation, the active hydroxylating species could not be identified in these previous studies, except for a recent example of a hs-FeIV(O) that undergoes intramolecular arene hydroxylation. 31 In this work we have demonstrated that intermediatespin (S = 1) FeIV(O) species are capable of mediating aromatic hydroxylation, even with inert, electron-deficient C–F substituents. We have also expanded the set of well-characterized FeIV(O) species that carry out aromatic hydroxylation with the synthesis of two new FeIV(O) complexes.
These results support the conclusion that a triplet FeIV(O) can carry out challenging oxidations on par with quintet FeIV(O), provided that the substrate can be positioned properly in the second coordination sphere. We believe this study also highlights the power of synthetic inorganic chemistry to address fundamental mechanistic questions of biological relevance through ligand design and trapping of metastable intermediates. Similar studies regarding electrophilic and nucleophilic reactivity of FeIV(O) intermediates would be very difficult to perform directly on an enzymatic system.
EXPERIMENTAL SECTION
Materials and Methods
All reagents and chemicals were purchased from commercial sources and were used without further purification. The solvents were purified via Pure-Solv Solvent Purification System from Innovative Technology, Inc. Solvents used for inorganic reactions involving iron(II) complexes were subjected to additional purification steps. Acetonitrile was distilled over calcium hydride. All solvents were degassed by freeze–pump–thaw cycles and stored in a N2-filled drybox until ready for use. All the reactions were performed under inert atmosphere (using either standard Schlenk techniques or in a N2-filled drybox) unless otherwise noted. Bis(6-bromopyridin-2-yl)-methanone,54 isopropyl 2-iodoxybenzoate (IBX-ester),55 [FeII(N4Py2Ar1) (CH3CN)](BF4)2/(ClO4)2, and [57FeIV(O)(N4Py2Ar1)](BF4)233 were also prepared according to previously reported procedures.
Instrumentation
UV–visible spectra were recorded on a Varian Cary 50 Bio spectrophotometer and on an Agilent 5453 diode-array spectrophotometer. Low-temperature UV–vis experiments were carried out in a custom-made Schlenk flask fitted with a fiber optical UV–vis immersion probe (2 mm path length). Stopped-flow experiments were performed in a HiTech SHU-61SX2 (TgK scientific Ltd.) with xenon light source. The data was processed using Kinetic Studio software. Variable-temperature kinetic experiments (Eyring analysis) were performed on a Cary bio-50 spectrophotometer equipped with a Unisoku USP-203A cryostat using a 1 cm cuvette. NMR data were collected on either a Bruker Avance 400 MHz FT-NMR spectrometer (1H) or a Bruker 300 MHz FT-NMR spectrometer (19F). Electron paramagnetic resonance (EPR) spectra were obtained on a Bruker EMX EPR spectrometer controlled with a Bruker ER 041 X G microwave bridge. The spectrometer was equipped with a continuous-flow liquid He cryostat and an ITC503 temperature controller made by Oxford Instruments, Inc. Mössbauer spectra were recorded on spectrometers from SEECO (Edina, MN). The spectrometer used to acquire the weakfield spectra of 1-O and 2-O is equipped with a Janis SVT-400 variabletemperature cryostat. The spectrometer used to acquire the strong-field spectra of 1-O and 2-O is equipped with a Janis 8TMOSS-OM-12SVT variable-temperature cryostat. The spectrometer used to acquire the spectra of 3-O is equipped with a closed-cycle refrigerator system from Janis Research Co. and SHI (Sumitomo Heavy Industries, Ltd.). The external magnetic field was applied parallel to the γ beam. All isomer shifts quoted are relative to the centroid of the spectrum of α-iron metal at room temperature. Simulation of the Mössbauer spectra was carried out by using the WMOSS spectral analysis software from SEECO (www.wmoss.org; Edina, MN). For simulation of spectra in applied external fields, the commonly used spin-Hamiltonian formalism was used (eq 1), in which the first three terms represent the electron Zeeman effect and zero field splitting (ZFS) of the electron-spin ground state, the fourth term represents the interaction between the electric field gradient and the nuclear quadrupole moment, the fifth term describes the magnetic hyperfine interactions of the electronic spin with the 57Fe nucleus, and the last term represents the 57Fe nuclear Zeeman interaction.
| (1) |
FAB-MS was obtained using a VG analytical VG-70SE magnetic sector mass spectrometer. Cryospray-ionization MS (CSI-MS) measurements were performed on a UHR-TOF Bruker Daltonik (Bremen, Germany) maXis plus, an ESI-quadrupole time-of-flight (qToF) mass spectrometer capable of resolution of at least 60 000 fwhm, which was coupled to a Bruker Daltonik Cryospray unit. Detection was in positive ion mode; the source voltage was 3.8 kV. The flow rates were 280 μL/hour. The drying gas (N2), to aid solvent removal, was held at −35 °C, and the spray gas was held at −40 °C. The machine was calibrated prior to every experiment via direct infusion of the Agilent ESI-TOF low concentration tuning mixture, which provided an m/z range of singly charged peaks up to 2700 Da in both ion modes. Elemental analyses were performed by Atlantic Microlab Inc., Norcross, GA.
Synthesis of the Ligands
Both ligands L2 (N4Py2Ar2; Ar2 = −2,6-difluoro-4-methoxyphenyl) and L3 (N4Py2Ar3; Ar3 = −2,6-difluoro- 3-methoxyphenyl) were prepared in a similar synthetic route shown in Scheme 2.
Bis(6-(2,6-difluoro-4-methoxyphenyl)pyridin-2-yl)methanone
Amounts of bis(6-bromopyridin-2-yl)methanone (1.2 g, 3.51 mmol), 2,6-difluoro-4-methoxyphenyl boronic acid (1.45 g, 7.72 mmol), and XPhos-Pd-G2 (166 mg, 0.21 mmol) were taken in a Schlenk flask. The reagents were dissolved in degassed THF (~35 mL). Degassed 0.5 M K3PO4 solution in H2O (28 mL) was added to the stirring solution and the reaction allowed to continue for 72 h at 45 °C. After cooling the reaction to room temperature, the organic layer was extracted with Et2O and concentrated in vacuo. The product was purified by silica gel column chromatography using ethyl acetate/hexanes as eluent and was obtained as yellow solid (1.08 g, 66%). 1H NMR (CDCl3) δ 8.21 (dd, J = 7.8, 1.1 Hz, 2H), 7.94 (t, J = 7.8 Hz, 2H), 7.64–7.61 (m, 2H), 6.54 (d, J = 9.8 Hz, 4H), 3.82 (s, 6H). 19F NMR (CDCl3, C6F6 reference standard) δ −116.6.
Bis(6-(2,6-difluoro-3-methoxyphenyl)pyridin-2-yl)methanone
After following the method for bis(6-(2,6-difluoro-4-methoxyphenyl)- pyridin-2-yl)methanone, utilizing 2,6-difluoro-3-methoxyphenyl boronic acid as the reagent for the Suzuki–Miyaura coupling reaction, the ketone was isolated as yellow solid (49%). 1H NMR (CDCl3) δ 8.27 (dd, J = 7.9, 1.0 Hz, 2H), 7.99 (t, J = 7.8 Hz, 2H), 7.66 (dd, J = 7.8, 1.2 Hz, 2H), 7.01–6.89 (m, 4H), 3.90 (s, 6H). 19F NMR (CDCl3, C6F6 reference standard) δ −128.0, −138.2.
Bis(6-(2,6-difluoro-4-methoxyphenyl)pyridin-2-yl)methanamine
Hydroxylamine hydrochloride (0.24 g, 3.42 mmol) and sodium acetate (0.28 g, 3.42 mmol) were dissolved in H2O (6 mL) and stirred at 60 °C for 1 h under ambient air conditions. Bis(6-(2,6-difluoro-4- methoxyphenyl)pyridin-2-yl)methanone (0.8 g, 1.71 mmol) was added to the solution followed by ethyl alcohol (~200 mL). The reaction was stirred at 60 °C for 20 h, at which time the temperature was raised to 80 °C. Amounts of ammonium acetate (0.22 g, 2.91 mmol) and ammonium hydroxide solution (5.5 mL, 28–30% solution) were added. After 2 h of stirring, Zn dust (0.50 g, 7.69 mmol) was added in portions over 10 min. Stirring was continued for another 16 h at 80 °C. After cooling at room temperature, the solution was filtered through Celite. The filtrate was concentrated in vacuo. The residue was made basic by NaOH solution and extracted with CH2Cl2. The organic layer was dried over anhydrous MgSO4 and finally concentrated in vacuo to obtain bis(6-(2,6-difluoro-4-methoxyphenyl)-pyridin-2-yl)methanamine as a pale brown-red solid (0.69 g, 86%). 1H NMR (CDCl3) δ 7.67 (t, J = 7.8 Hz, 2H), 7.39 (dd, J = 7.9, 1.0 Hz, 2H), 7.31 (dd, J = 7.8, 1.3 Hz, 2H), 7.60–6.53 (m, 4H), 5.44 (s, 1H), 3.82 (s, 6H), 2.76 (s, 2H).
Bis(6-(2,6-difluoro-3-methoxyphenyl)pyridin-2-yl)methanamine
Following the procedure described above, the compound was obtained as brown-orange solid in 96% yield. 1H NMR (CDCl3) δ 7.72 (t, J = 7.8 Hz, 2H), 7.45 (dd, J = 7.9, 1.0 Hz, 2H), 7.34 (dd, J = 7.7, 1.2 Hz, 2H), 6.99–6.93 (m, 4H), 5.47 (s, 1H), 3.90 (s, 6H), 2.48 (s, 2H).
Synthesis of N4Py2Ar2 (L2) and N4Py2Ar3 (L3)
Bis(6-(2,6-difluoro-4-methoxyphenyl)pyridin-2-yl)methanamine (0.14 g, 0.30 mmol) was taken in CH3CN (30 mL) under ambient conditions. An amount of 2-bromomethylpyridine hydrobromide (0.19 g, 0.75 mmol) was added to the solution followed by Cs2CO3 (0.58 g, 1.79 mmol) and NaI (0.13 g, 0.89 mmol). The reaction was stirred at 60 °C for 72 h. The solution was filtered through Celite, and the filtrate was concentrated in vacuo. Ligand L2 was purified by column chromatography on basic alumina using ethyl acetate/hexanes and was obtained as a yellowishbrown solid (98 mg, 50%). 1H NMR (CDCl3) δ 8.44 (ddd, J = 4.9, 1.9, 0.9 Hz, 2H), 7.74 (dt, J = 7.9, 1.1 Hz, 2H), 7.67 (t, J = 7.8 Hz, 2H), 7.59–7.50 (m, 4H), 7.30 (dq, J = 7.7, 1.3 Hz, 2H), 7.02 (ddd, J = 7.5, 4.9, 1.2 Hz, 2H), 6.56–6.47 (m, 4H), 5.45 (s, 1H), 4.11 (s, 4H), 3.79 (s, 6H). 19F NMR (CDCl3, C6F6 reference standard) δ −116.4. FAB-MS for [L2 + H]+ observed m/z 652.23353, calcd m/z 652.23356.
Ligand N4Py2Ar3 (L3) was prepared in the same way as L2 by alkylating bis(6-(2,6-difluoro-3-methoxyphenyl)pyridin-2-yl)-methanamine (0.65 g, 1.38 mmol) and was obtained as a yellow solid (60%). 1H NMR (CDCl3) δ 8.44 (ddd, J = 4.9, 1.9, 0.9 Hz, 2H), 7.74–7.61 (m, 6H), 7.51 (td, J = 7.7, 1.8 Hz, 2H), 7.33 (dq, J = 7.3, 1.3 Hz, 2H), 7.01 (ddd, J = 7.5, 4.9, 1.2 Hz, 2H), 6.92–6.80 (m, 4H), 5.50 (s, 1H), 4.14 (s, 4H), 3.81 (s, 6H). 19F NMR (CDCl3, C6F6 reference standard) δ −127.6, −138.1. FAB-MS for [L3 + H]+ observed m/z 652.23399, calcd m/z 652.23356.
Synthesis of 2 and 3
An amount of N4Py2Ar2 (L2, 150 mg, 0.23 mmol) was dissolved in CH3CN (10 mL), and Fe(ClO4)2·6H2O (84 mg, 0.23 mmol) was added. The reaction was stirred for 3 h, and the solvent was removed in vacuo. The residue was redissolved in CH3CN (4 mL) and filtered through Celite. Vapor diffusion of Et2O into the CH3CN solution yielded dark brown-red crystals of [FeII(N4Py2Ar2)(CH3CN)](ClO4)2 (2) along with some brown-red precipitate. The crystals were collected, and a second crystallization (CH3CN/Et2O) was performed with the precipitate. A second batch of crystals of 2 was obtained in a few days. Combined yield: 140 mg (71%). UV–vis (CH3CN, 23 °C) λmax 460 nm (ε = 2420 M−1 cm−1), 370 nm (ε = 3260 M−1 cm−1). ESI-MS observed m/z 353.5795 (calcd m/z for [Fe(N4Py2Ar2)]2+ 353.5798). 1H NMR (CD3CN, TMS reference standard) δ 100.3, 80.8, 64.8, 38.5, 37.1, 36.4, 30.2, 22.4, 6.7, 6.0, 4.7, 3.8, 2.1, −1.4.
Complex [FeII(N4Py2Ar3)(CH3CN)](ClO4)2 (3) was prepared via a method similar to that used to obtain 2, using N4Py2Ar3 (L3, 250 mg, 0.38 mmol) and Fe(ClO4)2·6H2O (140 mg, 0.38 mmol). Complex 3 was obtained as a brown-red powder (260 mg, 72%). UV–vis (CH3CN, 23 °C) λmax 460 nm (ε = 2330 M−1 cm−1), 373 nm (ε = 2850 M−1 cm−1). ESI-MS observed m/z 353.5798 (calcd m/z for [Fe(N4Py2Ar3)]2+ 353.5798). 1H NMR (CD3CN, TMS reference standard) δ 74.8, 60.5, 49.0, 30.8, 29.5, 28.8, 24.2, 18.4, 8.1, 8.0, 5.0, 4.6, 4.1, 2.7, 2.1, 1.8.
[FeII(N4Py2Ar1)(OH)](ClO4) (4)
FeII complex [FeII(N4Py2Ar1)-(CH3CN)](ClO4)2 (87.7 mg; 0.099 mmol) was dissolved in CH2Cl2 (4 mL), and NaOH (aqueous, 79 μL, 20% w/v) was added under Ar atmosphere. The reaction mixture was stirred for 1 h, and the color of the solution changed from pale yellow to dark red. The volatiles were removed in vacuo, and the red residue was redissolved in CH2Cl2 (3 mL). The solution was filtered through Celite, and layering of pentane over the CH2Cl2 solution yielded red crystals of [FeII(N4Py2Ar1)(OH)](ClO4)·H2O (4·H2O, 47 mg, 61%). 1H NMR (CD2Cl2, TMS reference standard) δ 153.7, 98.9, 58.6, 54.0, 45.7, 44.8, 34.6, 31.0, 30.5, 27.0, 25.0, 8.2. UV–vis (CH2Cl2 under N2) λmax = 436 nm, 538 nm. Elemental Anal. (calcd for C35H28ClO6F4FeN5, 4·H2O) C, 53.76, H, 3.61, N, 8.96; found: C, 53.66, H, 3.55, N, 8.92.
Generation of 2-O and 3-O: UV–Vis Spectroscopy
An amount of complex 2 or 3 in CH3CN (1.04 mM for 2 and 0.99 mM for 3) was taken in a custom-made Schlenk flask fitted with a UV–vis dip-probe. The setup was cooled to −20 °C (−35 °C for 3). After recording the initial UV–vis spectrum for the starting FeII complex, IBX-ester (1.5 equiv for 2 and 3.0 equiv for 3) was added to the solution, and the changes in the UV–vis spectra were recorded.
Freeze-Quench Mössbauer for 3 + IBX-ester
Freeze-quenching of 57Fe-enriched samples of 3 was carried out using a custom quench system. Samples of 3 dissolved in acetonitrile (7.5 mM in 57Fe) were mixed anaerobically with IBX-ester (75 mM in acetonitrile) using gas-tight Hamilton syringes with 2:1 mixing in order that the complexes had a final concentration of 5 mM. IBX-ester (5 equiv) was mixed with complex 3 at −5 °C. The reaction temperature was kept constant using a DC10-K10 Haake water circulator. Samples were manually ejected and frozen in an 2-methylbutane bath kept below −135 °C using liquid nitrogen.
Kinetic Studies
Rate Constant for 2-O to 2-OAr (k2)
In a typical experiment, reaction of IBX-ester with complex 2 in CH3CN was followed by UV–vis spectroscopy. Addition of IBX-ester resulted in rapid decay of the FeII peaks (370 and 460 nm) along with the concomitant formation of the FeIV(O) species 2-O (750 nm). The kinetics of the reaction was followed after the full conversion of the 750 nm peak (t = 0). The 750 nm peak was subsequently converted into a more intense peak at 780 nm, corresponding to 2-OAr. The absorbance at 780 nm was plotted versus time, and the fitting of the data with the first-order rate equation (Abst = Absf + (Abs0 − Absf) exp(−kt); Abst = absorbance at time t; Absf = final absorbance at the end of the reaction; Abs0 = initial absorbance at t = 0) provided the rate constant (k2) for the C–F hydroxylation reaction.
Rate Constant for 3-O to 3-OAr (k3)
In a single-mixing experiment, complex 3 in CH3CN was reacted with IBX-ester (5 equiv), taken in CH3CN at 22 °C. Complete decay of the FeII peak (460 nm) and the formation of the FeIV(O) (755 nm) was observed within 0.75 s. Subsequent formation of the 915 nm peak (corresponding to 3-OAr) was plotted versus time and fitted with first-order rate equation (Abst = Absf + (Abs0 −Absf) exp(−kt)) to obtain the rate constant (k3).
X-ray Crystallography
All reflection intensities were measured at 110(2) K using a SuperNova diffractometer (equipped with Atlas detector) with Cu Kα radiation (λ = 1.54178 Å) under the program CrysAlisPro (Version 1.171.36.32 Agilent Technologies, 2013). The program CrysAlisPro (Version 1.171.36.32 Agilent Technologies, 2013) was used to refine the cell dimensions and for data reduction. The structure was solved with the program SHELXS-2013 (Sheldrick, 2013) and was refined on F2 with SHELXL-2013 (Sheldrick, 2013). Analytical numeric absorption corrections based on a multifaceted crystal model were applied using CrysAlisPro (Version 1.171.36.32 or 1.171.37.27t, Agilent Technologies, 2013). The temperature of the data collection was controlled using the system Cryojet (manufactured by Oxford Instruments). The H atoms were placed at calculated positions (unless otherwise specified) using the instructions AFIX 13, AFIX 23, AFIX 43, or AFIX 137 with isotropic displacement parameters having values 1.2 or 1.5 times Ueq of the attached C atoms. For 1-O, the coordinates of the H atoms attached to C1S (from CH3CN) were refined freely, and the C–H bonds were restrained to be 0.98(3) Å using the DFIX instruction. For 4, the H atoms attached to O1A/O1B (coordinated OH−) and O1W/O2W (lattice water molecules) were approximately located from difference Fourier maps, and the coordinates were refined freely. The O–H and H···H distances (water molecules only) were restrained within reasonable ranges using the DFIX instructions.
[FeIV(O)(N4Py2Ar1)](BF4)2 (1-O)
Before the last set of refinement cycles, the occupancy factor for O1 was refined freely using a free variable. Its refined value was 1.009(6), which proves that the O2− site is fully occupied. In the final refinement, the occupancy factor for O1 was set to 1. The Fe1–O1 distance refines to 1.6600(16). The structure is mostly ordered. However, one of the two crystallographically Independent BF4− counterions is found to be disordered over two orientations, and the occupancy factor of the major component of the disorder refines to 0.565(9). Chemical formula = C35H25F4FeN5O·2(BF4)·C2H3N, Fw = 878.12, small yellow block, crystal size =0.092 × 0.050 × 0.045 mm3, monoclinic, P21/c (no. 14), a = 11.7881(2), b = 16.3973(2), and c = 19.7780(3) Å, β = 105.5388(18)°, V = 3683.22(10) Å3, Z = 4, Dx = 1.584 g cm−3, μ = 4.208 mm−1, Tmin–Tmax: 0.736–0.863. A total of 47 739 reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. A total of 7232 reflections were unique (Rint = 0.0584), of which 6017 were observed [I > 2σ(I)]. A total of 581 parameters were refined using 151 restraints. R1/wR2 [I > 2σ(I)]: 0.0379/0.0907. R1/wR2 [all refln]: 0.0499/0.0978. S = 1.024. Residual electron density was found to be between −0.44 and 0.62 e Å−3.
[FeII(N4Py2Ar1)(CH3CN)](ClO4)2 (2)
The structure is mostly ordered. The two crystallographically independent ClO4− counterions are found to be disordered over two orientations, and the occupancy factors of the major components of the disorder refine to 0.54(5) and 0.588(19). Chemical formula = C39H32F4FeN6O2·2(ClO4), Fw = 947.45, orange-red lath, crystal size =0.39 × 0.13 × 0.05 mm3, monoclinic, P21/n (no. 14), a = 12.6196(3), b = 20.6085(5), and c = 14.9361(3) Å, β = 96.679(2)°, V = 3858.08(15) Å3, Z = 4, Dx = 1.631 g cm−3, μ = 5.192 mm−1, Tmin–Tmax: 0.290–0.823. A total of 25 214 reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. A total of 7568 reflections were unique (Rint = 0.0445), of which 6270 were observed [I > 2σ(I)]. A total of 636 parameters were refined using 278 restraints. R1/wR2 [I > 2σ(I)]: 0.0393/0.0939. R1/wR2 [all refln]: 0.0521/0.1022. S = 1.014. Residual electron density was found to be between −0.40 and 0.49 e Å−3.
[FeIII(N4PyAr2,Ar2O)](ClO4)2 (2-OAr)
The structure is significantly disordered (except for the Fe complex which is ordered). All perchlorate counterions are found to be significantly disordered as they are found at special positions, i.e., at sites of either mirror or 2-fold axial symmetries. The sum of all occupancies factors of the components of the disorder is restrained to be 2 using the SUMP instruction. The two methoxy groups of the Fe complex are also disordered over two orientations, and the occupancy factors of the major components of the disordered refine to 0.791(9) and 0.545(10). One lattice CH3CN solvent molecule is found at sites of 2-fold axial symmetry, and its occupancy must be 0.5. The crystal lattice also contains some unresolved residual electron density, which most likely corresponds to some heavily disordered lattice solvent molecules. Their contribution has been taken out in the final refinement (SQUEEZE details are provided in the CIF file, Spek, 2009). Chemical formula = (C37H29F3FeN5O3)·2(ClO4)·0.5C2H3N, Fw = 923.93, crystal size =0.23 × 0.18 × 0.13 mm3, monoclinic, I2/m, a = 11.6550(2), b = 26.4005(6), and c = 28.4632 (5) Å, β = 91.2677 (15)°, V = 8755.9 (3) Å3, Z = 4, Dx = 1.402 g cm−3, μ = 4.54 mm−1, Tmin–Tmax: 0.527−0.690. A total of 36 960 reflections were measured up to a resolution of (sin θ/λ)max = 0.616 Å−1. A total of 8791 reflections were unique (Rint = 0.041), of which 7375 were observed [I > 2σ(I)]. A total of 934 parameters were refined using 1531 restraints. R1/wR2 [I > 2σ(I)]: 0.0576/0.1669. R1/wR2 [all refln]: 0.0679/0.1771. S = 1.03 e Å−3.
[FeII(N4Py2Ar1)(OH)](ClO4) (4)
Before the last set of refinement cycles, the occupancy factors for O1A and O1B were refined freely as O atoms using free variables, and their refined values were 0.960(12) and 1.052(12). When refined as F atoms, their refined values were 0.797(10) and 0.875(10). Consequently, the sites O1A and O1B are unambiguously occupied by coordinated OH− and not F−. In the final refinement, the occupancy factors for O1A and OAB were set to 1. The structure is mostly ordered. The two crystallographically independent perchlorate counterions are found to be disordered over two or three orientations (all occupancy factors are provided in the CIF file). The crystal that was mounted on the diffractometer was non-merohedrally twinned, and the two twin components are related by a 2-fold axis along the direct space vector [1 0 0]. The BASF scale factor refines to 0.4628(11). The structure was solved and refined in the noncentrosymmetric space group P21. The absolute configuration was established by anomalous-dispersion effects in diffraction measurements on the crystal. The Flack parameter refines to −0.005(2). Chemical formula = C35H26F4FeN5O·ClO4·H2O, Fw = 781.92, dark red block, crystal size =0.38 × 0.34 × 0.19 mm3, monoclinic, P21 (no. 4), a = 14.4312(4), b = 12.8954(3), and c = 18.3009(5) Å, β = 92.161(2)°, V = 3403.30(15) Å3, Z = 4, Dx = 1.526 g cm−3, μ = 4.946 mm−1, abs. corr. range: 0.255–0.511. A total of 22 245 reflections were measured up to a resolution of (sin θ/λ)max = 0.62 Å−1. A total of 12 531 reflections were unique (Rint = 0.0165), of which 12 165 were observed [I > 2σ(I)]. A total of 1077 parameters were refined using 584 restraints. R1/wR2 [I > 2σ(I)]: 0.0332/0.0904. R1/wR2 [all refln]: 0.0340/0.0909. S = 1.018. Residual electron density was found to be between −0.37 and 0.85 e Å−3.
Computations
All computations were carried out using ORCA version 3.0.2.56 Geometry optimization of the ferryl complex was begun from the crystal structure coordinates and employed the BP86 functional57,58 and def2-TZVP basis set59 on all atoms except Fe, which had the expanded CP(PPP) basis set60 and an increased integration accuracy. Solvation was simulated using the conductorlike screening model (COSMO)61 in an infinite dielectric, while dispersion corrections were included using Grimme’s correction.62 Spectroscopic parameters were calculated using these optimized coordinates. Isomer shift and quadrupole splitting parameters were calculated at the same level of theory using a calibration procedure similar to that reported in ref 63 (see Supporting Information for additional details), while hyperfine coupling tensors were calculated as described previously.63 Optimized coordinates and sample input files for all calculations are included in the Supporting Information.
Supplementary Material
Acknowledgments
The NIH (D.P.G., GM62309 and GM119374) is acknowledged for financial support. We are grateful to Prof. Kenneth Karlin (JHU) for the access to stopped-flow UV–vis and low-temperature (−70 °C) freezer. I.I.-B. and M.D. gratefully acknowledge support through the “Solar Technologies Go Hybrid” initiative of the State of Bavaria.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03346.
Crystallographic information file for 1-O (CIF)
Crystallographic information file for 2 (CIF)
Crystallographic information file for 2-OAr (CIF)
Crystallographic information file for 4 (CIF)
Spectroscopic (1HNMR, EPR, Mössbauer), mass spectrometric, kinetic and crystallographic data (PDF)
References
- 1.Bruijnincx PCA, van Koten G, Klein Gebbink RJM. Chem Soc Rev. 2008;37:2716–2744. doi: 10.1039/b707179p. [DOI] [PubMed] [Google Scholar]
- 2.Fitzpatrick PF. Biochemistry. 2003;42:14083–14091. doi: 10.1021/bi035656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kovaleva EG, Lipscomb JD. Nat Chem Biol. 2008;4:186–193. doi: 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eser BE, Barr EW, Frantom PA, Saleh L, Bollinger JM, Jr, Krebs C, Fitzpatrick PF. J Am Chem Soc. 2007;129:11334–11335. doi: 10.1021/ja074446s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panay AJ, Lee M, Krebs C, Bollinger JM, Jr, Fitzpatrick PF. Biochemistry. 2011;50:1928–1933. doi: 10.1021/bi1019868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ansari A, Kaushik A, Rajaraman G. J Am Chem Soc. 2013;135:4235–4249. doi: 10.1021/ja307077f. [DOI] [PubMed] [Google Scholar]
- 7.de Visser SP, Oh K, Han AR, Nam W. Inorg Chem. 2007;46:4632–4641. doi: 10.1021/ic700462h. [DOI] [PubMed] [Google Scholar]
- 8.Hegg EL, Ho RYN, Que L., Jr J Am Chem Soc. 1999;121:1972–1073. [Google Scholar]
- 9.Jensen MP, Lange SJ, Mehn MP, Que EL, Que L., Jr J Am Chem Soc. 2003;125:2113–2128. doi: 10.1021/ja028478l. [DOI] [PubMed] [Google Scholar]
- 10.Makhlynets OV, Rybak-Akimova EV. Chem - Eur J. 2010;16:13995–14006. doi: 10.1002/chem.201002577. [DOI] [PubMed] [Google Scholar]
- 11.Mehn MP, Fujisawa K, Hegg EL, Que L., Jr J Am Chem Soc. 2003;125:7828–7842. doi: 10.1021/ja028867f. [DOI] [PubMed] [Google Scholar]
- 12.Mekmouche Y, Ménage S, Toia-Duboc C, Fontecave M, Galey JB, Lebrun C, Pécaut J. Angew Chem, Int Ed. 2001;40:949–952. [PubMed] [Google Scholar]
- 13.Nielsen A, Larsen FB, Bond AD, McKenzie CJ. Angew Chem, Int Ed. 2006;45:1602–1606. doi: 10.1002/anie.200502656. [DOI] [PubMed] [Google Scholar]
- 14.Oh NY, Seo MS, Lim MH, Consugar MB, Park MJ, Rohde JU, Han J, Kim KM, Kim J, Que L, Jr, Nam W. Chem Commun. 2005;5644–5646 doi: 10.1039/b511302d. [DOI] [PubMed] [Google Scholar]
- 15.Sahu S, Widger LR, Quesne MG, de Visser SP, Matsumura H, Moënne-Loccoz P, Siegler MA, Goldberg DPJ. Am Chem Soc. 2013;135:10590–10593. doi: 10.1021/ja402688t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thibon A, Jollet V, Ribal C, Sénéchal-David K, Billon L, Sorokin AB, Banse F. Chem - Eur J. 2012;18:2715–2724. doi: 10.1002/chem.201102252. [DOI] [PubMed] [Google Scholar]
- 17.Makhlynets OV, Das P, Taktak S, Flook M, Mas-Balleste R, Rybak-Akimova EV, Que L., Jr Chem - Eur J. 2009;15:13171–13180. doi: 10.1002/chem.200901296. [DOI] [PubMed] [Google Scholar]
- 18.Harman WH, Chang CJ. J Am Chem Soc. 2007;129:15128–15129. doi: 10.1021/ja076842g. [DOI] [PubMed] [Google Scholar]
- 19.Jensen MP, Mehn MP, Que L., Jr Angew Chem, Int Ed. 2003;42:4357–4360. doi: 10.1002/anie.200351605. [DOI] [PubMed] [Google Scholar]
- 20.de Ruiter G, Thompson NB, Takase MK, Agapie T. J Am Chem Soc. 2016;138:1486–1489. doi: 10.1021/jacs.5b12214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Visser SP. J Am Chem Soc. 2006;128:15809–15818. doi: 10.1021/ja065365j. [DOI] [PubMed] [Google Scholar]
- 22.de Visser SP. J Am Chem Soc. 2006;128:9813–9824. doi: 10.1021/ja061581g. [DOI] [PubMed] [Google Scholar]
- 23.Geng CY, Ye SF, Neese F. Angew Chem, Int Ed. 2010;49:5717–5720. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- 24.Neidig ML, Decker A, Choroba OW, Huang F, Kavana M, Moran GR, Spencer JB, Solomon EI. Proc Natl Acad Sci U S A. 2006;103:12966–12973. doi: 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srnec M, Wong SD, England J, Que L, Jr, Solomon EI. Proc Natl Acad Sci U S A. 2012;109:14326–14331. doi: 10.1073/pnas.1212693109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson SA, Chen J, Hong S, Lee YM, Clémancey M, Garcia-Serres R, Nomura T, Ogura T, Latour JM, Hedman B, Hodgson KO, Nam W, Solomon EI. J Am Chem Soc. 2012;134:11791–11806. doi: 10.1021/ja3046298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong SD, Bell CB, III, Liu LV, Kwak Y, England J, Alp EE, Zhao J, Que L, Jr, Solomon EI. Angew Chem, Int Ed. 2011;50:3215–3218. doi: 10.1002/anie.201007692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirao H, Kumar D, Que L, Jr, Shaik S. J Am Chem Soc. 2006;128:8590–8606. doi: 10.1021/ja061609o. [DOI] [PubMed] [Google Scholar]
- 29.Bollinger JM, Jr, Chang W-c, Matthews ML, Martinie RJ, Boal AK, Krebs C. 2-Oxoglutarate-Dependent Oxygenases. The Royal Society of Chemistry; London: 2015. pp. 95–122. [Google Scholar]
- 30.Krebs C, Galonić Fujimori DG, Walsh CT, Bollinger JM., Jr Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. J Am Chem Soc. 2012;134:1536–1542. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]
- 32.McDonald AR, Guo Y, Vu VV, Bominaar EL, Münck E, Que L., Jr Chem Sci. 2012;3:1680–1693. doi: 10.1039/C2SC01044E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sahu S, Quesne MG, Davies CG, Dürr M, Ivanović-Burmazović I, Siegler MA, Jameson GNL, de Visser SP, Goldberg DP. J Am Chem Soc. 2014;136:13542–13545. doi: 10.1021/ja507346t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Puri M, Biswas AN, Fan R, Guo Y, Que L., Jr J Am Chem Soc. 2016;138:2484–2487. doi: 10.1021/jacs.5b11511. [DOI] [PubMed] [Google Scholar]
- 35.Biswas AN, Puri M, Meier KK, Oloo WN, Rohde GT, Bominaar EL, Münck E, Que L., Jr J Am Chem Soc. 2015;137:2428–2431. doi: 10.1021/ja511757j. [DOI] [PubMed] [Google Scholar]
- 36.McDonald AR, Que L., Jr Coord Chem Rev. 2013;257:414–428. [Google Scholar]
- 37.Pearson DM, Conley NR, Waymouth RM. Organometallics. 2011;30:1445–1453. [Google Scholar]
- 38.Terrier F. Modern Nucleophilic Aromatic Substitution. Wiley- VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2013. [Google Scholar]
- 39.England J, Guo Y, Farquhar ER, Young VG, Jr, Münck E, Que L., Jr J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L., Jr Angew Chem, Int Ed. 2005;44:3690–3694. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 41.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyer S, Klawitter I, Demeshko S, Bill E, Meyer F. Angew Chem, Int Ed. 2013;52:901–905. doi: 10.1002/anie.201208044. [DOI] [PubMed] [Google Scholar]
- 43.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 44.Thibon A, England J, Martinho M, Young VG, Jr, Frisch JR, Guillot R, Girerd JJ, Münck E, Que L, Jr, Banse F. Angew Chem, Int Ed. 2008;47:7064–7067. doi: 10.1002/anie.200801832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prakash J, Rohde GT, Meier KK, Münck E, Que L., Jr Inorg Chem. 2015;54:11055–11057. doi: 10.1021/acs.inorgchem.5b02011. [DOI] [PubMed] [Google Scholar]
- 46.Neese F. J Inorg Biochem. 2006;100:716–726. doi: 10.1016/j.jinorgbio.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 47.Lim MH, Rohde JU, Stubna A, Bukowski MR, Costas M, Ho RY, Münck E, Nam W, Que L., Jr Proc Natl Acad Sci U S A. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Prat I, Company A, Corona T, Parella T, Ribas X, Costas M. Inorg Chem. 2013;52:9229–9244. doi: 10.1021/ic4004033. [DOI] [PubMed] [Google Scholar]
- 49.Ye W, Ho DM, Friedle S, Palluccio TD, Rybak-Akimova EV. Inorg Chem. 2012;51:5006–5021. doi: 10.1021/ic202435r. [DOI] [PubMed] [Google Scholar]
- 50.Ye W, Staples RJ, Rybak-Akimova EV. J Inorg Biochem. 2012;115:1–12. doi: 10.1016/j.jinorgbio.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Soo HS, Komor AC, Iavarone AT, Chang CJ. Inorg Chem. 2009;48:10024–10035. doi: 10.1021/ic9006668. [DOI] [PubMed] [Google Scholar]
- 52.Ogo S, Yamahara R, Roach M, Suenobu T, Aki M, Ogura T, Kitagawa T, Masuda H, Fukuzumi S, Watanabe Y. Inorg Chem. 2002;41:5513–5520. doi: 10.1021/ic0200040. [DOI] [PubMed] [Google Scholar]
- 53.Benisvy L, Halut S, Donnadieu B, Tuchagues JP, Chottard JC, Li Y. Inorg Chem. 2006;45:2403–2405. doi: 10.1021/ic060100r. [DOI] [PubMed] [Google Scholar]
- 54.Nierengarten H, Rojo J, Leize E, Lehn JM, Van Dorsselaer A. Eur J Inorg Chem. 2002;2002:573–579. [Google Scholar]
- 55.Zhdankin VV, Koposov AY, Litvinov DN, Ferguson MJ, McDonald R, Luu T, Tykwinski RR. J Org Chem. 2005;70:6484–6491. doi: 10.1021/jo051010r. [DOI] [PubMed] [Google Scholar]
- 56.Neese F. Wiley Interdisciplinary Reviews: Computational Molecular Science. 2012;2:73–78. doi: 10.1002/wcms.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Becke AD. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 58.Perdew JP. Phys Rev B: Condens Matter Mater Phys. 1986;33:8822. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 59.Pantazis DA, Chen XY, Landis CR, Neese F. J Chem Theory Comput. 2008;4:908–919. doi: 10.1021/ct800047t. [DOI] [PubMed] [Google Scholar]
- 60.Neese F. Inorg Chim Acta. 2002;337:181. [Google Scholar]
- 61.Klamt A, Schüürmann G. J Chem Soc, Perkin Trans 2. 1993;2:799–805. [Google Scholar]
- 62.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 63.Sinnecker S, Slep LD, Bill E, Neese F. Inorg Chem. 2005;44:2245–2254. doi: 10.1021/ic048609e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.