

Abstract
Tuberculosis is a major global health problem for which improved therapeutics are needed to shorten the course of treatment and combat emergence of drug resistance. Mycobacterium tuberculosis, the etiologic agent of tuberculosis, is an intracellular pathogen of mononuclear phagocytes. As such, it is an ideal pathogen for nanotherapeutics because macrophages avidly ingest nanoparticles even without specific targeting molecules. Hence, a nanoparticle drug delivery system has the potential to target and deliver high concentrations of drug directly into M. tuberculosis-infected cells – greatly enhancing efficacy while avoiding off-target toxicities. We developed stimulus-responsive mesoporous silica nanoparticles of two different sizes, 100 nm and 50 nm, as carriers for the major anti-tuberculosis drug isoniazid in a prodrug configuration. The drug is captured by the aldehyde-functionalized nanoparticle via hydrazone bond formation and coated with PEI-PEG. The drug is released from the nanoparticles in response to acidic pH at levels that naturally occur within acidified endolysosomes. We demonstrate that isoniazid-loaded PEI-PEG-coated nanoparticles are avidly ingested by M. tuberculosis-infected human macrophages and kill the intracellular bacteria in a dose-dependent manner. We further demonstrate in a mouse model of pulmonary tuberculosis that the nanoparticles are well tolerated and much more efficacious than an equivalent amount of free drug.
Keywords: Mesoporous silica nanoparticles, multifunctional nanoparticle, mesoporous silica in vitro and in vivo, nanoparticle drug delivery, tuberculosis
TOC
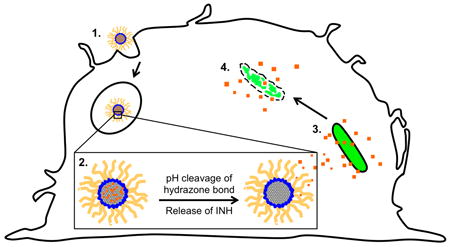
Depiction of novel pH-sensitive MSN-CHO-INH-PEI-PEG system (1) phagocytosed by a macrophage into the acidic endolysosome whereupon (2) INH is released with the cleavage of the hydrazone bond holding the cargo to the surface of the MSN inside the acidic vacuole; and (3 and 4) INH diffuses to and is delivered into the M. tuberculosis, thereby killing the bacterium.
Introduction
Tuberculosis (TB) is a devastating disease and global health problem that infects one third of the world’s population.[1] In 2013, worldwide, 9 million people fell ill with TB and 1.5 million died. Caused by Mycobacterium tuberculosis, TB ranks with HIV/AIDS as the greatest killers worldwide due to single infectious agents, and TB is a leading killer of HIV-positive people, causing one fourth of all HIV-related deaths.[1] While TB can be treated with antibiotics, the treatment duration is exceedingly long, requiring 6-9 months for drug-sensitive TB, and several times longer for drug-resistant TB. Thus improved therapeutic modalities are needed.
Nanotherapeutics have the potential to be more effective than free drug in treating TB. Nanotherapeutics can selectively deliver high concentrations of drug to macrophages, the primary host cells for TB, while avoiding off target effects, which limit the doses of many current TB drugs. For example, three of the first line drugs for treating TB – isoniazid (INH), rifampicin (RIF), and pyrazinamide (PZA) – are limited by hepatotoxicity.[2, 3] In particular, INH – a potent first line anti-TB drug used in standard regimens both for active and latent TB, at standard therapeutic dosages in humans, has adverse effects including neurotoxicity, optic neuritis, and severe hepatic injury.[3, 4] As these side effects are due to the action of the drug on hepatocytes and neuronal cells rather than on macrophages, selective delivery of these antibiotics into macrophages has the potential to increase greatly their therapeutic index by achieving higher drug concentrations locally where the M. tuberculosis replicate while limiting systemic toxicities. Moreover, because drug resistance develops when bacteria are treated with sub-therapeutic levels of antibiotics, a system that delivers high concentrations of antibiotic to the site where bacteria divide would facilitate sterilization of sites of infection and minimize emergence of drug resistance. Because host mononuclear phagocytes internalize particles more efficiently than other cells, encapsulation of anti-TB drugs within nanoparticles offers a mechanism for specific targeting of M. tuberculosis-infected cells. Indeed, intravenously (i.v.) injected nanoparticles are taken up by macrophages of the Mononuclear Phagocyte System and accumulate in liver, spleen, and lung,[5-7] hence, they are ideally suited to treat M. tuberculosis, which infects macrophages in these organs. An additional advantage of nanoparticle drug delivery vs. free drug is that nanoparticle delivery shields the antibiotics from hepatic degradation and renal clearance during delivery to infected tissues; hence, it has the potential to greatly improve the pharmacokinetic properties of the drug. Thus, a nanoparticle-based platform for TB drugs has the potential to be safer and more effective than free drug, reducing treatment duration and emergence of drug resistance.
A variety of different nanoparticles including liposomes, solid lipid particles, poly-L-lactide (PLGA), and biological materials such as gelatin, chitosan, and alginates have been tested in vitro and in vivo as delivery platforms for TB drugs[8-10] and shown therapeutic efficacy. While these nanoparticle delivery systems for TB drugs offer advantages over free drug, they have disadvantages compared with mesoporous silica nanoparticles (MSNs)[11] which offer improved structural and chemical stability, uniformity, inherent lack of toxicity, capacity to encapsulate exceptionally high concentrations of different types of cargo, and, most importantly, versatility in incorporating additional design features. With respect to stability, liposomes and solid lipid particles have intrinsically poor chemical stability and are degraded by serum,[12] decreasing drug delivery to target cells while increasing potential systemic toxicity; in contrast, the solid MSN framework provides intrinsic stability compared with existing liposome-, polymer-, and copolymer-based nano delivery platforms and MSNs show enhanced blood stability.[11] With respect to toxicity, biodegradable polymer based nanoparticles (e.g. PLGA), when ingested by macrophages, may cause cell damage, cytokine release, and inflammation,[13, 14] in contrast, MSNs display an inherent lack of toxicity. With respect to drug-loading capacity, MSNs have ultra-high internal surface area (~1000 m2 g-1), and consequently, loading capacities as high as 50 weight % have been achieved, exceeding by several orders of magnitude that of liposomal nanocarriers. Finally, with respect to functionalization, MSNs can be manufactured with a variety of different internal and surface design features,[15, 16] including those that allow for stimulus-responsive release of drug cargo under specific environmental conditions.[17-21] In addition, the MSNs can be manufactured with a variety of aspect ratios to optimize targeting of the particles to particular organs, tissues, cells, and intracellular environments.[22, 23] Finally, MSNs have demonstrated excellent biocompatibility and biodegradability—the silicon is excreted and does not accumulate after i.v. dosing; instead they are degraded to silicic acid and excreted in urine and feces.[24-26] MSNs have been studied extensively as delivery platforms for treatment of cancer, and more recently also have been studied for treatment of diabetes and atherosclerosis.[27-29]
Previously, we developed a pH-gated MSN loaded with INH for treatment of TB, and we demonstrated efficacy against M. tuberculosis in an in vitro study.[30] However due to the small size of the INH molecule (~ 1 nm), the drug was difficult to trap inside the 2 nm MSN pores. Because of its relatively low loading capacity, the system was not a prime candidate to pursue in an in vivo analysis. In the present study, we have developed an entirely new MSN-based INH delivery system and studied its efficacy against M. tuberculosis in vitro and in vivo. In this new design, INH is covalently bonded to MSNs via a hydrazone bond to form a pro-drug nanoparticle-based system.[31-33] The hydrazone bond is pH-sensitive and the unmodified INH can be reconstituted in acidic conditions, such as naturally occurs in the acidified endolysosomal compartment of macrophages after ingestion of particles. Finally, we coated the INH-loaded pH-responsive MSNs with a poly(ethylene imine)-poly(ethylene glycol) (PEI-PEG) co-polymer to improve their dispersibility and stability. With this newly designed pH-responsive INH-loaded MSN system, we demonstrate high loading of INH, excellent uptake by M. tuberculosis-infected macrophages, in vitro efficacy in killing intramacrophage M. tuberculosis, and in vivo efficacy in treating pulmonary tuberculosis in mice that is significantly greater than that achieved with equivalent amounts of free INH.
Results
Synthesis of MSNs Carrying INH as a Prodrug (MSN-CHO-INH and SMSN-CHO-INH)
To prepare a MSN delivery platform with high INH loading that retains the drug at neutral pH but releases it at the acidic pH of intracellular compartments, we tethered INH covalently to MSN as a “prodrug” with a pH-sensitive release mechanism. Our ‘prodrug’- MSN platform achieves high INH loading by taking advantage of the high external and internal surface areas of MSNs for drug bonding. MSNs first were synthesized by a simple, solution-based procedure that has been well established in literature.[34, 35] The MSNs were then aldehyde-functionalized (Figure 1A) by attaching aldehyde-terminated silane to the MSNs as described in Methods. Finally, the MSNs were loaded with INH as a prodrug by chemically bonding the hydrazine portion of INH to the aldehyde-functionalized MSNs, forming a pH-sensitive hydrazone bond. At neutral pH (7.4), the hydrazone bond is stable, but once exposed to acidic conditions (pH 5-6), the hydrazone is hydrolyzed and the INH is released from the MSN in its original form (Figure 1A).
Figure 1.
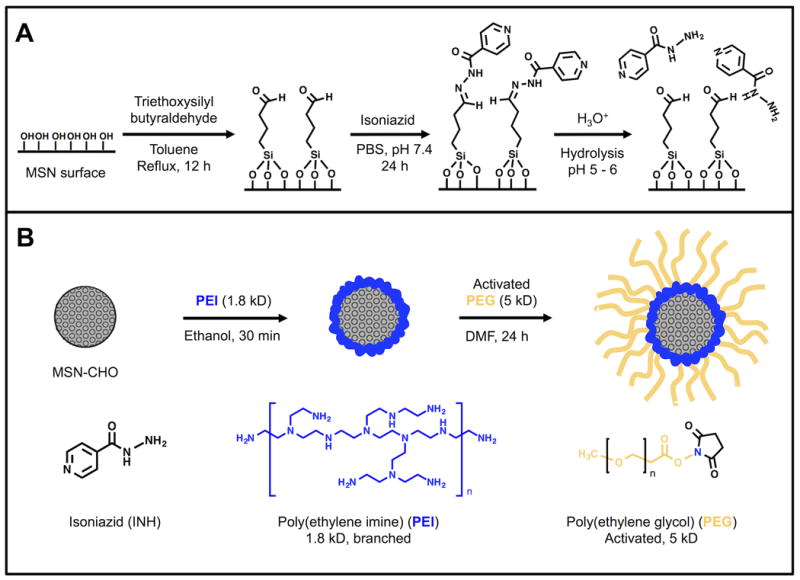
A) Synthesis of aldehyde-functionalized MSNs, yielding MSN-CHO (leftmost reaction); attachment of isoniazid (INH) onto the surface of the aldehyde-modified nanoparticles, yielding MSN-CHO-INH (middle reaction); and release of the INH from MSN-CHO-INH by hydrolysis at acid pH (rightmost reaction). B) Assembly of copolymer PEI-PEG on the pH-responsive INH-loaded MSNs, yielding MSN-CHO-INH-PEI-PEG.
As size is a major factor impacting the efficacy of MSN platforms, in this study, we synthesized two sizes (100 nm and 50 nm diameters) of MSNs. We synthesized both types of MSNs using CTAB as a templating agent for the pores; however, for the smaller, 50 nm MSNs (SMSNs), we added an additional co-surfactant, Pluronic F127. The copolymer serves to restrict the growth of the CTAB micelle structure and helps stabilize silica nucleation sites, keeping the silica structure small [36]. We evaluated the in vivo and in vitro efficacy of the pH-responsive INH-loaded MSNs – large 100 nm diameter MSNs (MSN-CHO-INH) and small 50 nm MSNs (SMSN-CHO-INH).
We followed the pH-stimulated release of INH from the MSN-CHO-INH and SMSN-CHO-INH by UV-vis spectroscopy, measuring INH absorbance (λmax = 262 nm) in the MSN supernate. We suspended nanoparticles in PBS and obtained the supernate by centrifugation to obtain the residual drug present. We then resuspended the nanoparticles in 0.1 N HCl (pH 1) for 15 minutes, pelleted the nanoparticles by centrifugation, and obtained the supernate under this condition (acid release) for measuring the absorbance of INH at 262 nm. There was negligible release of INH into the neutral pH solution, but substantial release of INH under acid conditions, with a release capacity of the INH-loaded nanoparticles as high as 11% wt (drug / MSN) (Figure 2).
Figure 2.
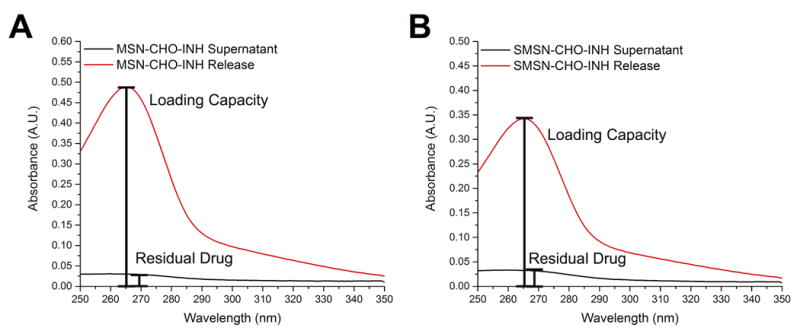
pH-stimulated release of INH from INH-loaded MSNs. UV-vis spectra of supernatants after washing INH-loaded nanoparticles with PBS (black trace) or with 0.1 N HCl (red trace) demonstrating release of INH under acidic conditions for both A) MSN-CHO-INH- and B) SMSN-CHO-INH. The analysis was repeated 2 times with MSN-CHO-INH and 2 times with SMSN-CHO-INH with similar results.
Synthesis of PEI-PEG Copolymer Coated MSN-CHO-INH and SMSN-CHO-INH
PEI-PEG copolymer coating of MSNs improves their dispersibility and promotes endosomal escape following uptake.[36, 37] To improve the dispersion and stability of the MSNs in aqueous media, we added the copolymer poly(ethylene imine)-poly(ethylene glycol) (PEI-PEG) to the surface of the MSN-CHO-INH and SMSN-CHO-INH in a two-step process (Figure 1B) following the attachment of the aldehyde-terminated silane to the MSNs and the bonding of INH to the aldehyde, as described above. First, we coated PEI polymer onto the surface via electrostatic attraction between the negatively charged silica surface and the cationic polymer (Figure 1B, left). The cationic polymer improves particle dispersion by providing electrostatic repulsion, however it can be toxic at high molecular weights above 25 kD.[38] We employed 1.8 kD PEI polymer throughout this study to ensure low cytotoxicity. The positive charge of the PEI has the additional advantage of conferring a ‘proton sponge effect’ wherein a high osmotic pressure induces endosomal rupture and release of drug cargo into the cell cytosol, thus avoiding endolysosomal entrapment. We then attached an amine-reactive form of PEG (5 kD) to the MSNs by utilizing the amines of the PEI (Figure 1B, right). PEG is a branched, hydrophilic polymer that helps decrease particle aggregation by adding steric hindrance to the nanoparticle.[39] Before and after all modifications of the MSNs, we determined the hydrodynamic radius by dynamic light scattering and zeta potential, and we examined their structure by TEM imaging (Supplemental Table 1 and Figure 3).
Figure 3.

TEM images of A) MSN-CHO-INH-PEI-PEG and B) SMSN-CHO-INH-PEI-PEG. The structural integrity of the MSNs is preserved after all surface modifications.
Loading and Stability of INH Prodrug on MSNs Coated with PEI-PEG Copolymer
We evaluated the INH loading and release capacity of the 100 nm and 50 nm MSNs by UV-Vis measurements of INH release into neutral and acidic aqueous solutions from the MSNs. At neutral pH, relatively low amounts of INH were detected as being released from the MSNs. In contrast, at acid pH, substantial amounts of INH were released (Figure 2). We also evaluated INH release capacity of the MSNs using a trans-cinnamaldehyde derivitization assay (Supplemental Figure 1) as described previously.[30] The INH release capacity for the 100 nm MSN-CHO-INH-PEI-PEG was 8.2% (wt/wt), and for the smaller 50 nm SMSN-CHO-INH-PEI-PEG was 11.3% (wt/wt).
We evaluated the stability of the hydrazone bond formed between the aldehyde and INH by repeating the release capacity evaluation of the MSN-CHO-INH-PEI-PEG after one month storage at 4 °C, again using the trans-cinnamaldehyde derivitization assay. The amount of INH released into the neutral solution remained very low, confirming the stability of the hydrazone bond on the MSN-CHO-INH-PEI-PEG. For this sample, we measured 8.3% (wt/wt) release of INH from the MSN-CHO-INH-PEI-PEG under acidic pH, similar to the 8.9% (wt/wt) release of INH from the same batch one month earlier (Supplemental Figure 2), demonstrating that the MSN-CHO-INH-PEI-PEG is stable for at least one month in storage with negligible INH leakage from the nanoparticle carrier.
MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG are Taken Up by Human Macrophages Infected with M. tuberculosis
To determine whether our MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG are taken up efficiently by M. tuberculosis-infected macrophages, we employed nanoparticles covalently labeled with Rhodamine B and PMA-differentiated human macrophage-like THP-1 cells infected with green fluorescence protein (GFP)-expressing M. tuberculosis (Mtb-GFP). THP-1 cells were infected with Mtb-GFP for 90 minutes, washed, and incubated with Rhodamine-labeled SMSN-CHO-INH-PEI-PEG (12.5 μg mL-1) for 3 hours prior to fixation and immunofluorescence imaging. As shown in Figure 4, the Rhodamine-labeled SMSN-CHO-INH-PEI-PEG is taken up avidly by M. tuberculosis-infected macrophages. We obtained similar results with the larger 100 nm MSN-CHO-INH-PEI-PEG in human THP-1 cells and with both sizes of MSN in human peripheral blood monocyte derived macrophages (MDM) (Supplemental Figure 3).
Figure 4.
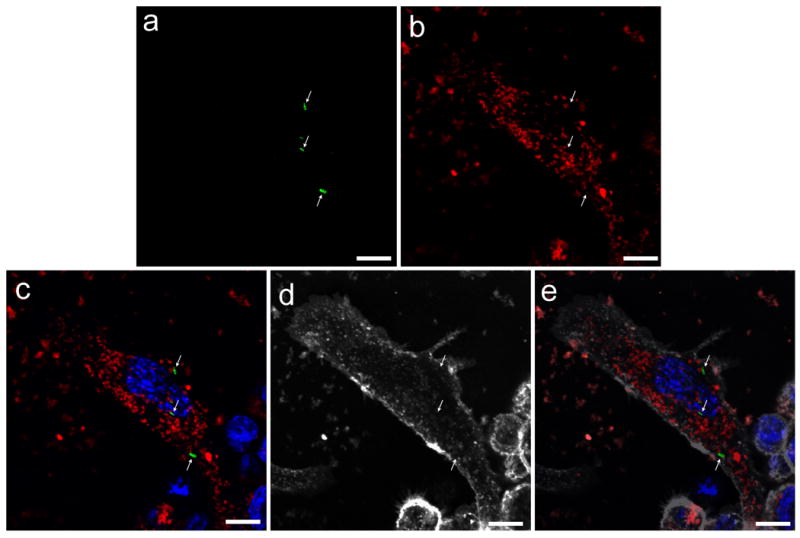
Confocal microscopy demonstrates avid uptake of RITC-labeled SMSN-CHO-INH-PEI-PEG by M. tuberculosis infected THP-1 macrophages. Human macrophage-like THP-1 cells were infected with GFP-expressing M. tuberculosis for 90 minutes, washed, and 12.5 μg mL-1 of RITC-labeled 50 nm SMSN-CHO-INH-PEI-PEG added. After 3 hours, the cells were washed, the plasma membrane was stained with WGA-AlexaFluor 633, the cells were fixed, and nuclei were stained with DAPI. a) Mtb-GFP (green, arrows); b) RITC-labeled SMSN-CHO-INH-PEI-PEG (red); c) Nuclei stain blue with DAPI in merged color image, d) contours of the cell are stained with WGA-AlexaFluor 633 (gray scale); e) gray scale image superimposed onto merged color image. Scale bars, 10 μm.
MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG Kill M. tuberculosis Growing in Macrophages in a Dose-dependent Fashion
To assess the efficacies of the two types of pH-responsive INH-loaded MSNs (100 nm and 50 nm), we infected differentiated THP-1 macrophage-like cells with virulent M. tuberculosis and left them untreated or treated them with a) control MSN or SMSN not loaded with INH but coated with PEI-PEG b) MSN-CHO-INH-PEI-PEG; c) SMSN-CHO-INH-PEI-PEG; or d) free INH. M. tuberculosis grew similarly in macrophages that were untreated or treated with control nanoparticles (no INH loading), indicating that the nanoparticle carrier by itself has no inhibitory effect on the bacterium. The amount of M. tuberculosis killing in macrophages treated with free INH, MSN-CHO-INH-PEI-PEG, or SMSM-CHO-INH-PEI-PEG increased in a dose dependent fashion (Figure 5, A-C).
Figure 5.
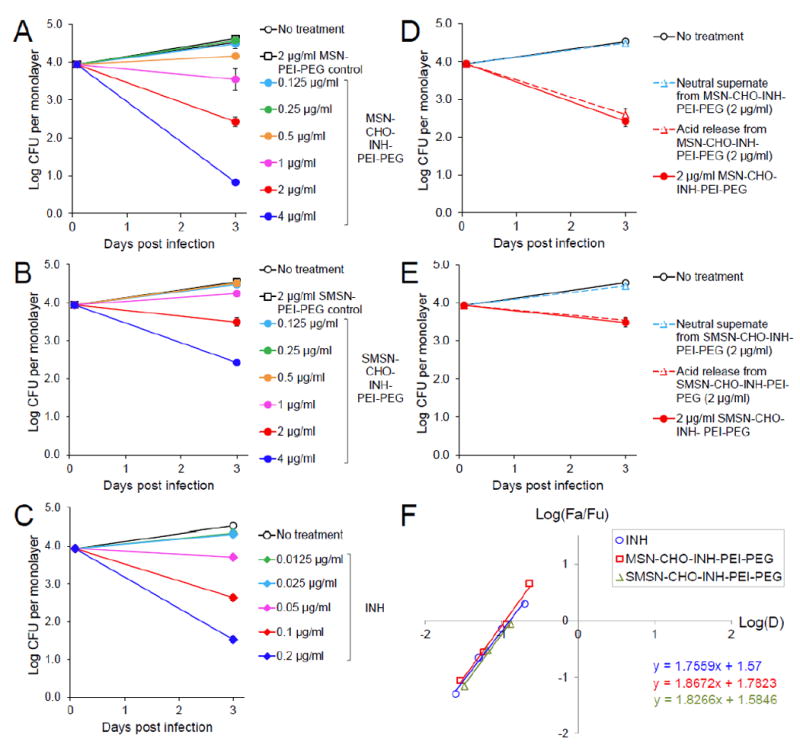
Killing of M. tuberculosis in human macrophages by free INH and the 100 nm and 50 nm INH-loaded pH-responsive nanoparticles coated with PEI-PEG. THP-1 macrophages were infected with M. tuberculosis and treated with various concentrations of A) MSN-CHO-INH-PEI-PEG, B) SMSN-CHO-INH-PEI-PEG or C) INH. The killing effect of supernates prepared from D) MSN-CHO-INH-PEI-PEG and E) SMSN-CHO-INH-PEI-PEG under neutral or acidic conditions on intracellular M. tuberculosis were compared with the killing effect of no treatment or treatment with INH-loaded pH-responsive nanoparticles. F) Median-effect plot, where D is the dose of INH; Fa/Fu is the Fraction of bacteria killed/Fraction of bacteria surviving. Data are represented as means ± standard errors (some are smaller than the symbol) with two biological and two technical repeats.
We prepared supernates from MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG resuspended in PBS, pH 7.4 (neutral supernate) or in 0.1 N HCl (acid release) and compared the potency of these supernates with the corresponding INH-loaded nanoparticles in killing intracellular M. tuberculosis. Our results showed that the neutral supernates prepared from MSN-CHO-INH-PEI-PEG or SMSN-CHO-INH-PEI-PEG had minimal killing effect on intramacrophage M. tuberculosis (Figure 5, D-E). In contrast, the acid released solution killed M. tuberculosis to the same extent as the corresponding MSN-CHO-INH-PEI-PEG or SMSN-CHO-INH-PEI-PEG from which they were prepared. These results confirm that the hydrazone bond formed between INH and the aldehyde-functionalized nanoparticle is hydrolyzed in the acidic macrophage endolysosomal environment, releasing INH from the nanoparticles, and that the acid-released INH is biologically active.
With a drug loading of 5.73% wt/wt, 1 μg mL-1 of MSN-CHO-INH-PEI-PEG carried 0.11 μg mL-1 of INH and killed 2.1 log of M. tuberculosis, close to the 1.9-log killing achieved by free INH at 0.1 μg mL-1. Similarly, 4 μg mL-1 of SMSN-CHO-INH-PEI-PEG, with 3.26% (wt/wt) drug loading, carried 0.13 μg mL-1 of INH and gave 2.1 log killing of M. tuberculosis. As shown in Figure 5F, the median-effect plot of the free INH and the two types of pH-responsive INH-loaded nanoparticles show similar slopes indicating that they have comparable anti-M. tuberculosis efficacy in this cell culture assay system. Thus, this study demonstrates that INH delivered by the MSN-CHO-INH-PEI-PEG or SMSN-CHO-INH-PEI-PEG is as effective as an equivalent amount of free INH in killing M. tuberculosis in infected human macrophages, indicating an efficacy ratio of 1 in the macrophage cell culture system.
INH-loaded pH-responsive PEI-PEG-coated MSNs are More Efficacious than Free INH in a Mouse Model of Pulmonary Tuberculosis
To evaluate the efficacy of our 100 nm MSN-CHO-INH-PEI-PEG and 50 nm SMSN-CHO-INH-PEI-PEG in a mouse model of pulmonary tuberculosis, we infected mice with virulent M. tuberculosis Erdman by aerosol with 250-500 colony-forming units (CFU) delivered into the lung. Over the ensuing two weeks, the bacteria grew 5-logs to approximately 107 CFU per lung (Figure 6, A and B). Two weeks after aerosol infection, mice were sham-treated or treated with pH-responsive INH-loaded MSNs or free INH with doses matching 1, 2 or 4 fold the equivalent amount of INH loaded on the nanoparticles, 3 times/week for 2 weeks (total of 6 injections) via the i.v. or subcutaneous (s.c.) route. The pH-responsive INH-loaded PEI-PEG-coated MSNs were well tolerated by the infected mice as evidenced by the fact that all treated mice, regardless of nanoparticle size or route of administration, maintained their body weight over the course of treatment (Supplemental Figure 4).
Figure 6.
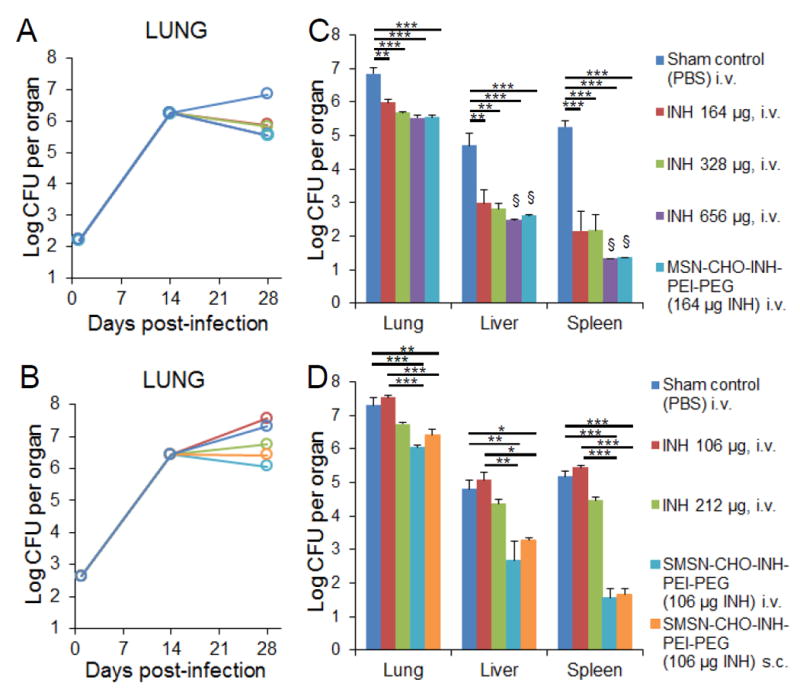
In vivo efficacy of MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG. Mice were infected with A) 250 and B) 500 live M. tuberculosis bacilli by aerosol. Bacterial burdens in the lung were monitored throughout the course of infection. The effect of C) MSN-CHO-INH-PEI-PEG and D) SMSN-CHO-INH-PEI-PEG treatments on M. tuberculosis burden in lung, liver, and spleen was determined by assaying M. tuberculosis CFU three days after the final treatment. The equivalent amount of free INH for each type of nanoparticle is shown in parenthesis. Statistics were analyzed using one-way ANOVA with Bonferroni post-test correction. *p< 0.1, **p< 0.01, ***p< 0.001. Error bars represent standard errors with 3 mice per group. §Organ bacterial CFU below the experimental limit of detection.
The bacterial burden in mice treated with MSN-CHO-INH-PEI-PEG, compared with sham-treated mice, was reduced by 1.3 logs in the lung, 2.1 logs in the liver, and 3.9 logs in the spleen (Figure 6C). The reduction in bacterial CFU in the lung achieved by MSN-CHO-INH-PEI-PEG was significantly greater than that achieved by an equivalent amount of free INH (t-test, p < 0.005). Of note, bacterial burden in the organs of mice treated with MSN-CHO-INH-PEI-PEG was as low as that in mice treated with 4-fold the equivalent amount of free INH, consistent with an efficacy ratio of 4 for the MSN-CHO-INH-PEI-PEG vs. free drug. In the liver and spleen, both the MSN-CHO-INH-PEI-PEG and the 4-fold amount of free INH lowered bacterial CFU to a level below the limit of detection. Mice treated with SMSN-CHO-INH-PEI-PEG by either the i.v. or s.c. route had significantly fewer bacteria in the lung, liver, and spleen than mice treated with 2-fold the amount of free INH (Figure 6D), consistent with an efficacy ratio of 2. These results demonstrate that both 100 nm MSN-CHO-INH-PEI-PEG and 50 nm SMSN-CHO-INH-PEI-PEG are more effective than two to four times the equivalent amount of INH (Supplemental Figure 5). Interestingly, SMSN-CHO-INH-PEI-PEG showed comparable efficacies when delivered s.c. or i.v. Consistent with the reduced bacterial organ burden, mice treated with pH-responsive INH-loaded MSNs also exhibited reduced numbers of surface lesions on their lungs (Figure 7; Supplemental Figure 6).
Figure 7.
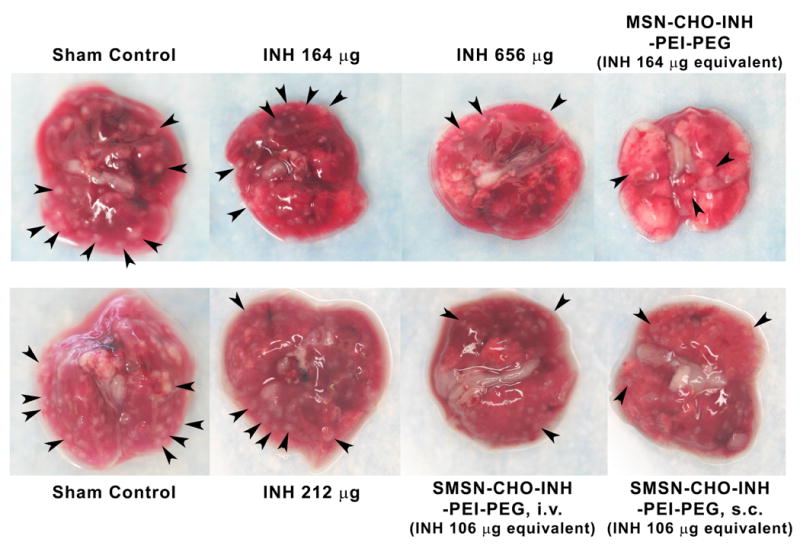
Gross pathology of lungs dissected from sham-treated or mice treated with INH or pH-responsive INH-loaded PEI-PEG-coated MSNs (100 nm) or SMSNs (50 nm) by intravenous (i.v.) or subcutaneous (s.c.) injection. Some of the granulomas are indicated by arrowheads.
Distribution of MSNs In Vivo
We evaluated the biodistribution and clearance of nanoparticles in vivo over time by inductively coupled plasma optical emission spectrometry (ICP-OES) analysis of the silicon content in the animal organs. In short-term studies, we administered nanoparticles to mice in a single dose by i.v. (tail vein) injection and euthanized the mice 24 hours after injection (24 hour time point). In long-term studies, we injected mice three times a week for two weeks and euthanized the mice 72 hours after the last injection (2 weeks, 72 hour time point). Figure 8 shows that the biodistribution of nanoparticles varies as a function of MSN size and time after injection.
Figure 8.
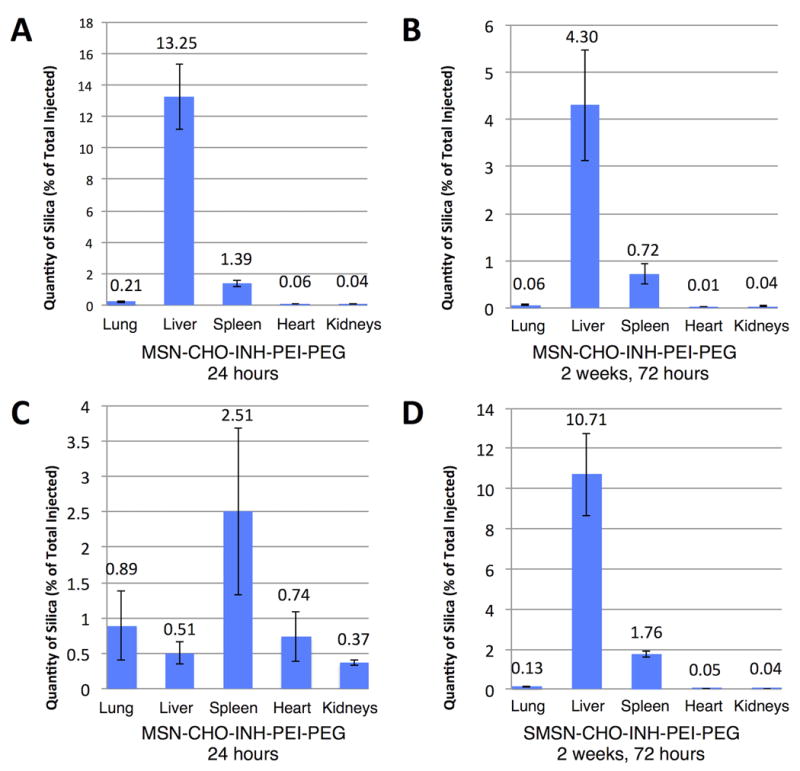
Distribution of MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG in animal organs. The distribution of i.v. administered (A and B) MSN-CHO-INH and (C and D) SMSN-CHO-INH in lung, liver, spleen, heart and kidney 24 hours after a single injection (A and C) or 72 hours after six injections over 2 weeks (B and D) was determined by ICP-OES analysis of silica. (A and C) At 24 hours after a single injection, MSN-CHO-INH-PEI-PEG are primarily in the liver (A), while SMSN-CHO-INH-PEI-PEG are well distributed throughout the body (C). At 72 hours after six injections spread over 2 weeks, MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG show a similar distribution pattern – primarily in the liver, spleen and lung – but mice injected with SMSN-CHO-INH-PEI-PEG have more than twice as much silica in these organs as mice injected with MSN-CHO-INH-PEI-PEG. Data represent means ± standard errors of results from 3 mice per experimental condition with 3 technical repeats per mouse.
In the short-term study, the larger nanoparticles (MSN-CHO-INH-PEI-PEG) preferentially localized to the liver followed by the spleen, whereas the smaller nanoparticles (SMSN-CHO-INH-PEI-PEG) preferentially localized to the spleen followed by the lung (Figure 8, A and C). Interestingly, at the 24-hour time point, the SMSNs showed a 4-fold greater delivery to the lungs than the larger MSNs. In the long-term study, the larger MSNs showed a localization pattern similar to that in the short-term study. However, the quantities of silica present were much lower, indicating that most of the silica had been cleared by 72 hours after the last of six injections (Figure 8B). On the other hand, the SMSNs showed a different localization pattern in the long-term study with the liver now the primary location (Figure 8D). This study demonstrates that the majority of the silica was cleared from animal organs by three days after the last of six doses, as only a small percentage of the total injected silica remained (5.13% for the five organs for MSN-CHO-INH-PEI-PEG and 2.69% for SMSN-CHO-INH-PEI-PEG).
Discussion
Current regimens for treatment of tuberculosis are prolonged, requiring 6 – 9 months of multidrug therapy, and are complicated by toxicities, noncompliance, and emergence of drug resistance. Nanoparticle delivery platforms that deliver high concentrations of antibiotic directly to the site of infection have the potential to improve treatment by sterilizing infected tissues more rapidly, reducing the duration of treatment while also reducing systemic drug exposure and off-target toxicities. To achieve this goal, we have developed a stimulus-responsive MSN drug delivery platform for INH with high (up to 11%) drug loading that releases INH intracellularly after uptake into acidified compartments. Stimulus-responsive release was achieved by bonding the INH to the MSN via acid-reversible hydrazone bonds. The INH loading is stable at neutral pH for at least one month under refrigeration. We have shown that the MSN are tolerated well with repeated i.v. administration in mice and that they target tissues of the Mononuclear Phagocyte System that are the sites of tuberculosis infection. Moreover, our ICP-OES silica analysis demonstrates that the MSNs do not accumulate and are cleared, consistent with previous demonstrations of MSN biodegradability.[24-26]
In our macrophage cell culture model, the MSN-CHO-INH-PEI-PEG is as effective as equivalent doses of free INH, whereas in vivo, the MSN-CHO-INH-PEI-PEG given i.v. or s.c. kill more M. tuberculosis in lung, liver and spleen than 2 – 4 times the equivalent dose of free INH. The higher efficacy ratio of the MSN-delivered drug in vivo vs. in vitro could reflect a combination of several factors: (1) direct uptake of the MSN by M. tuberculosis-infected macrophages with release of high concentrations of INH inside these macrophages, (2) uptake of drug into cells nearby infected macrophages with release of high concentrations of drug in the vicinity of the infected macrophages, (3) uptake of drug into uninfected mononuclear phagocytes elsewhere in the host that migrate to the site of infection and ingest M. tuberculosis (see below), and (4) delayed drug clearance and prolonged drug release by virtue of the drug being encapsulated in MSNs, resulting in improved pharmacokinetics, such as a higher AUC/MIC (Area Under the Curve/ Minimal Inhibitory Concentration) ratio. With respect to the last factor, in the in vitro macrophage model, free INH in the culture medium bathes the infected cells at a constant concentration for the duration of the experiment, whereas after in vivo administration, free INH is cleared rapidly by hepatic metabolism. The half-life of INH after oral administration in mice is 1.7 hours, similar to that of human rapid acetylators.[40] The half-life of INH in humans is similar after i.v. and oral administration (1.8 hours in rapid acetylators and 2.9 hours in slow acetylators).[41] Enclosing the INH within MSNs that release the drug only after uptake into acidified compartments shields the drug from immediate metabolism and hepatic clearance and likely provides for a delayed release of drug with more favorable pharmacokinetics.
Both s.c. and i.v. administered SMSN-CHO-INH-PEI-PEG were more effective than i.v. administered free drug. While it is likely that the improved efficacy of nanoparticle-delivered drug vs. free drug is due in large part to improved pharmacokinetics (i.e. greater AUC due to sustained drug release), delivery of the drug-loaded nanoparticle to M. tuberculosis-infected lung tissue may occur via both routes of administration, thereby providing for higher tissue levels of drug and enhanced therapeutic efficacy. Subcutaneously administered MSN could either enter the blood directly or be taken up by uninfected macrophages that subsequently migrate to sites of M. tuberculosis infection, including the lung. For example, Fenaroli et al.[42] observed that PLGA nanoparticles injected into M. marinum infected Zebrafish embryos were taken up by blood macrophages that rapidly migrated into M. marinum granulomas. Similarly, Kourtis et al. observed blood bioavailability of 54% and 83%, respectively, for intramuscularly and intradermally administered 30 nm nanoparticles.[43] Dynamic influx of macrophages into BCG granulomas in rabbits has also been demonstrated, supporting the concept that uninfected macrophages from distant sites may carry nanoparticles into infected mycobacterial granulomas.[44]
We utilized PEI-PEG coating of our MSN primarily to improve their dispersibility and uptake properties. The PEI coating may also disrupt endolysosomal membranes, facilitating access of drug cargo to the cytosol, although this may not be of much consequence in the case of INH, which crosses membranes freely. While it might be thought that the PEI-coating, by virtue of the proton sponge effect, would prevent acidification of the endolysosomal compartments containing the MSNs and thereby interfere with hydrolysis of the hydrazone bond, the high anti-mycobacterial efficacy of our MSN-CHO-INH-PEI-PEG nanoparticles argues strongly that the hydrazone bond is cleaved after uptake of these MSNs into macrophages. Furthermore, we and others have shown that pH sensitive release mechanisms are not inhibited by the presence of PEI and PEI-PEG coatings on the MSNs.[45] Indeed, it has been shown by use of nanoparticles with a pH sensor that PEI-coating of the nanoparticles does not block acidification of their compartment.[46]
The in vivo efficacy of MSN-platforms is greatly impacted by the size of the MSN, because size impacts both tissue penetration and cellular uptake of the MSN. In the case of tumors, 100 nm MSNs are suitable to transit the vasculature by virtue of the enhanced permeability and retention (EPR) effect, but show poor diffusion into the collagen matrix of the interstitial space, whereas smaller nanoparticles are able to penetrate further.[47] Lu et al. demonstrated that for MSNs of 30 nm – 200 nm, optimum endocytic uptake by HeLa cells was achieved with MSNs of 50 nm size[48] and Zhang et al. identified 30 nm as the optimal size for endocytic uptake of nanoparticles based on thermodynamic considerations.[49] We have observed that smaller MSNs (SMSNs) with 50 nm diameters achieve a higher distribution than traditional MSNs (100 nm) into tumor tissue and lower distribution into other vital organs (liver, kidneys, spleen, lung, heart).[17] While macrophages are known to phagocytose a wide range of sizes, including objects much larger than themselves, their discriminatory behavior towards different nanoscale sizes is less well characterized, but Walkey et al. demonstrated greater uptake efficiency by J774 mouse macrophages for 90 nm gold nanoparticles than 60 nm, 30 nm, or 15 nm nanoparticles.[50] In this study, we examined two sizes of MSNs (100 nm and 50 nm diameters) for the delivery of INH in vitro and in vivo. These two MSNs had somewhat different tissue distribution, but both were highly effective against M. tuberculosis both in vitro and in vivo.
Our nanoparticle delivery system for treatment of TB differs from previous systems that have been tested in vivo[9] in that it is engineered to be stimulus responsive to release drug only after uptake into acidified intracellular compartments as opposed to being a passive biodegradable drug encapsulation system. While our present demonstration that a stimulus responsive nanoparticle INH delivery system has greater in vivo therapeutic efficacy than free drug is an important proof of principle, incorporation of additional rational design features has the potential for even greater improvement in efficacy. For example, modifications of the MSN to enhance pulmonary targeting or to optimize the drug release profile would be expected to further enhance efficacy. Aerosol delivery of the MSN represents another important modification for potential enhancement of therapeutic efficacy. Hamblin et al. recently demonstrated that liposomal ciprofloxacin administered by aerosol was more effective than even a 50-fold greater dose of oral or intranasal antibiotic in a mouse Schu S4 tularemia model.[51] Likewise, delivering the pH-responsive INH-loaded MSNs by aerosol inhalation would immediately target the lung, avoid systemic toxicities, and potentially provide high concentrations of drug directly to infected lung tissue, likely greatly enhancing efficacy.
Conclusions
We have developed stimulus-responsive PEI-PEG-coated mesoporous silica nanoparticles that bind INH covalently through hydrazone bonds that are stable at neutral pH but have high INH release capacity at acid pH. We show that these MSN are avidly ingested by M. tuberculosis-infected human macrophages and that they kill the intracellular bacteria in a dose-dependent manner. We further demonstrate in a mouse model of pulmonary tuberculosis that the nanoparticles are well tolerated and that they are much more efficacious than an equivalent amount of free drug. Further optimization of this MSN platform has the potential to provide improved treatment of tuberculosis with regard to safety, efficacy, treatment duration, and prevention of the emergence of drug resistance.
Experimental Section
Reagents
All reagents were used as received without further purification. Cetyltrimethylammonium bromide (CTAB, 90%), Pluronic F127 (F127), isoniazid (INH, 99%), rhodamine B isothiocyanate (RITC, 90%), polyethyleneimine (PEI, 1.8 kD, tetraethyl orthosilicate (TEOS, 90%), and 3-(trihydroxysilyl)propyl methylphosphonate monosodium aqueous solution (HTMP, 42% in H2O) were purchased from Sigma-Aldrich. Triethoxysilylbutyraldehyde (CHO-silane, 90%) poly(ethylene glycol) methyl ether (m-PEG, MW 5 kD) were obtained from Gelest.
Synthesis of 100 nm and 50 nm MSN
MSNs were synthesized by a simple, solution-based procedure that has been well established in literature.[17, 25, 34-36] Surfactant (cetyltrimethylammonium bromide, CTAB) was dissolved in a basic aqueous solution; the resulting micelles assembled into a 2-dimensional hexagonal structure that served as templating agent for the porous structure. The solution was heated and the silica precursor (tetraethyl orthosilicate, TEOS) condensed around the surfactant template in a base-catalyzed reaction. The solution was aged and the surfactant extracted to produce 100 nm diameter MSNs with ~2 nm diameter pores. Adding an additional surfactant, a triblock copolymer Pluronic F127, to the solution yielded smaller MSNs (SMSNs) with 50 nm diameters, while maintaining the ~2 nm diameter pore size.[36] The nonionic co-surfactant limited the growth of the CTAB micelle structure, yielding smaller nanoparticles.[52, 53]
Functionalization of MSNs and Loading with Isoniazid to Yield MSN-CHO-INH and SMSN-CHO-INH
To functionalize the surface of the nanoparticles with aldehyde groups (CHO) for binding drugs, we suspended MSNs (100 mg) in dry toluene (10 ml) and added triethoxysilylbutyraldehyde (200 μl) to the solution. The reaction was heated overnight under nitrogen, cooled, and washed sequentially with toluene, methanol, and water. Aldehyde-functionalized MSNs (CHO-MSNs) were suspended in a concentrated INH solution [40 mg mL-1, Dulbecco’s Phosphate Buffered Saline (PBS)] and left to load for up to 48 hours. The INH-loaded nanoparticles (MSN-CHO-INH and SMSN-CHO-INH) were extensively washed with PBS. UV-vis spectra of INH loading were collected by a Cary 500 UV-vis-NIR spectrophotometer. DLS and zeta potentials were measured by ZetaSizer Nano (Malvern Instruments Ltd., Worcestershire, U.K.).Transmission electron microscopy (TEM) images of SMSNs and MSNs were obtained using a JEM1200-EX (JEOL) instrument.
Addition of Copolymers (PEI and PEG) to MSN-CHO-INH and SMSN-CHO-INH, yielding MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG
Stability and dispersion of the MSNs were improved by using copolymers poly(ethylene imine) (PEI) and poly(ethylene glycol) (PEG). Due to the positively charged amines in PEI, the polymer was electrostatically attached to the negatively charged silica surface. MSN-CHO-INH and SMSN-CHO-INH (10 mg) were suspended in a PEI solution (2.5 mg mL-1, 1.8 kD) for 30 minutes, and the coating was repeated to ensure full coverage. MSNs were collected by centrifugation, and washed sequentially with ethanol and water. The PEG coating was prepared by first synthesizing the activated form of PEG, using poly(ethylene glycol) methyl ether (m-PEG) to prevent crosslinking. The hydroxyl group on m-PEG was replaced with an NHS-ester to react with the amine groups on PEI.[37] PEG was attached to the amine groups of the PEI coating by suspending the PEI-coated MSNs (10 mg) in dry DMF (1.5 ml), adding 50 mg of activated m-PEG, and stirring for 24 hours. Fourier transform infrared spectroscopy (FTIR) was carried out with a JASCO FT/IR-420 spectrometer averaging 64 scans in the range of 4000–400 cm-1 at a resolution of 2 cm-1. KBr discs were prepared by mixing approximately 2 mg of nanoparticles with approximately 200 mg of KBr and forming the disc under pressure. The spectra (Supplemental Figure 7) are background corrected with a linear baseline with 3 segments, normalized to the symmetric Si–O–Si stretching vibration around ν = 800 cm-1, and vertically offset by 0.4 units.
Fluorescence Labeling of MSN
Nanoparticles were labeled with fluorescent dye molecules for imaging purposes. The dye was co-condensed with the silica precursor to ensure labeling throughout the silica matrix. Rhodamine B isothiocyanate (RITC, 2 mg) was dissolved in dry ethanol (1.5 ml), 3-aminopropyltrimethoxysilane (6 μL, APTES) added, and the molecules left to react under nitrogen for 2 hours. TEOS was then added to the solution, and the solution was added drop wise to a heated (80 °C), aqueous solution of CTAB, NaOH, and aged for 2 hours. The nanoparticles were washed with water and collected by centrifugation.
Trans-cinnamaldehyde Derivitization Assay
To assess the amount of INH loading on nanoparticles, we suspended 1 mg MSN-CHO-INH-PEI-PEG or SMSN-CHO-INH-PEI-PEG in 1 ml of PBS or 0.1 N HCl, mixed the suspension on a nutator for 1 hour at room temperature, and centrifuged at 10,000 g for 10 minutes to pellet the nanoparticles. The supernate was diluted in the range of 1:20 to 1:1000 in 0.1 N HCl to a final volume of 1 ml and mixed with 0.15 ml trans-cinnamaldehyde reagent (0.04%). Reactions with known amount of INH standard were also carried out at the same time. After 15-minute incubation at room temperature, the absorbance at 340 nm of the reactions was measured, and the amount of INH in each sample was calculated based on the INH standard curve.
Bacteria
The M. tuberculosis virulent strain Erdman (35801; American Type Culture Collection) was used in this study. For the macrophage infection experiments, a single suspension of bacteria was prepared as described previously.[30] For the in vivo studies, a pre-titered stock of M. tuberculosis Erdman was used to infect mice by aerosol. This stock was prepared by plating guinea pig passaged bacteria one time only on Middlebrook 7H11 agar. A GFPuv expressing M. tuberculosis Erdman strain (Mtb-GFP) was used in MSN uptake experiments.[54] Mtb-GFP was grown on Middlebrook 7H11 agar containing 50 μg mL-1 hygromycin and prepared in the same way as the non-fluorescent M. tuberculosis for use in infection experiments.
Macrophages
Peripheral blood monocytic cells were isolated from donor blood by Ficoll-Hypaque centrifugation and incubated in Teflon wells for 5 days to differentiate them into monocyte-derived macrophages (MDM) before use in infection and MSN uptake experiments. Human monocytic cell line THP-1 (ATCC TIB202) was maintained in RPMI-1640 (Lonza) with 10% fetal bovine serum (Mediatech). Absence of mycoplasma contamination was verified by polymerase chain reaction (Universal Mycoplasma Detection Kit, ATCC). Prior to use in infection experiments, THP-1 cells were treated with phorbol 12-myristate 13-acetate (PMA) to differentiate them into a macrophage-like cell type as described.[30] Briefly, THP-1 cells were pelleted by centrifugation at 200 g for 10 minutes at room temperature, resuspended in culture medium containing 100 nM PMA, and seeded in 96-well plates (Matrical) at a density of 1 × 105 cells/200 μl/well for 3 days at 37 °C in a 5% CO2-95% air atmosphere.
Uptake of MSNs by M. tuberculosis-infected Macrophages
Prior to infection with M. tuberculosis, THP-1 cells were added to 24-well plates containing poly-L-lysine coated glass coverslips at a concentration of 3 × 105 cells mL-1 per well and differentiated to a macrophage-like cell type with PMA (100 nM) in RPMI-1640 with 10% fetal bovine serum for 3 days at 37 °C in air containing 5% CO2. The THP-1 cells were infected with M. tuberculosis at a ratio of 10 bacteria per cell in RPMI-1640 containing 10% human serum type AB for 90 minutes at 37 °C, 5% CO2. The cells were washed and the medium was replaced with fresh medium containing rhodamine isothiocyanate (RITC)-labeled MSNs (12.5 μg mL-1). After incubation for 3 hours at 37 °C, the cells were incubated with Wheat Germ Agglutinin (WGA)-Alexa Fluor 633 (5 μg mL-1, Life Technologies) for 5 minutes at room temperature to stain plasma membranes, washed, fixed with 4% paraformaldehyde in PBS for 30 minutes, cell nuclei were stained with DAPI (1 μg mL-1) and the coverslips were mounted with Prolong Gold Anti-fade mounting medium (Molecular Probes). Confocal images of MSNs uptake by M. tuberculosis–infected THP-1 macrophages were acquired by Leica confocal SP2 1P-FCS microscope and Leica confocal software in the UCLA/CNSI Advanced Light Microscopy/Spectroscopy Core Facility.
To assess the uptake efficiency of MSNs by infected macrophages, we seeded MDMs in Poly-D-Lysine coated μClear 384-well sterile polystyrene cell culture plates (Greiner Bio-One) and infected them with GFP-expressing M. tuberculosis as described above. At the end of the 90-minute incubation period, half of the medium was replaced with fresh medium containing RITC-labeled MSNs. The monolayers were fixed at 3 hours and 1 day in 4% paraformaldehyde in PBS. Macrophages were incubated with WGA-Alexa Fluor 633 (5 μg mL-1) for 5 minutes at room temperature to stain plasma membranes. Cell nuclei were stained for 10 minutes with Hoechst 33342 (1 μg mL-1l) in PBS containing 0.1% Tween 20. Monolayers were washed twice with PBS and viewed by epifluorescence microscopy with an Eclipse TE2000-S microscope equipped with an X-Cite 120 light source (Nikon) and images were acquired with a SPOT RT-KE monochrome camera and SPOT software (Diagnostic Instruments, Sterling Heights, MI).
In Vitro Efficacy of MSN-CHO-INH-PEI-PEGs and SMSN-CHO-INH-PEI-PEGs in Killing M. tuberculosis in Macrophages
A single bacterial suspension of M. tuberculosis was used to infect PMA-differentiated, human macrophage-like THP-1 cells at a multiplicity of infection ratio of 10:1 for 90 minutes at 37 °C, 5% CO2-95% air. The infected macrophages were washed to remove extracellular bacteria and fresh medium with or without INH or MSNs was added. The cultures were incubated in the continued presence of INH or MSNs for 3 days after which the infected macrophage monolayers were lysed with 0.1% SDS. Control infected THP-1 monolayers not treated with INH or MSNs were harvested at 2 hours and 3 days post infection to assess bacterial growth. Culture supernatant and lysed THP-1 cells were combined, serially diluted, plated on 7H11 agar, and bacterial CFU enumerated after 2 weeks of incubation at 37 °C, 5% CO2-95% air.
In Vivo Efficacy of MSN-CHO-INH-PEI-PEGs and SMSN-CHO-INH-PEI-PEGs in Treating Pulmonary Tuberculosis
Eight-week old, female, pathogen-free Balb/c mice (Taconic) were infected with 250-500 CFU of M. tuberculosis Erdman via exposure for 30 minutes within an aerosol chamber to an aerosol generated by a Collison Type 6-jet nebulizer (BGI, Inc. Waltham, MA) at 20 psi from a suspension containing ~2 ×106 CFU mL-1 M. tuberculosis in PBS. The precise number of bacteria used in the aerosol was determined by plating serial dilutions of the stock on Middlebrook 7H11 agar plates and enumerating bacterial CFU. One day later, two mice were euthanized to determine the initial number of bacteria delivered to their lungs. Two weeks later, three mice were euthanized to determine the number of bacteria in the lungs prior to treatment. The mice were then sham-treated, treated with MSN-CHO-INH-PEI-PEG, SMSN-CHO-INH-PEI-PEG, or free INH 3 days a week (Monday, Wednesday, and Friday) for 2 weeks (total of 6 doses) by tail vein injection. Three days after the last treatment dose, all mice were euthanized and the lung, liver, and spleen removed aseptically. The organs were homogenized in PBS, and the homogenates serially diluted and plated on 7H11 agar containing ampicillin (12.5 μg mL-1), amphotericin B (5 μg mL-1), and polymyxin B (20 units mL-1). The plates were incubated at 37 °C in 5% CO2-95% air atmosphere for 3 weeks and CFU enumerated.
Biodistribution of MSN-CHO-INH-PEI-PEGs and SMSN-CHO-INH-PEI-PEGs In Vivo
Organs harvested from M. tuberculosis-infected mice that were either sham-treated or treated with pH-responsive INH-loaded MSNs were homogenized in PBS, digested with 0.1% HNO3, and analyzed by ICP-OES (ICPE-9000, SHIMADZU, Japan). The amount of silica recovered from each organ was normalized to the total dose of MSNs injected.
Median-Effect Plots
We used median effect plots[55] to compare the relative efficacies of free INH, MSN-CHO-INH-PEI-PEG and SMSN-CHO-INH-PEI-PEG. The fraction of inhibition for samples treated with different amounts of INH was calculated using bacterial CFU in base-10 logarithm (log CFU) with the equation: Fraction of inhibition = 1 - (log CFU from sample treated with INH or pH-responsive INH-loaded MSNs/log CFU from untreated sample). A median-effect plot for INH or pH-responsive INH-loaded MSNs was generated using INH or INH equivalent (MSNs) dose in base-10 logarithm as the X-axis and the fraction of surviving bacteria divided by the fraction of killed bacteria in base-10 logarithm as the Y-axis.
Statistics
All statistical analyses were performed using GraphPad Prism software (version 5.01). Experimental comparisons with multiple groups used ANOVA analysis with Bonferroni’s post-test correction. Two-tailed Student’s t tests were performed for some comparisons, as indicated in the figure captions. A P value of 0.05 or less was considered statistically significant.
Study approval
All experiments with mice were conducted within the guidelines and according to the protocol approved by the UCLA institutional animal care and use committee.
Supplementary Material
Acknowledgments
This work was supported by NIH NIAID Award Number UM1AI068636 Brigham and Women’s Hospital ACTG Novel Formulations EOY Supplement. The authors thank the Advanced Light Microscopy Facility in the UCLA/CNSI for providing confocal microscopy equipment, Bastian Ruehle for helpful discussions, and Saša Masleša-Galić and Susana Nava for expert technical assistance.
Footnotes
Supporting Information:
Supporting Information is available from the Wiley Online Library or from the author. It includes additional data on MSN size distribution and zeta potential; MSN drug loading and stability; uptake of MSNs by human monocyte-derived macrophages; weight charts of sham-treated, MSN-treated, or free drug-treated infected mice; median-effect plots comparing efficacy of INH-loaded MSNs vs. free INH; quantitation of lung granuloma lesions of sham-treated, free INH-treated, or MSN-treated infected mice; and FITR spectra of MSN-CHO-INH.
Conflict of interest: The authors have declared that no conflict of interest exists.
Contributor Information
Dr. Angela Hwang, Department of Chemistry & Biochemistry, University of California, Los Angeles, California, USA.
Dr. Bai-Yu Lee, Division of Infectious Diseases, Department of Medicine, University of California, Los Angeles, California, USA.
Prof. Daniel L. Clemens, Division of Infectious Diseases, Department of Medicine, University of California, Los Angeles, California, USA.
Ms. Barbara Jane Dillon, Division of Infectious Diseases, Department of Medicine, University of California, Los Angeles, California, USA
Prof. Jeffrey I. Zink, Department of Chemistry & Biochemistry, University of California, Los Angeles, California, USA California NanoSystems Institute, University of California, Los Angeles, California, USA.
Prof. Marcus A. Horwitz, Division of Infectious Diseases, Department of Medicine, University of California, Los Angeles, California, USA
References
- 1.WHO. 2014 http://apps.who.int/iris/bitstream/10665/137094/1/9789241564809_eng.pdf?ua=1.
- 2.van Hest R, Baars H, Kik S, van Gerven P, Trompenaars MC, Kalisvaart N, Keizer S, Borgdorff M, Mensen M, Cobelens F. Clin Infect Dis. 2004;39:488. doi: 10.1086/422645. [DOI] [PubMed] [Google Scholar]
- 3.Tostmann A, Boeree MJ, Aarnoutse RE, De Lange WCM, Van Der Ven AJAM, Dekhuijzen R. J Gastroenterol Hepatol. 2008;23:192. doi: 10.1111/j.1440-1746.2007.05207.x. [DOI] [PubMed] [Google Scholar]
- 4.Goldman AL, Braman SS. Chest. 1972;62:71. doi: 10.1378/chest.62.1.71. [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Wang L, Yeh J, Wu X, Cao Z, Wang YA, Zhang M, Yang L, Mao H. Biomaterials. 2010;31:5397. doi: 10.1016/j.biomaterials.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. J Am Chem Soc. 2010;132:552. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]
- 7.He Q, Zhang Z, Gao F, Li Y, Shi J. Small. 2011;7:271. doi: 10.1002/smll.201001459. [DOI] [PubMed] [Google Scholar]
- 8.Gelperina S, Kisich K, Iseman MD, Heifets L. Am J Respir Crit Care Med. 2005;172:1487. doi: 10.1164/rccm.200504-613PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Griffiths G, Nystrom B, Sable SB, Khuller GK. Nat Rev Microbiol. 2010;8:827. doi: 10.1038/nrmicro2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sosnik A, Carcaboso AM, Glisoni RJ, Moretton MA, Chiappetta DA. Adv Drug Deliv Rev. 2010;62:547. doi: 10.1016/j.addr.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 11.Barbé C, Bartlett J, Linggen K, Finnie K, Qiang H, Larkin M, Calleja S, Bush A, Calleja G. Adv Mater. 2004;16:1959. [Google Scholar]
- 12.Foradada M, Pujol MD, Bermudez J, Estelrich J. Chem Phys Lipids. 2000;104:133. doi: 10.1016/s0009-3084(99)00118-8. [DOI] [PubMed] [Google Scholar]
- 13.Lam KH, Schakenraad JM, Esselbrugge H, Feijen J, Nieuwenhuis P. J Biomed Mater Res. 1993;27:1569. doi: 10.1002/jbm.820271214. [DOI] [PubMed] [Google Scholar]
- 14.Marques AP, Reis RL, Hunt JA. J Biomed Mater Res A. 2004;71:419. doi: 10.1002/jbm.a.30155. [DOI] [PubMed] [Google Scholar]
- 15.Liong M, Angelos S, Choi E, Patel K, Stoddart JF, Zink JI. J Mater Chem. 2009;19:6251. [Google Scholar]
- 16.Liong M, Lu J, Kovochich M, Xia T, Ruehm SG, Nel AE, Tamanoi F, Zink JI. ACS Nano. 2008;2:889. doi: 10.1021/nn800072t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meng H, Xue M, Xia T, Zhao YL, Tamanoi F, Stoddart JF, Zink JI, Nel AE. J Am Chem Soc. 2010;132:12690. doi: 10.1021/ja104501a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coti KK, Belowich ME, Liong M, Ambrogio MW, Lau YA, Khatib HA, Zink JI, Khashab NM, Stoddart JF. Nanoscale. 2009;1:16. doi: 10.1039/b9nr00162j. [DOI] [PubMed] [Google Scholar]
- 19.Du L, Liao S, Khatib HA, Stoddart JF, Zink JI. J Am Chem Soc. 2009;131:15136. doi: 10.1021/ja904982j. [DOI] [PubMed] [Google Scholar]
- 20.Ferris DP, Zhao YL, Khashab NM, Khatib HA, Stoddart JF, Zink JI. J Am Chem Soc. 2009;131:1686. doi: 10.1021/ja807798g. [DOI] [PubMed] [Google Scholar]
- 21.Zhao Y, Vivero-Escoto JL, Slowing II, Trewyn BG, Lin VS. Expert Opin Drug Deliv. 2010;7:1013. doi: 10.1517/17425247.2010.498816. [DOI] [PubMed] [Google Scholar]
- 22.Gratton SE, Ropp PA, Pohlhaus PD, Luft JC, Madden VJ, Napier ME, DeSimone JM. Proc Natl Acad Sci U S A. 2008;105:11613. doi: 10.1073/pnas.0801763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang X, Teng X, Chen D, Tang F, He J. Biomaterials. 2010;31:438. doi: 10.1016/j.biomaterials.2009.09.060. [DOI] [PubMed] [Google Scholar]
- 24.Lin VS. Nat Mater. 2009;8:252. doi: 10.1038/nmat2413. [DOI] [PubMed] [Google Scholar]
- 25.Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JA, Mou CY, Lo LW. Biomaterials. 2010;31:5564. doi: 10.1016/j.biomaterials.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J, Liong M, Li Z, Zink JI, Tamanoi F. Small. 2010;6:1794. doi: 10.1002/smll.201000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackowiak SA, Schmidt A, Weiss V, Argyo C, von Schirnding C, Bein T, Brauchle C. Nano Lett. 2013;13:2576. doi: 10.1021/nl400681f. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, Trewyn BG, Slowing II, Lin VS. J Am Chem Soc. 2009;131:8398. doi: 10.1021/ja901831u. [DOI] [PubMed] [Google Scholar]
- 29.Aznar E, Villalonga R, Gimenez C, Sancenon F, Marcos MD, Martinez-Manez R, Diez P, Pingarron JM, Amoros P. Chem Commun (Camb) 2013;49:6391. doi: 10.1039/c3cc42210k. [DOI] [PubMed] [Google Scholar]
- 30.Clemens DL, Lee BY, Xue M, Thomas CR, Meng H, Ferris D, Nel AE, Zink JI, Horwitz MA. Antimicrob Agents Chemother. 2012;56:2535. doi: 10.1128/AAC.06049-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li ZY, Liu Y, Wang XQ, Liu LH, Hu JJ, Luo GF, Chen WH, Rong L, Zhang XZ. ACS Appl Mater Interfaces. 2013;5:7995. doi: 10.1021/am402082d. [DOI] [PubMed] [Google Scholar]
- 32.Fan J, Fang G, Wang X, Zeng F, Xiang Y, Wu S. Nanotechnology. 2011;22:455102. doi: 10.1088/0957-4484/22/45/455102. [DOI] [PubMed] [Google Scholar]
- 33.Lee CH, Cheng SH, Huang IP, Souris JS, Yang CS, Mou CY, Lo LW. Angew Chem Int Ed Engl. 2010;49:8214. doi: 10.1002/anie.201002639. [DOI] [PubMed] [Google Scholar]
- 34.He Q, Shi J. J Mater Chem. 2011;21:5845. [Google Scholar]
- 35.Trewyn BG, Slowing II, Giri S, Chen HT, Lin VS. Acc Chem Res. 2007;40:846. doi: 10.1021/ar600032u. [DOI] [PubMed] [Google Scholar]
- 36.Meng H, Xue M, Xia T, Ji Z, Tarn DY, Zink JI, Nel AE. ACS Nano. 2011;5:4131. doi: 10.1021/nn200809t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S, Zink JI, Nel AE. ACS Nano. 2009;3:3273. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI, Nel AE. ACS Nano. 2010;4:4539. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bagwe RP, Hilliard LR, Tan W. Langmuir. 2006;22:4357. doi: 10.1021/la052797j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosset J, Ji B. Experimental chemotherapy of mycobacterial diseases In Mycobacteria: II Chemotherapy. Chapman and Hall; 1998. p. 51. [Google Scholar]
- 41.Hutchings AD, Monie RD, Spragg BP, Routledge PA. Br J Clin Pharmacol. 1988;25:585. doi: 10.1111/j.1365-2125.1988.tb03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenaroli F, Westmoreland D, Benjaminsen J, Kolstad T, Skjeldal FM, Meijer AH, van der Vaart M, Ulanova L, Roos N, Nyström B, Hildahl J, Griffiths G. ACS Nano. 2014;8:7014. doi: 10.1021/nn5019126. [DOI] [PubMed] [Google Scholar]
- 43.Kourtis IC, Hirosue S, de Titta A, Kontos S, Stegmann T, Hubbell JA, Swartz MA. PLoS One. 2013;8:e61646. doi: 10.1371/journal.pone.0061646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dannenberg AM., Jr Tuberculosis (Edinb) 2003;83:251. doi: 10.1016/s1472-9792(03)00048-9. [DOI] [PubMed] [Google Scholar]
- 45.Dong J, Xue M, Zink JI. Nanoscale. 2013;5:10300. doi: 10.1039/c3nr03442a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjaminsen RV, Mattebjerg MA, Henriksen JR, Moghimi SM, Andresen TL. Mol Ther. 2013;21:149. doi: 10.1038/mt.2012.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Nano Lett. 2009;9:1909. doi: 10.1021/nl900031y. [DOI] [PubMed] [Google Scholar]
- 48.Lu F, Wu SH, Hung Y, Mou CY. Small. 2009;5:1408. doi: 10.1002/smll.200900005. [DOI] [PubMed] [Google Scholar]
- 49.Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Adv Mater. 2009;21:419. doi: 10.1002/adma.200801393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walkey CD, Olsen JB, Guo H, Emili A, Chan WC. J Am Chem Soc. 2012;134:2139. doi: 10.1021/ja2084338. [DOI] [PubMed] [Google Scholar]
- 51.Hamblin KA, Wong JP, Blanchard JD, Atkins HS. Front Cell Infect Microbiol. 2014;4:79. doi: 10.3389/fcimb.2014.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikari K, Suzuki K, Imai H. Langmuir. 2006;22:802. doi: 10.1021/la0525527. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki K, Ikari K, Imai H. J Am Chem Soc. 2004;126:462. doi: 10.1021/ja038250d. [DOI] [PubMed] [Google Scholar]
- 54.Clemens DL, Lee BY, Horwitz MA. Infect Immun. 2002;70:5800. doi: 10.1128/IAI.70.10.5800-5807.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chou TC. Pharmacol Rev. 2006;58:621. doi: 10.1124/pr.58.3.10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.