

Abstract
The dynamics of transcription factors play important roles in a variety of biological systems. However, the mechanisms by which these dynamics are decoded into different transcriptional responses are not well understood. Here we focus on the dynamics of the tumor suppressor protein p53, which exhibits a series of pulses in response to DNA damage. We performed time-course RNA-Seq and ChIP-Seq measurements to determine how p53 pulses are linked with gene expression genome wide. We discovered multiple distinct patterns of gene expression in response to p53 pulses. Surprisingly, p53 binding dynamics were uniform across all genomic loci even for genes that exhibited distinct mRNA dynamics. Using a mathematical model, supported by additional experimental measurements in response to a sustained p53 input, we determined that p53 binds to and activates transcription of its target genes uniformly, while posttranscriptional mechanisms are responsible for the differences in gene expression dynamics.
Introduction
Many transcription factors are known to respond to multiple signals and to activate various downstream programs1–6. Specificity between a particular input and output can be achieved through regulation of transcription factors’ abundance, posttranslational modifications, or binding to cofactors. Another potential level of regulation is through the temporal dynamics of transcription factors. Indeed, recent single cell studies pointed to a few examples in which the dynamics of transcription factors differ for different stimuli5–7 and lead to distinct gene expression and phenotypic responses3,8,9. Understanding the molecular mechanisms that detect time-dependent features of transcription factors, and decode them into distinct gene expression profiles, remains a challenge. Here we focused on the dynamics of the tumor suppressing transcription factor p53 and studied how its stereotypic pulses in response to DNA damage10,11 affect the activation of its downstream target genes genome wide.
The tumor suppressor protein p53 has a major role in the control of cell fate decisions. p53 is induced in response to stress and activates the transcription of ample stress-response genes. Several mechanisms of how p53 selectively activates target genes have been proposed. For example, a recent “affinity model” suggests that p53’s affinity to the promoters of cell cycle arrest genes is stronger compared to apoptotic genes. In this model the choice between alternative downstream programs depends on the abundance of p5312–15. Specific posttranslational modifications (PTMs) on p53, and interactions with specific cofactors, have also been proposed to confer preference for some promoters versus others16–20. In addition, the dynamics of p53 have recently been shown to play an important role in the response to DNA damage; different stimuli trigger different p53 dynamics, and p53 dynamics were suggested to carry information that affect gene expression and cell fate3,4,7,10. However, recent ChIP-Seq studies showed substantial overlap between the strongly bound p53 regions independently of the treatment or cell line21,22. The results from these studies challenge the idea of context specific regulation by p53, and repose the demand to further understand how different regulations of p53 control the induction of its target genes.
Here we studied how p53 dynamics are linked with gene expression. Recent studies have looked at the response of a few known p53 target genes to double strand break induced p53 pulses and found that not all the tested genes showed a response, and within the responders, there was a diversity in the dynamics and amplitudes of expression 3,23. The stability of mRNA was suggested to correlate with the differences in dynamic behavior between the tested genes 23,24. However the specific role of p53 dynamics in regulating the expression of genes across the genome, and the relationship between p53 dynamics and binding to its direct targets, have not been explored. In this work, we used RNA sequencing to measure global gene expression in response to p53 pulses allowing us to quantify the dynamics of p53’s downstream target genes in an unbiased way across the genome. We compared our findings in p53 wild-type cells with cells expressing p53 shRNA and focused on the p53-dependent genes. We also performed ChIP-Seq to identify direct targets of p53. Our genome-wide quantitative dynamical data allowed us to identify p53-dependent target genes, to determine the specific parameters describing their kinetics, and to develop a quantitative model that captures these dynamics and predicts the response under a new p53 dynamical input.
Results
p53 oscillations lead to multiple distinct patterns of gene expression
To establish a comprehensive connection between p53 dynamics and gene expression, we focused on the response to DNA damage caused by γ irradiation, which triggers stereotypic p53 pulses (Figure 1A)10,11,25. We measured gene expression by mRNA-Seq every hour during the first two pulses (12hr) and again at 24 hours. Our analysis revealed 229 differentially expressed genes with at least a two-fold change in expression relative to basal condition at one or more time-points (FDR<0.2, t-test Benjamini and Hochberg corrected). We clustered these genes based on their normalized expression within the first 12 hours (z-scores) and defined five main dynamical patterns (Figure 1B, C), including three activated (red) and two repressed (blue) clusters (Supplemental data set 1). Cluster 1 included genes that followed the p53 protein levels and pulsed with approximately one-hour delay (Figure 1C, D). Cluster 2 included genes that were activated during the first five hours and retained high expression levels throughout the time-course. The genes in cluster 3 showed a slow continuous increase in expression throughout the time of measurement. Repressed genes were clustered into either transient repression (cluster 4) or continuous repression (cluster 5). To look at the dependence of these dynamical behaviors on p53, we performed a time-course mRNA-Seq in the same cell line expressing a p53 shRNA. We found that the fold changes (FC) of activation or repression of genes in clusters 1, 2, 3 and 5 were significantly attenuated in the p53 knockdown line (Figure 1E, S1A). Repressed genes in cluster 4 were not significantly affected under p53 knockdown, suggesting that their repression in response to irradiation is independent of p53 (Figure 1E, S1A). The reverse approach (first eliminating genes that were not affected by p53 shRNA and then clustering them) also led to elimination of cluster 4, while the other clusters were largely unchanged (see details and statistics in the Methods section).
Figure 1.
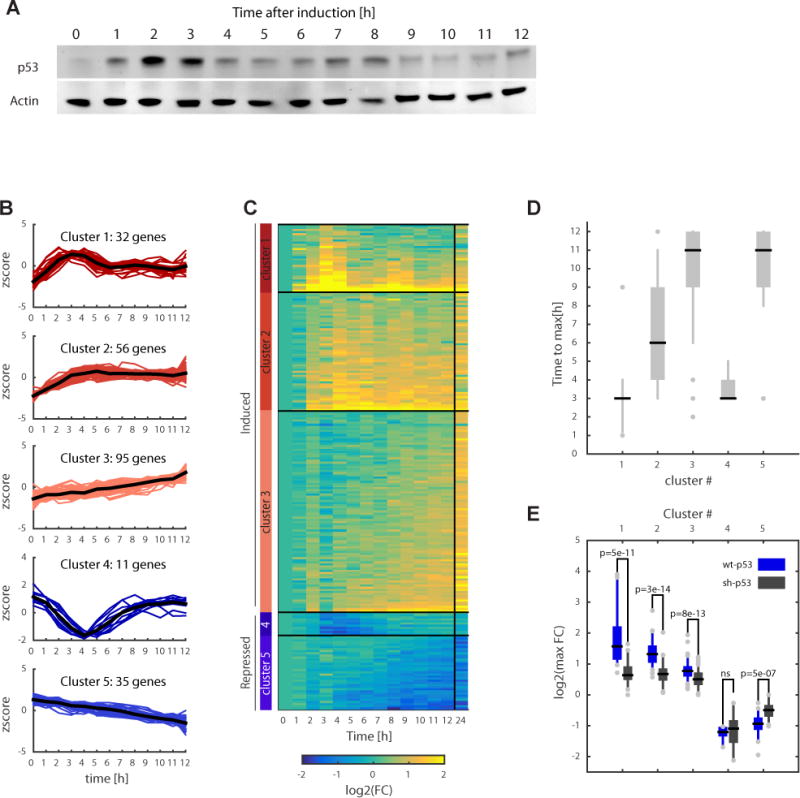
Time course mRNA-seq reveals distinct clusters of gene expression dynamics in response to γ-irradiation. (a) Western Blot for p53, with actin as a control, following 10 Gy γ-irradiation, representative of three independent experiments. Samples were taken every hour for the first 12 h, during which the p53 protein undergoes two pulses. Note that while p53 pulses in individual cells are not damped, at the population level they appear damped owing to the loss of synchrony between cells10,11,25. Uncropped gel images are shown in Supplementary Data Set 5. (b) Genome-wide expression profiling following γ-irradiation. Samples were taken every hour for the first 12 h, as well as 24 h post irradiation. 229 genes were found to be differentially expressed relative to the basal level. Differential expression was defined as fold change > 2 and FDR < 0.2 (t test, Benjamini-Hochberg corrected) based on two independent experiments. Clustering was based on normalized time trace for each gene (z score) into five expression clusters, three induced (red) and two repressed (blue). Mean expressions of each cluster are shown in black. (c) Heat map of the clustered genes. Clusters are ordered based on the median activation time (shown in d). (d) The distribution of times to reach the maximal fold change per cluster. Black lines indicate the median, and the box edges and whiskers extend to the 25–75% and the 5–95% quantiles, respectively. (e) Distributions of maximal log2 (fold change) in the first 10 h post irradiation per cluster for p53 wild type cells (blue, WT-p53) and p53 knockdown cells (gray, sh-p53). FC, fold change. Box edges represent the interquartile range, black midlines indicate the median, and the lower and upper whiskers extend to the 5th and 95th percentiles, respectively. P values were calculated using a two-sided t test. Individual dots represent outliers. n.s., not significant.
The time it took for each gene to reach a maximum fold change varied between the p53 dependent clusters. Among the activated genes, cluster 1 had the fastest induction time of 3hr. Cluster 2 showed an intermediate induction time with a median of 6hr. Genes in clusters 3 and 5 took 11 hours to reach their maximal fold change (Figure 1D). Cluster 1 had the highest fraction of literature described p53 target genes (Figure S1B), and was enriched in GO categories associated with DNA damage response and the p53 signal transduction pathway (Figure S1C). Known p53 target genes were also significantly enriched in clusters 2 and 3 although they represented a much lower fraction of the total genes in these groups (Figure S1B). Taken together our gene expression profiling analysis showed that γ irradiation leads to five distinct patterns of mRNA dynamics, and four of these patterns (clusters 1, 2, 3 and 5) are p53 dependent.
Time-course p53 ChIP-Seq reveals pulsatile p53 DNA binding
To identify directly bound p53 genes and determine if the distinct patterns of gene expression (Figure 1B, C) result from differential binding of p53, we performed ChIP-Seq of p53 during the first and second pulses following irradiation (Figure 2A). We first looked at the well-established p53 binding site in the promoter of CDKN1A26,27, a canonical p53 target gene from cluster 1 (Figure 2B). We detected the expected promoter proximal p53 ChIP peak (Figure 2B) and a smaller adjacent peak that has also been seen in other p53 ChIP datasets21,22,28. Quantification of the stronger p53 ChIP peak after DNA damage revealed pulsatile binding dynamics, consistent with p53 protein levels and the pulses in CDKN1A mRNA level (Figure 2C).
Figure 2.
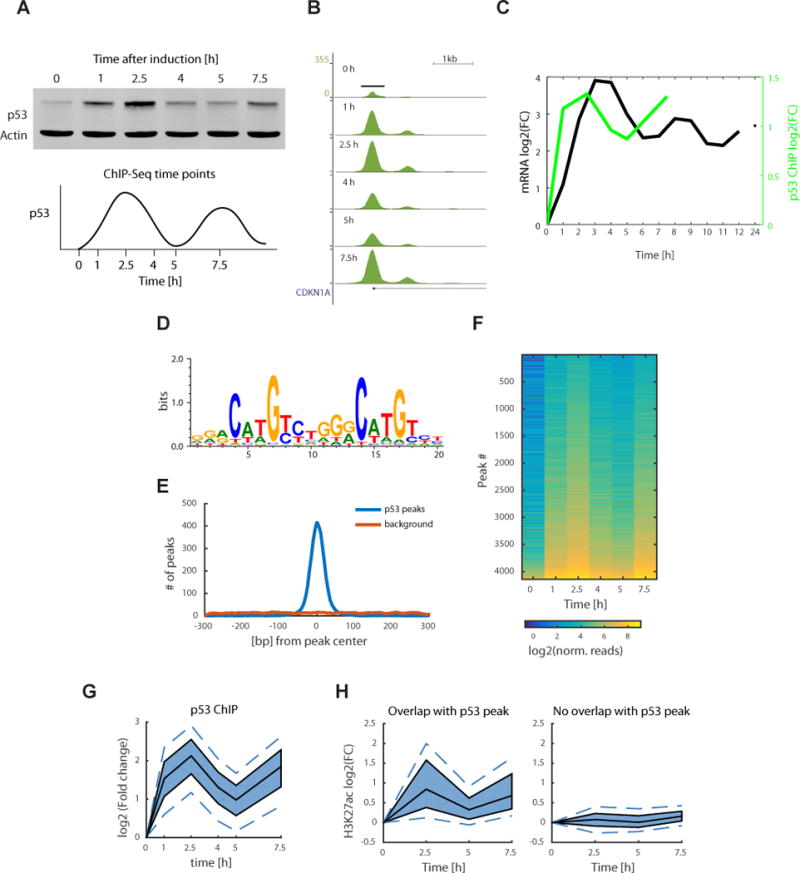
Time course p53 ChIP-seq experiment shows pulsatile dynamics genome wide. (a) Western blot of p53 protein levels, with actin as a control, at the selected time points for the ChIP-seq experiment sampling the first two p53 pulses (uncropped gel image in Supplementary Data Set 5). (b) p53 ChIP peak at the CDKN1A promoter with signal normalized to the total number of reads in each sample (UCSC tracks generated by HOMER software). (c) Quantification of the log2(FC) of p53 ChIP peak in b plotted with p21 mRNA dynamics from the mRNA-seq experiment. FC, fold change. (d) De novo motif found using the HOMER software present in 86.6% of the peaks (1.5% of background genomic regions) and plotted using WebLogo. (e) Distribution of motif (from d) start positions relative to the p53 peak centers. (f) Heat map of normalized reads in a total of 4141 p53 ChIP peaks, ordered by peak intensity. (g) Quantile plot of p53 ChIP peak log2(FC). The area shaded in dark blue represents the interquartile range, the black lines indicate the median, and the lower and upper dashed blue lines show the 10th and 90th percentiles, respectively. (h) H3K27ac ChIP was done at 0, 2.5, 5 and 7.5 h time points. Quantile plot shows H3K27ac ChIP peaks that overlap with the p53 ChIP peaks (left, 1,075 peaks) and of H3K27ac peaks that do not overlap with p53 (right, 26,153 peaks).
We next looked at p53 binding across the genome. We found a total of 4141 peaks that were bound by p53 across all time points (Supplemental data set 2). The top de novo found motif corresponded to the known p53 binding site26,29,30. It was present in 86.6% of the peaks (compared to 1.5% of background genomic regions) and distributed around the peak center (Figure 2D: HOMER motif, p=10−2119, Figure 2E). To determine if p53 shows different binding dynamics at different genomic locations as suggested by the distinct clusters of gene expression (Figure 1B, C), we quantified the ChIP signal over time for all peaks. We observed pulsatile dynamics of p53 ChIP for all genomic sites (Figure 2F, G). We tested if p53 pulses cause global chromatin changes by performing H3K27ac ChIP and found a total of 27,228 peaks (Supplemental data set 2). Only the peaks that overlapped with p53 peaks (4%) showed pulsatile dynamics (Figure 2H), suggesting that the pulsatile ChIP dynamics are specific to p53.
Taken together our data showed that in response to γ irradiation, pulses of p53 protein levels are converted into pulses of p53 DNA binding across the genome. This suggests that the differential expression patterns we observed at the mRNA level (Figure 1) do not result from distinct binding dynamics at different loci but rather from other mechanisms.
p53 target genes with different dynamics show similar p53 binding
To directly associate p53 binding with gene expression, we assigned each peak to the closest gene and imposed a distance cutoff of 20kb from the transcription start site (TSS). Cluster 1 was the most enriched in p53-bound genes; 69% of genes in this cluster had a p53 peak (binomial statistic, p<1×10−16, Figure 3A). The other two activated clusters also showed enrichment in p53-bound genes, with 36% of genes in cluster 2 (p=1.8×10−11) and 13% in cluster 3 (p=0.01). Cluster 5 had <10% of genes that were bound by p53 (not significant, p=0.4). The small fraction of differentially repressed genes that are bound by p53 is consistent with previous studies suggesting that p53 does not directly act as a repressor of transcription22,31–33. In agreement with our p53 shRNA data (Figure 1E, S1A) there were no p53 ChIP peaks in cluster 4.
Figure 3.
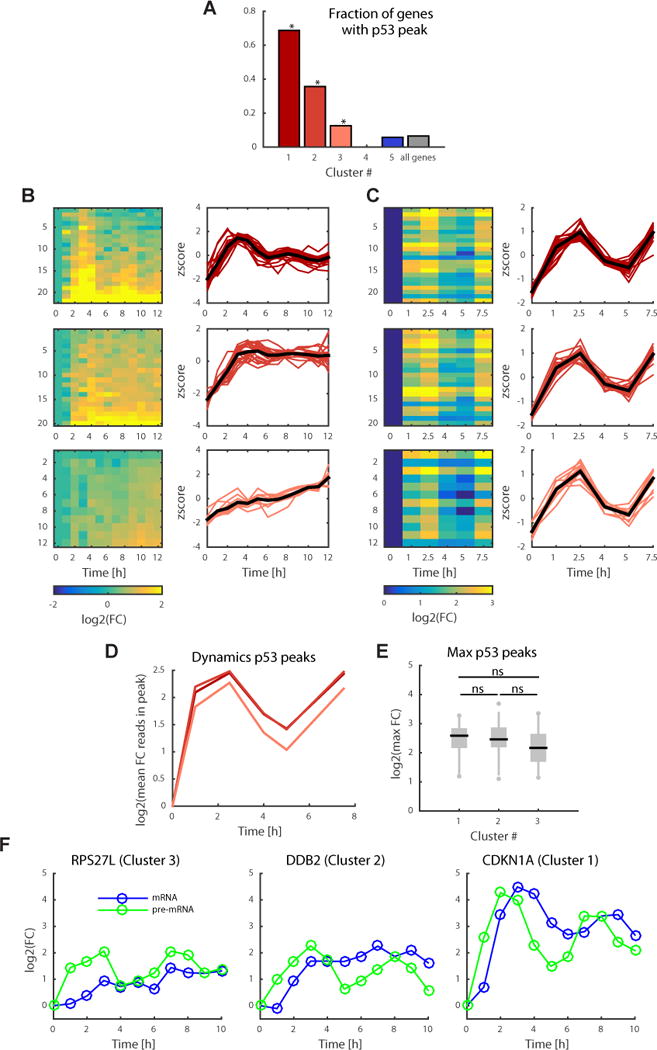
p53 target genes with distinct expression dynamics show similar dynamics of p53 binding. (a) Fraction of genes in each of the five gene expression clusters (Fig. 1) with a p53 peak within 20 kb of the TSS. P values were calculated using the binomial statistic; * for P < 0.01 probability of containing a peak. (b) Gene expression dynamics of the induced genes that contain a p53 peak within 20 kb of the TSS with log2(FC) on the left and normalized expression (z scores) on the right. FC, fold change. (c) Heat map of p53 ChIP log2 (fold change) of peaks corresponding to genes in b on the left and corresponding z scores of the p53 ChIP signal on the right. (d) Log2(fold change) of mean p53 ChIP dynamics per cluster. (e) Distribution of maximal fold change of p53 peaks per cluster. Black lines indicate the median, and box edges and whiskers extend to the 25–75% and 5–95% quantiles, respectively. A two-sided t test was done to evaluate significance; ns stands for not significant (P > 0.2 for all comparisons). (f) Quantitative RT-PCR on CDKN1A, DDB2 and RPS27L genes, in clusters 1, 2 and 3, respectively, using primers specific for pre-mRNA (green) and mRNA (blue) and averaged over two independent experiments.
We next focused on p53 bound genes in clusters 1, 2 and 3. Note that the expression levels of all p53 bound genes in these clusters were significantly lower in the p53 shRNA cell line relative to wild-type cells, consistent with these genes being direct targets of p53. We determined the quantitative differences in p53 binding to genes in these clusters by plotting the average ChIP signal for each cluster over time after irradiation. Surprisingly, we found that the three activated clusters, which showed distinct mRNA dynamics, had similar dynamics of p53 binding (Figure 3B, C). In addition, no significant difference was observed in the fold changes or absolute ChIP peak signal between clusters (Figure 3D, E and S2). The observation that p53 binds similarly to all induced genes suggests that the distinct dynamics of p53 target genes do not result from distinct binding, but rather from either additional regulation of transcription (e.g. p53 co-factors) or through post-transcriptional mechanisms. To distinguish between these options we measured the prem RNA dynamics of three p53 target genes representing the three clusters with distinct mRNA dynamics. We found similar pre-mRNA pulsatile profiles albeit distinct mature mRNA dynamics (Figure 3F), suggesting that post-transcriptional mechanisms contribute to the differential dynamics observed between clusters.
The dynamics of p53-dependent target genes across the genome are determined by their mRNA half-lives
How can pulsatile p53 give rise to different mRNA dynamics despite similar DNA binding and transcription? To answer this, we built a two-parameter model describing mRNA dynamics with p53 protein levels as input:
| (Equation 1) |
We used p53 levels at t-1hr as the input for p53 (Figure 1A quantified in Figure 4A) and fit the production and degradation rate constants, kp and kd to the measured mRNA-Seq data of p53 bound induced genes (see Methods and Supplemental data set 3). We obtained a fit with R2=0.92 for the CDKN1A gene (Figure 4A) and a median R2=0.75 for all genes compared to R2=0.46 for genes without a p53 peak (Figure 4B, S3A, Wilcoxon rank sum test p=2.23×10−7).
Figure 4.
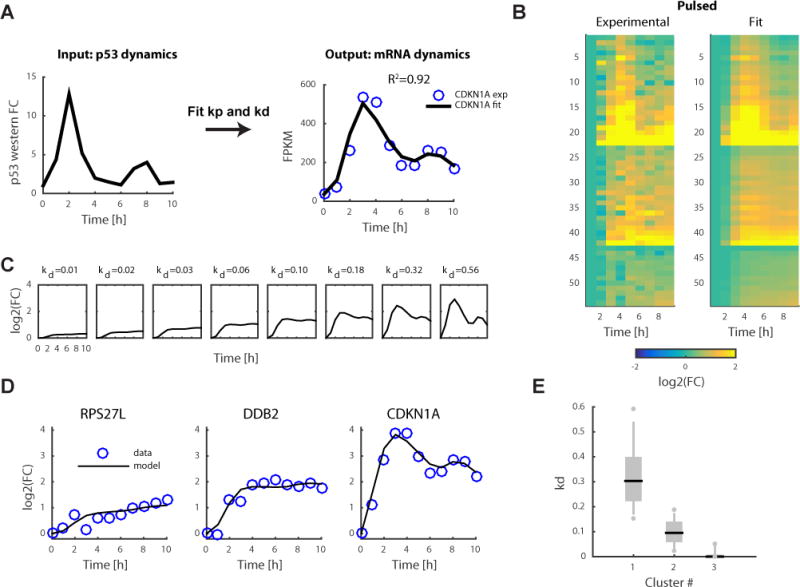
Mathematical model links the input–output relationship between p53 protein dynamics and the dynamics of its target genes genome wide. (a) Using p53 protein dynamics (quantified from Fig. 1a) as the input, we fit the production and degradation rates, kp and kd, respectively, to the mRNA-seq data. The results for the CDKN1A gene is shown as an example. FPKM, fragments per kilobase of transcript per million mapped reads. FC, fold change. (b) Heatmap of mRNA-seq data of p53 target genes (same as Fig. 3b) and corresponding fit data from the model. (c) Model simulation varying only the kd parameter. (d) Examples of mRNA-seq measurements (blue circles) and model (black lines) of three genes from clusters 1 (CDKN1A), 2 (DDB2), and 3 (RPS27L). (e) Distributions of kd values per cluster. Black lines indicate the median, and box edges and whiskers extend to the 25–75% and 5–95% quantiles, respectively. Individual dots represent outliers.
The activation rate kp showed a significant correlation with the maximal p53 ChIP peak amplitude on a per gene basis (Spearman’s ρ=0.54, p=3.7×10−5, Figure S3B). The degradation rate constant kd was significantly correlated with previously published mRNA half-life data collected in the same cell line (Spearman’s ρ=0.55, p=8.4×10−4, Figure S3C)34. The significant correlations between parameters extracted from our model and independent datasets (p53 ChIP and mRNA half-life) further validate the model and suggest that our mathematical formulation can be used for explaining p53 dependent gene expression.
We ran a simulation for a range of kp and kd parameters to explore their contribution to specific dynamical properties of gene expression (defined in Figure S3D). We observed that maximal induction of gene expression increased with kp values (Figure S3E). This dependency was lost when we looked at the maximal fold change, which was only dependent on the degradation rate kd (Figure S3F). The degradation rate kd also determined the pulsatility and the time to maximal induction, with higher degradation rates leading to pulsatile behavior and faster induction (Figure S3G, H). Our simulation therefore suggests that p53 DNA binding strength determines how strongly a gene is induced; however, the dynamics and timing of activation are controlled through mRNA half-life. In this scenario, different dynamics of target genes in response to p53 pulses can be achieved by solely varying the kd values while maintaining kp constant (Figure 4C). Indeed, the three genes representative of the three expression clusters with pulsatile pre-mRNA (Figure 3F) had different kd values (Figure 4D). At the global level, genes in cluster 1 were had higher kd values compared to genes in clusters 2 and 3 (p<10−5, Wilcoxon rank sum test, Figure 4E).
A mathematical model can predict the dynamics of p53-dependent genes under different p53 dynamical inputs
Can our model of gene regulation by p53 (Equation 1) be applied for a different dynamical input? To test the predictive power of the model we perturbed p53 dynamics from pulses to sustained using an established protocol3 (Figure 5A), and used the parameters derived from the pulsed condition to predict mRNA dynamics under sustained condition. The predicted mRNA dynamics for p53 target genes were in strong agreement with the levels measured by mRNA-Seq (median R2=0.91 versus R2=0.72 for genes without a p53 peak, Wilcoxon rank sum test p=2.27×10−7, Figure 5B, 5C, S3I, S3J and Supplemental data set 4). Thus, the kp and kd parameters that were derived from the pulsed condition can predict the expression of target genes under different p53 input dynamics.
Figure 5.
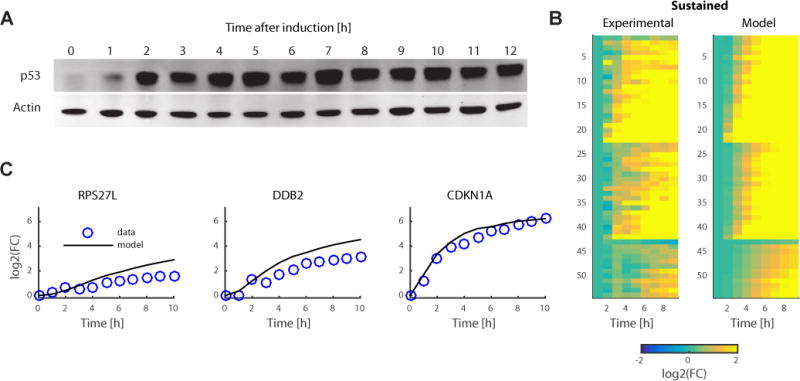
Mathematical model can predict gene expression for sustained p53 input dynamics. (a) Western blot for the sustained-p53 condition. Data is representative of three independent experiments (uncropped gel image shown in Supplementary Data Set 5). (b) Using sustained p53 protein dynamics derived from a, and kp and kd derived from the pulsed data, gene expression can be predicted under the sustained-p53 condition. Results are shown as a heat map for experimental data and model prediction. FC, fold change. (c) Examples of model prediction and mRNA measurements for the RPS27L, DDB2 and CDKN1A genes.
Discussion
How transcription factors’ dynamics affect the dynamics of their target genes is a fundamentally important question. Here, we focused on the stereotypic pulses of the transcription factor protein p53 and sought to determine how different target genes respond to variations in its dynamics. Using high-temporal resolution mRNA-Seq we identified three clusters of p53-dependent induced genes, with distinct gene expression dynamics: pulses, induction and a plateau, or continuous accumulation. However, when we measured the dynamics of p53 binding to its target genes using ChIP-Seq we observed pulsatile dynamics genome wide. Thus, p53 pulses were translated into pulsatile DNA binding but were not predictive of the output gene expression. We used mathematical modeling to understand how a uniform pulsatile input can give rise to different dynamics of gene expression. Using a two-parameter model with p53 protein levels as input, we found that we can recapitulate the different activation dynamics of p53 target genes. Moreover, our model parameters provide a mechanistic understanding of gene regulation by p53, where the strength of p53 binding contributes to the maximal induction of gene expression, and the mRNA degradation rate is important for the dynamic pattern of activation. We further used the model to predict gene expression dynamics for sustained p53 input and experimentally validated these predictions across the genome, thus demonstrating that our model can be used to predict gene expression for a new dynamical input of p53.
The contribution of mRNA half-life to the dynamics of gene expression has previously been observed for a subset of p53 target genes23,24. Our study, looking at genome wide mRNA dynamics, in a wild type and p53 knock down context, as well as integrating gene expression with time-course p53 ChIP-Seq data, allowed us to identify all p53 dependent and directly bound genes among the differentially expressed genes in response to γ irradiation, and to formulate a general model that explains and predicts mRNA dynamics of these target genes as a function of p53 input dynamics. We demonstrated the utility of our model for predicting gene expression in response to a different dynamical input of p53. A similar framework can be applied for future studies to determine the input-output relationship between p53 dynamics and its target genes under additional conditions and cellular backgrounds. Analogously to their role in the p53 system, mRNA half-lives were also found to be important for tuning the timing and pattern of gene expression in response to NFkB oscillations35,36. Further studies are required to determine if a similar general model of gene regulation could be used to predict expression of NFkB target genes in response to different NFkB input dynamics.
How p53 chooses between its target genes is a longstanding fundamental question. Our study suggests that, in the context of DNA damage, p53 itself does not choose which target gene to activate; it globally binds to DNA in a temporal pattern that mimics the dynamics of its protein levels, while the degradation rate of mRNA dominates the timing, profile and fold change of gene induction. Although we cannot exclude the contribution of other co-factors on the induction of specific p53 targets, our global observations are in agreement with a recent study, which used a high throughput enhancer assay to test p53 bound genomic regions, and determined that p53 can act in isolation22.
Lastly, our study provides new insights into the question of why p53 pulses in response to DNA damage. Expression dynamics of genes with short mRNA half-life, for example CDKN1A, follow p53 protein dynamics and thus have different dynamics under pulsed and sustained p53 inputs (Figure 4D and 5C). On the other hand, genes with long mRNA half-lives, like RPS27L, low pass filter the p53 signal and act as integrators (Figure 4D and 5C). Different effectors of p53 signaling may therefore act as integrators or instantaneous readouts of p53 activity. In addition, under sustained p53, gene expression dynamics show continuous accumulation and are less variable between genes (Figure 5B, 5C, S3J). On a global scale, sustained p53 dynamics result in a more uniform pattern of gene expression than does pulsatile p53 input, with significantly smaller Euclidean distances between genes in different clusters (Supplemental Table). These findings suggest that p53 pulses may provide a mechanism for diverse gene expression patterns which cannot be achieved under sustained conditions. Future studies are required for understanding why p53-target genes in the different clusters have different mRNA half-lives and the molecular mechanisms that control this differential mRNA stability. In addition, the impact of mRNA half-life on protein levels, and consequently on cell fate decision in response to different p53 inputs, remains an important open direction for future investigation.
Online methods
Cell culture and DNA damage treatment
MCF7 + p53shRNA, from Reuven Agami group37, and MCF7 cells were grown in RPMI + 10% fetal bovine serum (FBS) supplemented with 100 mL/L FBS, 100 U/mL penicillin, 100 mg/mL streptomycin, and 250 ng/mL fungizone (Gemini Bio-Products). The identity of MCF7 and MCF7 p53shRNA cell lines was confirmed by DNA fingerprinting with small tandem repeat (STR) profiling. MCF7 tested negatively for mycoplasma contamination. Irradiation and irradiation with Nutlin 3a treatment, for the sustained p53 dynamics were performed as described in 3.
RNA-Seq
RNA was collected using the TRIZOL reagent and purified using the Zymo RNA Clean-up kit. 500ng of RNA was used as input for the Illumina TruSeq Stranded mRNA Library Prep Kit. Barcoded libraries were pooled and single-end sequenced on Illumina Next-Seq.
ChIP-Seq
MCF7 cells were cross-linked in 1% formaldehyde for 5 min at room temperature, quenched with glycine, washed in ice cold PBS with Protease Inhibitors (Roche). Pellets were flash frozen in liquid nitrogen and stored at −80°C. Cross-linked cells were thawed on ice and lysed for 10 minutes in ice cold lysis buffer (10mM Tris-HCl pH 8.0, 10mM NaCl, 0.5% NP-40 with Protease Inhibitors (Roche) and 500uM DTT added just before use). Following a 5-min spin, pellets were washed in ice cold 1×PBS followed by a second spin, supernatant was discarded and nuclei were lysed for 10′ in nuclei lysis buffer (50mM Tris-HCl pH 8.0, 10mM EDTA, 0.32 % SDS) and sonicated using Diagenode Bioruptor for 15 minutes using high power and 30 seconds on, 30 seconds off cycle. The remainder of the ChIP protocol was done according to the Broad ChIP protocol from the Roadmap Epigenomics project (www.roadmapepigenomics.com) using 5M cells, 3ul of X-DO1 (Santa Cruz, sc-126 X) anti p53 antibody and anti H3K27ac antibody (Abcam ab4729). Illumina library preparation was done using the Nugen Ovation Ultralow Library Systems V2 kit. Barcoded libraries were pooled and sequenced on Illumina Next-Seq. For p53 ChIP, cells were crosslinked at the following timepoints post irradiation: 0h, 1h, 2.5h, 5h and 7.5h. For H3K27ac ChIP and ChIP input samples the following time points were used: 0h, 2.5h, 5h and 7.5h.
Quantitative RT-PCR
Same RNA samples as used RNA sequencing were used to generate complementary DNA using the high-capacity cDNA reverse transcription protocol (Applied Biosystems). Quantitative RT-PCR was then performed using 10ng of cDNA, 100 nM primer, and SYBR Green reagent (Applied Biosystems). Normalization was done to the average of both ACTB and GAPDH genes.
qPCR Primers used:
- CDKN1A mRNA
- FW: TGTCACTGTCTTGTACCCTTG
- REV: GGCGTTTGGAGTGGTAGAA
- CDKN1A pre-mRNA
- FW: CCCGGCCAGGTAACATAGT
- REV: CATGGGTTCTGACGGACATC
- DDB2 mRNA
- FW: TCGTCAGGACCCTCCAC
- REV: CGCCAAGGATGTAGCCC
- DDB2 intron
- FW: CGCTAGAGTGCAGTGATTCG
- REV: GGTGGTAGGTGCATGTGGTT
- RPS27L mRNA
- FW: CGTCCTTGGAAGAGGAAAAG
- REV: ACCGTGGTGATCTTGTAGCA
- RPS27L pre-mRNA:
- FW: GGGATTGCTAGTGTGGTGTG
- REV: TGTCCCTGACATTTCCAATTC
- GAPDH:
- FW: ACATCGCTCAGACACCATG
- REV: TGTAGTTGAGGTCAATGAAGGG
- ACTB:
- FW: ACCTTCTACAATGAGCTGCG
- REV: CCTGGATAGCAACGTACATGG
RNA-Seq data analysis
RNA-seq reads were mapped and analyzed by TopHat and Cufflinks RNA-seq analysis pipeline38 using Tophat version 2.1.0 and cufflinks version 2.1.1. Alignment was done against hg19 genome and hg19 RefSeq.gtf transcript annotations were used.
Selection of differentially expressed genes was done by calculating the fold change (FC) and significance relative to basal expression on the two biological replicates and selecting the genes that show a fold change above 2 with FDR (Benjamini and Hochberg) below 0.2 at any time point. Clustering was done on z-scores using Fuzzy c-means clustering with an exponent for the fuzzy partition matrix of 1.3 and number 5 clusters. Small variation in parameter values does not qualitatively affect the results and their interpretation.
We also performed a parallel analysis, selecting genes that were significantly different between wild-type and p53sh conditions. To be called significantly different, a gene had to pass at least one of two criteria: (1) the difference in fold change per time point between wild-type and p53sh had to be significantly different from zero using a Wilcoxon signed-rank test and P < 0.05, or (2) The dynamics of expression between wild-type and p53sh had to be uncorrelated (Spearman and P > 0.05). The expression of 92.6% of the genes, based in their fold change in expression in the p53 wild-type cell line, led to four clusters with clusters 1, 2, 3 and 5 remaining largely unchanged and retaining their cluster identity. Cluster 4 disappeared and the remaining p53-dependent genes in this cluster were assigned to clusters 3 and 5.
GO Enrichment analysis was done using Enrichr software39,40.
ChIP-Seq data analysis
Reads were aligned to the human reference genome (hg19) using Bowtie 1 algorithm and only uniquely aligned reads were retained41. Duplicate reads were removed for downstream analyses. For peak calling the IDR framework42,43 with MACS244 algorithm was used. Briefly, reads from all time points were pooled together and peaks were called using the MACS2 software using p=1e−3 cutoff on the pooled reads file and on pseudo-replicate files where reads from the pooled file were randomly split in half. We identified 4141 peaks with 0.0025 IDR threshold. For these 4141 peaks, we defined regions of + and − 300bp around the peak center to count the number of reads per peak for each condition which were then normalized to the total number of reads in the sample. For the analysis looking at p53 peak amplitude, the average number of input reads over all time-points and mapping to the corresponding peak locations was subtracted from the p53 ChIP reads.
The HOMER package45 was used for de novo motif discovery. WebLogo was used to generate the motif plot46 in Figure 2 for the top enriched motif. The top enriched motif (Figure 2) was then used to re-scan and score all peaks and background regions. Background regions were generated by selecting 600bp regions adjacent to either side of the peak and excluding regions that overlap with p53 peak regions.
Mapping of peaks to genes was done using the HOMER package to find the closest gene. Only peaks with 20kb relative to the TSS of each gene were used for the analyses. For genes with multiple p53 ChIP peaks, the signal from the closest p53 ChIP peak was used.
Model
Because of the discrete time points of our data, we implemented the model in Equation 1 by calculating the mRNA level at each step for a given set of kp and kd parameters at each time point.
Quantification of p53 Western blot (Figure 1A quantified in Figure 4A) was used as the input for p53 levels for the pulsed condition. For the sustained p53 condition, we quantified the Western blot (Figure 5A) and used it as the model input.
We fit the model under the pulsed p53 condition as to minimize the square of the Pearson’s correlation between the predicted and the measured mRNA levels for each gene.
Statistical analyses
In the cases were t-tests were used, the Anderson-Darling test was used to test for normality. Binomial statistics were used to calculate p-values in Figures 3A, S1A. Wilcoxon signed rank tests were used for non-normal distributions.
A Life Sciences Reporting Summary for this article is available online.
Data availability statement
All sequencing data for ChIP-Seq and RNA-Seq experiments have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE100099.
Source data for Figure 1, containing FPKM values and log2(Fold Change) per time point for 229 differentially expressed genes as well as their cluster assignments are available as Supplemental data set 1. p53 ChIP-Seq peaks with their genomic coordinates, corresponding gene assignments and normalized read counts are available in Supplemental data set 2. H3K27ac ChIP-Seq peaks with their genomic coordinates, distance to the closest p53 ChIP peak and normalized read counts are available in Supplemental data set 2. The fitting parameters for the 54 p53 bound genes in Figure 4 are provided in Supplemental data set 3. Data for Figure 5 containing FPKM values under the sustained p53 condition for 54 genes as well as the result of the model fit are available in Supplemental data set 4.
Source code for the mathematical model can be made available upon request.
Supplementary Material
Acknowledgments
We thank Sarah Boswell for help with RNA-Seq experiments and Xiaolan Zhang (Broad Institute) for help with p53 ChIP experiments; Mike Springer for helpful discussions and advice on modeling; members of the Lahav lab for comments and discussion. This research was supported by Novartis Institutes for Biomedical Research and NIH grant GM083303. A.H. was supported by Boehringer Ingelheim Fonds, PhD fellowship. J.S-O. was supported by NIH grant CA207727. J.E.P. was supported by NIH grant GM102372. M.L.B was supported by NIH grant HG003985.
Footnotes
Author contributions
A.H., J.E.P., W.C.F. and G.L. conceived experiments. A.H., M.L.B. and G.L. conceived analyses. A.H. performed experiments and A.H. and J.S-O. performed analyses. A.H. and G.L. wrote the paper.
Competing interests
The authors declare that they have no conflict of interest.
References
- 1.Cai L, Dalal CK, Elowitz MB. Frequency-modulated nuclear localization bursts coordinate gene regulation. Nature. 2008;455:485–90. doi: 10.1038/nature07292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IkappaB-NF-kappaB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 3.Purvis JE, et al. p53 dynamics control cell fate. Science. 2012;336:1440–4. doi: 10.1126/science.1218351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paek AL, Liu JC, Loewer A, Forrester WC, Lahav G. Cell-to-Cell Variation in p53 Dynamics Leads to Fractional Killing. Cell. 2016;165:631–42. doi: 10.1016/j.cell.2016.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hao N, O’Shea EK. Signal-dependent dynamics of transcription factor translocation controls gene expression. Nat Struct Mol Biol. 2011;19:31–9. doi: 10.1038/nsmb.2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–7. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 7.Batchelor E, Loewer A, Mock C, Lahav G. Stimulus-dependent dynamics of p53 in single cells. Mol Syst Biol. 2011;7:488. doi: 10.1038/msb.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen AS, O’Shea EK. Encoding four gene expression programs in the activation dynamics of a single transcription factor. Curr Biol. 2016;26:R269–71. doi: 10.1016/j.cub.2016.02.058. [DOI] [PubMed] [Google Scholar]
- 9.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–61. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 10.Lahav G, et al. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet. 2004;36:147–50. doi: 10.1038/ng1293. [DOI] [PubMed] [Google Scholar]
- 11.Lev Bar-Or R, et al. Generation of oscillations by the p53-Mdm2 feedback loop: a theoretical and experimental study. Proc Natl Acad Sci U S A. 2000;97:11250–5. doi: 10.1073/pnas.210171597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weinberg RL, Veprintsev DB, Bycroft M, Fersht AR. Comparative binding of p53 to its promoter and DNA recognition elements. J Mol Biol. 2005;348:589–96. doi: 10.1016/j.jmb.2005.03.014. [DOI] [PubMed] [Google Scholar]
- 13.Qian H, Wang T, Naumovski L, Lopez CD, Brachmann RK. Groups of p53 target genes involved in specific p53 downstream effects cluster into different classes of DNA binding sites. Oncogene. 2002;21:7901–11. doi: 10.1038/sj.onc.1205974. [DOI] [PubMed] [Google Scholar]
- 14.Kracikova M, Akiri G, George A, Sachidanandam R, Aaronson SA. A threshold mechanism mediates p53 cell fate decision between growth arrest and apoptosis. Cell Death Differ. 2013;20:576–88. doi: 10.1038/cdd.2012.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008;9:702–12. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 16.Flores ER, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–4. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 17.Samuels-Lev Y, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell. 2001;8:781–94. doi: 10.1016/s1097-2765(01)00367-7. [DOI] [PubMed] [Google Scholar]
- 18.Oda K, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–62. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- 19.Smeenk L, et al. Role of p53 serine 46 in p53 target gene regulation. PLoS One. 2011;6:e17574. doi: 10.1371/journal.pone.0017574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sykes SM, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–51. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nikulenkov F, et al. Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ. 2012;19:1992–2002. doi: 10.1038/cdd.2012.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verfaillie A, et al. Multiplex enhancer-reporter assays uncover unsophisticated TP53 enhancer logic. Genome Res. 2016;26:882–95. doi: 10.1101/gr.204149.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter JR, Fisher BE, Batchelor E. p53 Pulses Diversify Target Gene Expression Dynamics in an mRNA Half-Life-Dependent Manner and Delineate Co-regulated Target Gene Subnetworks. Cell Syst. 2016;2:272–82. doi: 10.1016/j.cels.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melanson BD, et al. The role of mRNA decay in p53-induced gene expression. Rna. 2011;17:2222–34. doi: 10.1261/rna.030122.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batchelor E, Mock CS, Bhan I, Loewer A, Lahav G. Recurrent initiation: a mechanism for triggering p53 pulses in response to DNA damage. Mol Cell. 2008;30:277–89. doi: 10.1016/j.molcel.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei CL, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell. 2006;124:207–19. doi: 10.1016/j.cell.2005.10.043. [DOI] [PubMed] [Google Scholar]
- 27.el-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–25. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 28.Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW. Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle. 2011;10:4237–49. doi: 10.4161/cc.10.24.18383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet. 1992;1:45–9. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 30.Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9:402–12. doi: 10.1038/nrm2395. [DOI] [PubMed] [Google Scholar]
- 31.Fischer M, Steiner L, Engeland K. The transcription factor p53: not a repressor, solely an activator. Cell Cycle. 2014;13:3037–58. doi: 10.4161/15384101.2014.949083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlereth K, et al. Characterization of the p53 cistrome–DNA binding cooperativity dissects p53’s tumor suppressor functions. PLoS Genet. 2013;9:e1003726. doi: 10.1371/journal.pgen.1003726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rashi-Elkeles S, et al. Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci Signal. 2014;7:rs3. doi: 10.1126/scisignal.2005032. [DOI] [PubMed] [Google Scholar]
- 34.Schueler M, et al. Differential protein occupancy profiling of the mRNA transcriptome. Genome Biol. 2014;15:R15. doi: 10.1186/gb-2014-15-1-r15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zambrano S, De Toma I, Piffer A, Bianchi ME, Agresti A. NF-kappaB oscillations translate into functionally related patterns of gene expression. Elife. 2016;5:e09100. doi: 10.7554/eLife.09100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hao S, Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. 2009;10:281–8. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 38.Trapnell C, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–78. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–7. doi: 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen EY, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, Brown JB, Huang H, Bickel PJ. Measuring reproducibility of high-throughput experiments. 2011:1752–1779. [Google Scholar]
- 43.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data for ChIP-Seq and RNA-Seq experiments have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE100099.
Source data for Figure 1, containing FPKM values and log2(Fold Change) per time point for 229 differentially expressed genes as well as their cluster assignments are available as Supplemental data set 1. p53 ChIP-Seq peaks with their genomic coordinates, corresponding gene assignments and normalized read counts are available in Supplemental data set 2. H3K27ac ChIP-Seq peaks with their genomic coordinates, distance to the closest p53 ChIP peak and normalized read counts are available in Supplemental data set 2. The fitting parameters for the 54 p53 bound genes in Figure 4 are provided in Supplemental data set 3. Data for Figure 5 containing FPKM values under the sustained p53 condition for 54 genes as well as the result of the model fit are available in Supplemental data set 4.
Source code for the mathematical model can be made available upon request.