

Abstract
4-Oxo-2-nonenal (ONE) derived from lipid peroxidation modifies nucleophiles and transduces redox signaling by its reactions with proteins. However, the molecular interactions between ONE and complex proteomes and their dynamics in situ remain largely unknown. Here we describe a quantitative chemoproteomic analysis of protein adduction by ONE in cells, in which the cellular target profile of ONE is mimicked by its alkynyl surrogate. The analyses reveal four types of ONE-derived modifications in cells, including ketoamide and Schiff-base adducts to lysine, Michael adducts to cysteine, and a novel pyrrole adduct to cysteine. ONE-derived adducts co-localize and exhibit crosstalk with many histone marks and redox sensitive sites. All four types of modifications derived from ONE can be reversed site-specifically in cells. Taken together, our study provides much-needed mechanistic insights into the cellular signaling and potential toxicities associated with this important lipid derived electrophile.
Reactive oxygen species generated from biological processes or environmental insults can result in damage to biomacromolecules including proteins and DNA (1, 2). The polyunsaturated fatty acyl chains found in biological membranes and lipoproteins are particularly susceptible to reactive oxygen species, leading to free radical chain autoxidation and the formation of a variety of unsaturated lipid hydroperoxides and their electrophilic decomposition products, such as 4-hydroxy-2-nonenal (HNE)1 and 4-oxo-2-nonenal (ONE) (3). These lipid derived electrophiles (LDE) can react with nucleophiles on proteins, including cysteine, lysine, and histidine (4). Chemical modification induced by the lipid derived electrophiles (LDEs) has emerged an important mechanism for cells to regulate redox signaling and drive cytotoxic responses (5). Dysregulation triggered by these LDE-protein interactions is associated with inflammation, diabetes, neurodegenerative disorders, and cardiovascular diseases (6–9).
Identifying the protein targets of LDEs is critical for better understanding of their functional impact on specific signaling pathways and cellular functions. Recent advances in proteomics have improved the detection of LDE-induced protein modifications and greatly expanded the global inventories of targeted proteins and/or sites of LDEs both in vitro and in situ, especially for HNE (10–14). Although ONE and HNE share a nearly identical chemical structure (supplemental Scheme S1), ONE is more reactive and cytotoxic than HNE in neuronal cells (15). Unlike HNE, which preferentially reacts with proteomic cysteines (10, 12), ONE displays a broader range of adduction chemistry because of differences in its stereoelectronic properties (3). For instance, ONE exhibits more potent chemical reactivity with lysine residues, as compared with HNE. Of interest, Galligan et al. recently showed that ONE forms stable ketoamide adducts with several lysine residues on histones and blocks nucleosome assembly, thereby suggesting a potential link between oxidative stress and epigenetic effects (16). In addition, ONE renders more likely intra- or intermolecular cross-linking of its targets, which has been implicated in many diseases associated with protein aggregation. For example, ONE facilitates the formation of more stable α-synuclein oligomers than those induced by HNE (17). More recently, Marnett and coworkers showed that ONE, rather than HNE, forms cross-links and alters the activities of pyruvate kinase M2 and peptidylprolyl cis/trans isomerase A1 in cells (18, 19). Despite these interesting findings, the molecular interactions between ONE and complex proteomes and their dynamics remain uncertain with respect to the following issues. First, the full nature of in situ adduction chemistry of ONE is still unknown, although the chemical reactivity of ONE with nucleophilic residues has been analyzed in chemical model systems (3, 20, 21). Second, the site-specific target profile and selectivity of ONE across native proteomes are still unexplored. Third, it is unclear whether ONE-derived adductions are reversible in cells, though two recent studies have shown that one of these modifications on histones can be removed by deacylase Sirt2 (22, 23).
Here we present the first global survey of ONE adduct chemistry, targeting sites, and dynamics in intact cells using a generalized quantitative chemoproteomic platform (10), in which the cellular target profile of ONE is mimicked by its alkynyl surrogate (aONE, Fig. 1). This analysis not only greatly expand the inventory of ONE-adducts in cells but also identify a novel pyrrole adduct to cysteine. Biochemical analyses further show that these ONE-derived adducts co-localize and exhibit crosstalk with many histone marks and redox sensitive sites. Moreover, quantitative analyses reveal that all four types of modifications derived from ONE are reversible in cells in a site-specific manner, which may be controlled by Sirt2-mediated deacylation and other unknown mechanisms.
Fig. 1.
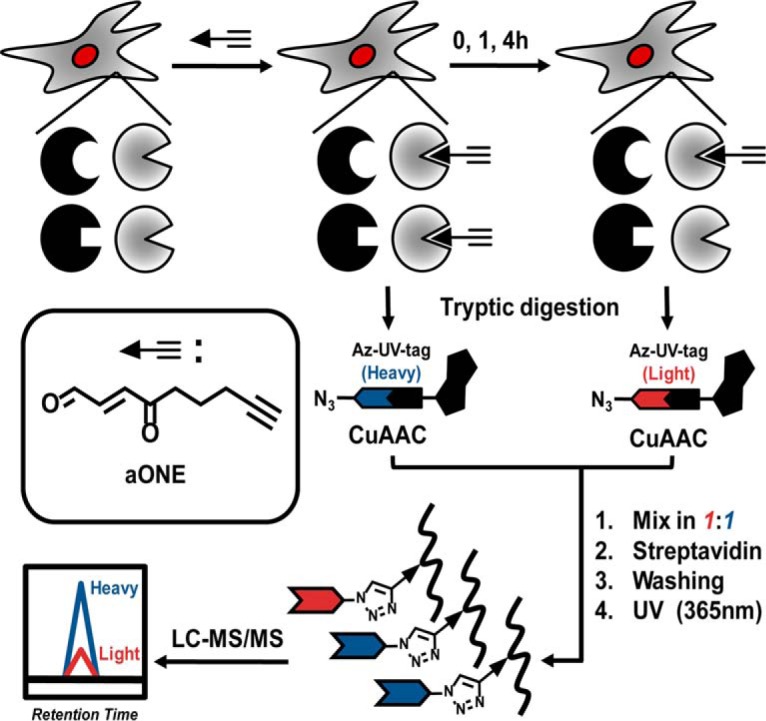
Workflow for quantitative chemoproteomic analysis of dynamic aONE-derived protein adducts in cells.
EXPERIMENTAL PROCEDURES
Chemicals
Alkynyl-ONE (aONE), 12C and 13C labeled azido-UV-biotin reagents (Azido-l-biotin and azido-H-biotin) were synthesized as described previously. ONE was purchased from Cayman (10185, Ann Arbor, MI). Unmodified PGHLQEGFGCVVTNR and LAHCEELR were purchased from Chinese Peptide Company (Hangzhou, China). Model peptide PDFAQELLCR was obtained from ONTORES (Hangzhou, China). HPLC-grade water, ACN, and MeOH were purchased from J.T. Baker, Valley, PA. Other chemicals and reagents were obtained from Sigma-Aldrich St. Louis, MO, unless otherwise indicated.
Antibodies
Anti SIRT2 (Abcam, Cambridge, MA, ab191383, diluted at 1:1000); anti ubiquityl-Histone H2BK120 (Cell Signaling Technology, Danvers, MA, #5546, diluted at 1:1000); anti Histone H2B (Proteintech, 15857–1-AP, diluted at 1:500); anti PARK7/DJ-1 (Proteintech, Rosemont, IL, 11681–1-AP, diluted at 1:500); anti oxDJ-1(Cys106) (Millipore, Billerica, MA, MABN1773, diluted at 1:500); anti β-Actin (ZSGB-BIO, Beijing, China, PR-0255, diluted at 1:1000); goat anti mouse-HRP(ZSGB-BIO, ZDR5307, diluted at 1:2500); goat anti-rabbit-HRP(ZSGB-BIO, ZDR5306, diluted at 1:2500); Anti V5 (Yeasen, Shanghai, China, diluted at: 1:5000).
Synthesis of aONE-derived Adducts
1 mm unmodified peptide (PGHLQEGFGCVVTNR or LAHCEELR) dissolved in 40 μl (final volume) 1× PBS was incubated in the presence of 1 mm aONE and 100 mm glycine for 2 h at 37 °C. The resulting peptide adduct mixture was desalted as previously described (24). Click chemistry then was performed by the addition of 1 mm Az-UV-biotin, 10 mm sodium ascorbate, 1 mm TBTA, and 8 mm CuSO4. Click reactions could proceed at RT for 2 h in the dark with rotation. The reaction was stopped by adding 400 μl 25 mm ammonium bicarbonate (pH 8.0) and transferred to glass tubes and irradiated with 365 nm UV light (Entela, Upland, CA) for 2 h at room temperature with stirring. The resulting peptide mixtures were desalted by 200 μl tips (AXYGEN, Tewksbury, MA T-400) containing Durashell C18 (3 μm, 150 Å, Algela, DC930010-L) filled on the C18 membrane (Empore Bioanalytical Technologies 3 m, St. Paul, MN, 2215-C18).
Cell Culture and Treatment
RKO cells (American Type Culture Collection, ATCC) were maintained at 37 °C in a 5% CO2, humidified atmosphere and were cultured in McCoy medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Invitrogen). HeLa cells (National Infrastructure of Cell Line Resource, Beijing, China) were maintained at 37 °C in a 5% CO2, humidified atmosphere and were cultured in DMEM medium (HyClone, Logan, UT) containing 10% fetal bovine serum (Life technologies, Gibco). Cells were grown until 80–90% confluency, rinsed with 1× PBS quickly, and treated with 50 μm aONE prepared in serum-free medium for 2 h. Treatments were stopped by removing the medium. Cells were then lifted with 0.25% trypsin-EDTA (Invitrogen) and harvested by centrifugation at 1500 × g for 3 min. For recovery experiments, cells were cultured as above, labeled for 2h with 50 μm aONE, either harvested immediately (control) or recovered after 1 h and 4 h time points in serum-free medium without aONE. For oxDJ-1 detection, HeLa cells were grown until 70–80% confluent in six well plates. After overnight serum deprivation, cells were incubated with indicated concentration of ONE in serum-free medium for 2 h at 37 °C. Cells were then treated with or without 5 mm H2O2 for 10 min.
siRNA Transfection
The siRNA duplexes were obtained from Genepharma (Suzhou, China). siRNA transfections were performed using RNAiMAX (Invitrogen, 13778–075), according to the manufacturer's instructions. In brief, HeLa cells were seeded onto 6 cm diameter dishes to reach 70–80% confluence. Cells were transfected twice with 10 nm control siRNA or Sirt2 siRNA (8 μl of a 20 μm stock) using 12 μl RNAiMAX reagent. After 48 h of transfection, the Sirt2 knockdown cells were used for relative stimulation. The siRNA sequences were as follows: Control siRNA (UUCUCCGAACGUGUCACGUTT); Sirt2 siRNA (UCUCCACAUCCGCAGGCAUTT).
Expression of V5-tagged CFL Proteins
Full-length cDNA encoding human CFL1 (NM_005507) in pDONR223 was purchased from YouBao and subcloned into pLX304 (Addgene, Cambridge, MA). C139A mutant was generated by QuikChange site-directed mutagenesis using the primer GAATTGCAAGCAAACGCCTACGAGGAGGTCAAG and then were also cloned into pLX304 vector. Transfection was performed by incubating 30 μg of each of plasmids and 270 μl of polyethylenimine with 80%-confluent HeLa cells on a 10-cm dish. After 6 h of transfection, cells were cultured in regular DMEM medium with 10% FBS for another 48 h.
Ionizing Radiation
Cells were grown until 70–80% confluent in 6 cm diameter cell culture flasks, rinsed with 1× PBS quickly and placed overnight (14–h) in serum-free medium. After serum deprivation, cells were irradiated with 10 Gy X-rays at 160 kV and a dose rate of 180 cGy/min using the XRAD160 X-ray device (Precision X-Ray, North Branford, CT). The field size for the X-ray irradiation was 20 × 20 cm to fully cover the flasks and the source to surface distance was 50 cm. After irradiation, cells were treated with or without 25 μm ONE in serum-free medium for 2 h at 37 °C. Treatments were stopped by removing the medium and washing the plates quickly with cold PBS.
Histone Extraction
EpiQuik™ Total Histone Extraction Kit (Epigentek group, Farmingdale, NY, # OP-0006) was used to produce histone extracts. The method was performed as described in the manufacturer's instructions. In brief, soluble chromatinized histones were extracted with H2SO4, and centrifuged to separate supernatant and pellet fractions. The supernatants were collected as histone extracts and neutralized to pH7–8.
Sample Preparation for Proteomic Analysis
Cell pellets were lysed on ice in NETN lysis buffer (50 mm HEPES, 150 mm NaCl, 1% Igepal, pH 7.5) containing inhibitor mixture. The lysate was first reduced with 4 mm NaBH4 for 1 h at room temperature. The lysate was further incubated with 8 mm DTT (Research Products International, Mount Prospect, IL) at 75 °C for 15 min to reduce the reversibly oxidized cysteines. Reduced cysteines then were alkylated with 32 mm IAM for 30 min in the dark at the room temperature. Proteins were then precipitated with methanol-chloroform (aqueous phase/methanol/chloroform, 4:4:1 (v/v/v)) as previously described (24, 25). The precipitated protein pellets were resuspended with 50 mm ammonium bicarbonate containing 0.2 m urea. Resuspended protein concentrations were determined with the BCA assay (Thermo Fisher Scientific, Waltham, MA) and adjusted to a concentration of 2 mg/ml. Resuspended proteins were first digested with sequencing grade trypsin (Promega, Madison, WI) at a 1:50 (enzyme/substrate) ratio overnight at 37 °C. A secondary digestion was performed by adding additional trypsin to a 1:100 (enzyme/substrate) ratio, followed by incubation at 37 °C for additional 4 h. The tryptic digests were desalted with HLB extraction cartridges (Waters, Milford, MA). The desalted samples were then evaporated to dryness under vacuum.
Click Chemistry, Capture, and Enrichment
Desalted tryptic digests were reconstituted in a solution containing 30% ACN at pH 6. Click chemistry was performed by the addition of 0.8 mm either Azido-l-biotin or Azido-H-biotin (2.5 μl of a 40-mM stock), 8 mm sodium ascorbate (10 μl of a 100 mm stock), 1 mm TBTA (2.5 μl of a 50 mm stock, and 8 mm CuSO4 (10 μl of a 100 mm stock). Samples were allowed to react at room temperature for 2 h in the dark with rotation. The Azido-l-biotin and Azido-H-biotin samples were then mixed together immediately following click chemistry. Excess reagents were removed by SCX chromatography as previously described (25) and then the extracts were allowed to interact with prewashed streptavidin Sepharose for 2 h at room temperature. Streptavidin Sepharose then was washed with 50 mm NaAc, 50 mm NaAc containing 2 m NaCl, and water twice each with vortexing and/or rotation to remove nonspecific binding peptides, and resuspended in 25 mm ammonium bicarbonate. The suspension of streptavidin Sepharose was transferred to several glass tubes (VWR, Radnor, PA), irradiated with 365 nm UV light (Entela, Upland, CA) for 2 h at room temperature with stirring. The supernatant was collected, evaporated to dryness under vacuum, and stored at −20 °C until analysis.
LC-MS/MS Analysis
LC-MS/MS analyses were performed on Q Exactive plus or HF instruments (Thermo Fisher Scientific) operated with an Easy-nLC1000 system (Thermo Fisher Scientific). For analyzing aONE-adducts in RKO cells, samples were reconstituted in 0.1% formic acid and pressure-loaded onto a 360 μm outer diameter ×75 μm inner diameter microcapillary precolumn packed with Jupiter C18 (5 μm, 300 Å, Phenomenex, Torrance, CA) and then washed with 0.1% formic acid. The precolumn was connected to a 360 μm outer diameter ×50 μm inner diameter microcapillary analytical column packed with the ReproSil-Pur C18-AQ (3 μm, 120 Å, Dr. Maisch, GebH, Germany) and equipped with an integrated electrospray emitter tip. The spray voltage was set to 1.5 kV and the heated capillary temperature to 250 °C. LC gradient condition consisted of 0–15 min, 2% B; 35 min, 15% B; 40 min, 20% B; 50 min, 30% B; 55 min, 35% B; 59–65 min, 90% B; 80–85 min, 2% B (A = water, 0.1% formic acid; B = ACN/0.1% formic acid) at a flow rate of 300 nL/min. HCD MS/MS spectra were recorded in the data-dependent mode using Top 20 method for quantitative analysis, respectively. MS1 spectra were measured with a resolution of 70,000, an AGC target of 3e6, and a mass range from m/z 300 to 1800. HCD MS/MS spectra were acquired with a resolution of 17,500, an AGC target of 2e5, and normalized collision energy of 28. Peptide m/z that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 20 s. For analyzing aONE-adducts in HeLa cells, samples were reconstituted in 0.1% formic acid and pressure-loaded onto a 2 cm microcapillary precolumn packed with C18 (3 μm, 120 Å, SunChrom, Friedrichsdorf, Germany). The precolumn was connected to a 12 cm 150-μm-inner diameter microcapillary analytical column packed with C18 (1.9 μm, 120 Å, Dr. Maisch) and equipped with a homemade electrospray emitter tip. The spray voltage was set to 2.1 kV and the heated capillary temperature to 320 °C. For histones, LC gradient consisted of 0 min, 7% B; 14 min, 10% B; 51 min, 20% B; 68 min, 30% B; 69–75 min, 95% B (A = water, 0.1% formic acid; B = Acetonitrile, 0.1% formic acid) at a flow rate of 600 nL/min. HCD MS/MS spectra were recorded in the data-dependent mode using a Top 20 method. MS1 spectra were measured with a resolution of 70,000, an AGC target of 3e6, a max injection time of 20 ms, and a mass range from m/z 300 to 1400. HCD MS/MS spectra were acquired with a resolution of 17,500, an AGC target of 1e6, a max injection time of 60 ms, a 1.6 m/z isolation window and normalized collision energy of 30. Peptide m/z that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 18 s. For model peptide analysis, samples were dissolved in HPLC buffer A (0.1% formic acid in water, v/v) and loaded onto a home-made microcapillary precolumn (360 μm outer diameter, 100 μm inner diameter) packed with SP C18 (3 mm, 120 Å, Sunchrom) and then washed with 0.1% formic acid. The precolumn was connected to a microcapillary analytical column (360 μm outer diameter, 150 μm inner diameter) packed with the ReproSil-Pur C18-AQ (1.9 mm, 120 Å) and equipped with an integrated electrospray emitter tip. The spray voltage was set to 2.0 kV and the heated capillary temperature to 320 °C. Gradient consisted of 0 min, 21%; 10 min, 30%; 30 min, 45%; 31 min, 95%; 36 min, 95% B (A = water, 0.1% formic acid; B = acetonitrile/0.1% formic acid) at a flow rate of 600 nL/min. HCD MS/MS spectra were recorded in the data-dependent mode using a Top 20 method for quantitative analysis. MS1 spectra were measured with a resolution of 70,000, an AGC target of 3e6, and a mass range from m/z 300 to 1200. HCD MS/MS spectra were acquired with a resolution of 17,500, an AGC target of 1e6, and normalized collision energy of 30. Peptide m/z that triggered MS/MS scans were dynamically excluded from further MS/MS scans for 18 s.
Peptide Identification and Quantification
Raw data files were searched against Homo sapiens Uniprot canonical database (Dec 2, 2016, 20,130 entries). Blind search and targeted search were performed with TagRecon (Version 1.4.47) (26, 27) and MS-GF+ (Version 2016.10.26) algorithm (28), respectively. For TagRecon based blind PTM search, the maximum modification mass was 500 Da, precursor ion mass tolerance was 0.01 Da, and fragmentation tolerance was 0.1 Da. For MS-GF+ analysis, precursor ion mass and fragmentation tolerance were 10 ppm for the database search. A specific-tryptic search was employed with a maximum of three missed cleavages allowed. The maximum number of modifications allowed per peptide was three. Modifications of 15.9949 Da (Methionine oxidation, M), + 57.0214 Da (iodoacetamide alkylation, C), +289.1426 (C15H19N3O3, Light Schiff-base adduct, K), +295.1628 (Heavy Schiff-base adduct, K), +307.1532 (C15H21N3O4, Light ketoamide adduct, K), +313.1733 (Heavy ketoamide adduct, K), +311.1845 (C15H25N3O4, Light Michael adduct, C), +317.2047 (Heavy Michael adduct, C), +346.1641 (C17H22N4O4, Light pyrrole adduct, C), +352.1842 (Heavy pyrrole adduct, C) were searched as dynamic modifications. No fixed modifications were searched. The maximum Q value of PSMs was set as 0.01 using IDPicker 3.0 software to achieve either a peptide or a protein FDR no greater than 5%. Additional assessments of the FDR were performed (described later), resulting in a final FDR below 1%.
Quantification of light/heavy ratios (RL:H) was performed using Skyline software (version 3.1) as previously described (24, 25). In brief, spectral libraries generated from the mzID files as well as a subset FASTA database containing all the identifications (with and/or without aONE-derived modification) were imported into Skyline. The peptide list was refined in Skyline by removing all unmodified peptides. To further reduce the final FDR of identification and enable reliable quantification of the remaining aONE-adducted peptides in the list, all the automatically annotated spectra in the Skyline libraries were manually evaluated. Peptides with wrongly annotated spectra and/or lacking diagnostic fragment ions were excluded. Following application of these filters, the FDR was recalculated and found to be less than 1% in all cases. After refining the peptide list, Raw files were directly imported into Skyline for automatic peak picking and MS1 filtering using the following criteria. First, the retention time of the PSM-identified peptide was used to position a retention time window (± 2.0 min) across the run lacking the same peptide identification. Second, the resolution for extracting the MS1 filtering chromatogram of the target precursor ions with both light and heavy labeled peptides was set to 60,000 at 400 Th. Following data extraction, graphical displays of chromatographic traces for the top three isotopic peaks were manually inspected for proper peak picking of MS1 filtered peptides and those with isotopic dot product scores lower than 0.8 were rejected. Several additional criteria were used to further ensure the high accuracy and precision of quantification: (1) S/N> 3.0; (2) baseline separation was required between the isotopic peaks of a quantifiable peptide and unknown isobaric interference; (3) manual integration was applied if necessary. The ratios of peptide areas of light peptides to their heavy isotopes (RL:H) were calculated automatically. Quantification results were obtained from two biological replicates with two technical LC-MS/MS runs for each.
Analysis of V5-tagged CFL1 Proteins
HeLa cells expressing V5-tagged WT and C139A CFL1 proteins were treated with 25 μm aONE at 37 °C for 2 h, washed twice with ice cold 1× PBS, and harvested. Cells then were lysed in HEPES lysis buffer (50 mm HEPES, 150 mm NaCl, 1% Igepal, pH 7.5) containing inhibitor mixture. Cell lysates (3 mg/ml, 0.2 ml) were incubated with 0.2 mm azido-tagged biotin, 1 mm sodium ascorbate, 0.1 mm TBTA and 1 mm CuSO4 at RT for 2 h. The proteins were precipitated with chloroform/methanol system as described above. The resulting protein precipitate then was resolubilized in 0.1 ml 0.3% SDS and diluted with 1 ml 1× PBS and subjected to 150 μl streptavidin pull-down. After 16 h incubation at 4 °C, beads were washed with 1% SDS (2×), 4 m urea (2×), PBS (2×), and H2O (2×). Beads were then resuspended in loading buffer and boiled to release biotin-conjugated proteins. The collected proteins were resolved on SDS-polyacrylamide gel electrophoresis gels and immunoblotted with anti-V5 antibody.
Western Blot
Proteins from cell lysates, histone extracts, and immunoprecipitants were then resolved by SDS-PAGE and transferred to PVDF membranes (Merck Millipore, IPVH00010), blocked with 5% milk in TBST at RT for 1 h and probed with the indicated primary antibodies overnight at 4 °C. After incubation, the membranes were washed three times with TBST and incubated with the appropriate HRP-conjugated specific secondary antibodies. Blots were visualized on a Tanon 5200 scanner (Shanghai, China), and analyzed with GelCap software using ECL chemiluminescence (CWBIO, Beijing, China, CW0049S).
Immunofluorescence
HeLa cells transfected with control or Sirt2 siRNA were cultured onto confocal dishes. Cells were exposed to 10 Gy IR and then incubated with or without 25 μm ONE. After 2 h incubation at 37 °C, cells were fixed in methanol at −20 °C for 15 min and blocked in 5% goat serum containing 0.3% TritonX-100. Fixed cells were then incubated with the anti-phospho-Histone H2A.X (S139) antibody, (Millipore, clone JBW301, diluted at 1:400) and anti-ubiquityl-Histone H2B (K120) antibody, (Cell Signaling Technology, #5546, diluted at 1:400), followed by exposure to the appropriate fluorescent secondary antibody: Alexa-488-conjugated anti-mouse IgG (ZSGB, ZF-0512, diluted at 1:400). Finally, stain nucleus with DAPI (ZSGB, ZLI-9557, diluted at 1:1000). Fluorescence was observed with a laser-scanning microscope (ZEISS, LSM880 ELYRAS.1).
Molecular Docking
Molecular docking studies were performed using the CDOCKER of Receptor-ligand Interactions protocol in Discovery studio 2.5. The 3D structure of ONE was generated with ChemBioOffice 2014 and optimized with AM1 method. Thereafter, the ligand was treated with Prepare Ligands protocol in DS 2.5. All the duplicate structures were removed and the options for ionization change, tautomer generation, isomer generation and 3D generator have been set true. FABP5 protein (PDB ID#: 5HZ5) was defined as a total receptor and the site sphere was built with diameter of 10 Å based on C43, C47, C67, C87, C120, and C127. The molecular docking was performed with a simulated annealing method to minimize the CDOCKER energy (ECD) for obtaining an optimum pose. 50 molecular docking poses saved for ligand were ranked according to -CDOCKER energy (-ECD). The pose with the highest -ECD was chosen as the most suitable pose for the subsequent pose analysis. Finally, the docking results were visualized using PyMol. Percent RSA (%) was calculated with a default probe radius of 1.4 Å.
Bioinformatics
GO term classification and INTERPRO domain (p ≤ 0.005, FDR≤0.01) analyses were performed by using the functional annotation tool DAVID (29).
Experimental design and statistical rationale. All chemoproteomic analyses were performed in at least two biological replicates to evaluate reproducibility. Values of Standard Deviation (S.D.) for each quantifiable peptide/site were provided in supplementary tables. The ratios obtained from control and Sirt2 siRNA transfected HeLa cells were compared with pair-sample t test using Origin 9 software. All biochemical experiments were performed in at least three biological replicates and showed consistent results.
RESULTS
Chemoproteomic Analysis of aONE-derived Adductions in Cells
To systematically investigate ONE-derived adducts and their dynamics in cells, we first treated intact RKO cells with a click-able ONE (aONE) for 2 h and then placed cells in aONE-free medium for another 0, 1, and 4 h recovery period. The cells from 2 h of aONE treatment without recovery were used as controls. The aONE labeled proteomes were digested with trypsin and then biotinylated by click chemistry with the light or heavy labeled UV-cleavable azido biotin, followed by streptavidin enrichment, photorelease, and mass spectrometry-based shotgun proteomics (Fig. 1). Quantification was achieved with the use of isotopically encoded light and heavy signals on the “clicked” aONE-modified peptides as previously described (10).
It is known that ONE forms three types of modifications on proteins, including ketoamide adducts of lysine, Schiff-base adducts of lysine, ketoaldehyde Michael adducts of cysteine, histidine, and lysine (supplemental Scheme S1) (3, 20, 30). Thus, an initial proteomic database search was targeted to these modifications using MS-GF+ algorithm (28). The isotopic signature of the aONE adducted peptides with light/heavy tags and extensive manual evaluation allowed us to unequivocally identify 31 ketoamide adducted lysine sites, 3 Michael adduct cysteine sites, and 21 Schiff base adduct lysine sites (supplemental Table S1–S2). In concert with several previous findings (16, 31), aONE-derived lysine adducts mainly were found on the lysine-rich histones. In addition to a known ONE-derived ketoamide adduct of H3K23, seven new histone lysine sites modified by aONE as either ketoamide or Schiff base structures were detected in RKO cells (supplemental Fig. S1). Interestingly, these aONE-derived adducts co-localized with many other functional histone marks (supplemental Fig. S2). For example, monoubiquitination of H2BK120 in chromatin has been implicated in diverse cellular functions, such as transcriptional regulation (32, 33), DNA replication (34), stem cell differentiation (35, 36), and DNA damage response (37, 38).
We asked if there is potential crosstalk between ONE-derived adducts and this histone mark. We found that ONE treatment significantly decreased the level of monoubiquitination of H2BK120 induced by ionizing radiation (IR), whereas absence of ONE abolished this effect (Fig. 2A). Also, we found that depletion of Sirt2, an “eraser” enzyme that can remove ONE-derived histone lysine modification, led to the sustained low level of this histone mark after removal of ONE (Fig. 2A). Because monoubiquitination of H2BK120 is required for repair of IR-induced DNA damage (37, 38), we next asked whether ONE can promote IR-induced DNA damage through inhibition of this histone mark. We found that ONE treatment after exposure to IR significantly induced formation of phosphorylated H2A.X (γH2A.X) foci, a well-known marker for DNA damage, whereas knockdown of Sirt2 further enhanced this phenotypical change (Fig. 2B). These results confirmed the functional crosstalk between ONE-derived lysine adduction and monoubiquitination on H2BK120.
Fig. 2.
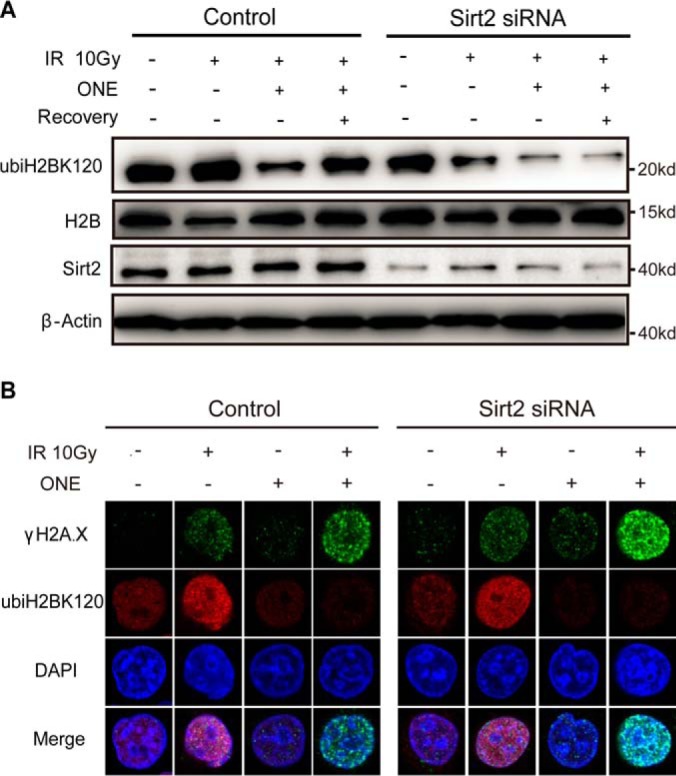
Functional crosstalk between ONE-derived adduction and monoubiquitination on H2BK120. A, HeLa cells were transfected with control and Sirt2 siRNA, followed by 10 Gy IR exposure. Cells were then treated with or without 25 μm ONE. After 2h incubation, cells were either harvested immediately or subjected to 1h recovery in medium without ONE. Histone and/or cell extracts were resolved by SDS-PAGE and subjected to Western blotting with indicated antibodies. B, Immunostaining of HeLa cells transfected with control and Sirt2 siRNA after IR exposure, followed by additional 2 h of ONE treatment (25 μm).
Surprisingly, the aONE-derived cysteinyl Michael adducts were mostly presented on C43 and C120 of epithelial fatty acid-binding protein FABP5 (supplemental Fig. S3A and S3B), which also functions as an antioxidant protein by scavenging HNE-like molecules (39). Molecular docking of ONE into the binding pocket of FABP5 further confirmed the site interaction preference of this LDE (supplemental Fig. S3C). Notably, although both C43 and C120 are considered as partially buried sites on FABP5 with percent RSA (%) of 7.3 and 12.3, respectively, the other four ONE-insensitive cysteines display even lower RSA values (C47: 6.5, C67: 5.0, C87: 0.44, C127: 2.9). This finding suggests that ONE, like most other small molecule thiol reactive chemicals (25, 40), appears more likely to target solvent accessible cysteines. C106 of PARK7/DJ-1, represented another interesting site modified through aONE-derived Michael addition, because it was previously identified as a target of sulfinic acid modification (41). Notably, we found that pre-incubation with ONE prevents sulfinic acid formation on this redox-active site in HeLa cells, suggesting potential crosstalk between electrophilic adduction and oxidation (supplemental Fig. S4).
Identification and Validation of a Novel ONE-derived Pyrrole Adduct to Cysteine
The relatively low numbers of identified cysteine Michael adducts let us ask whether there are more potential ONE derived cysteine modifications in cells. We used a tool that enables the identification of all possible PTMs, called TagRecon (26, 27), to re-analyze the obtained MS/MS data. Notably, we identified many light and heavy isotopic labeled cysteine-containing peptides with the mass shift of 346.1641Da and 352.1842 Da, respectively, located at cysteine residues (Fig. 3A and supplemental Table S2). Together with the mass difference between light and heavy peptides, the isotopic-encoded diagnostic fragment ions (m/z 381.16 and 387.18) in their MS/MS spectra confirmed that this is a cysteine modification derived from aONE (Fig. 3A). Based on elemental composition inferred from the m/z of the intact peptide adduct ions and a previously reported mechanism of ONE adduction, the most likely structure for this isotopic encoded mass shift is a tri-substituted pyrrole (C17H22N4O4). As shown in Fig. 3B, 4-ketoaldehyde Michael adduct formed by adduction of 4-ONE to protein cysteinyl thiol further undergoes a Paal Knorr condensation with the amino group of glycine, affording the pyrrole adduct. In a previously reported model study, ONE has also been shown to form tri-substituted pyrrole crosslinks through Paal Knorr condensation (20). To further verify the proposed mechanism, a Cys-containing model peptide (PDFAQELLCR) was synthesized and incubated with ONE in the presence or absence of glycine. As expected, the pyrrole adduct appeared only in the glycine-containing reaction mixture, and higher concentration of glycine further increased the yield of this product (Fig. 3C and supplemental Fig. S5). Moreover, we synthesized two aONE-derived pyrrole adduct peptides (LAHCPyEELR and PGHLQEGFGCPyVVTNR) and found that their MS/MS spectra and elution times matched exactly with the endogenous adducts on the same peptide sequences containing EIF5B C852 and SERBP1 C11 (Fig. 3D and supplemental Fig. S6). In total, we identified pyrrole adducts on 55 cysteine sites in RKO cells (supplemental Table S2). Among these was a well-known redox sensitive site, C139 of CFL1 (42, 43). To validate this finding, we first expressed V5-tagged WT and C139A mutant of CFL1 in HeLa cells and treated the cells with aONE. As a result, the C139A mutant of CFL1 was almost completely devoid of reactivity with aONE (supplemental Fig. S7), which confirmed the essential role of C139 of CFL1 for ONE adduction.
Fig. 3.

Identification and verification of a novel ONE-derived pyrrole adduct to cysteinyl residues. A, Representative MS/MS spectra generated from triply charged light- and heavy-tagged parent ions of an aONE-derived pyrrole adduct peptide from BCAT2 C342. B, Possible mechanism for the formation of ONE-derived cysteine pyrrole adduct in the presence of glycine. C, Extracted ion chromatograms of a model peptide (PDFAQELLCR) bearing ONE-derived Michael adduct and glycine-dependent pyrrole adduct. D, Extracted ion chromatograms (XICs) of the endogenous aONE-derived peptide adducts and the synthetic peptides bearing the same peptide sequence and modification.
Comparison of the In Situ Reactivity Profiles of ONE and HNE
The reactions between ONE and HNE with cellular proteins differ and these electrophiles produce different targeting profiles in cells (11). We previously found that aHNE predominantly targets protein cysteines, but produces little or no modification of protein lysine sites (10). On contrary, approximately half of the identified aONE adducts appeared on lysine (supplemental Fig. S8A). Nonetheless, 45.6% of aONE adducted cysteines are also reactive to aHNE in RKO cells (supplemental Fig. S9), including many redox sensitive cysteines, such as TXN C73 (44), BCAT2 C342 (45), and S100A2 C22 (46). The in vitro reactivity order of amino acids toward ONE was found to be cysteine ≫ histidine > lysine (21). We considered that lysine represents one of the most common amino acids in vertebrates (7.2%), whereas the frequency of cysteine and histidine are only 3.3% and 2.9%, respectively. Thus, the order determined in a model system might not reflect adduction in complex proteomes. Ranking the spectral counts for all the peptides modified by aforementioned four types of adducts revealed the reactivity order of ONE-derived adduction in RKO cells was cysteine Michael addition > lysine Schiff base > lysine ketoamide ≫ others (i.e. Michael addition to lysine or histidine) (supplemental Fig. S8B).
Dynamics of ONE-derived adductions in cells
Quantitative analysis revealed the site- and modification-specific stability of aONE adducts at 1 and 4h of recovery (Fig. 4A andsupplemental Table S2). Only ∼2% of aONE adduction events showed at least a 1.5-fold decrease over the course of 1 h (RL/H≤0.67), whereas such percentage dramatically increased to ∼68% at 4 h of recovery. Among four types of aONE-derived modifications, cysteine Michael adducts were most unstable during the recovery period (Fig. 4A). In principle, these initial products of Michael addition reaction by ONE were still chemically reactive, which could allow further reaction with biological nucleophiles to afford more stable end products (e.g. pyrrole adducts) or to form crosslinks (18). Interestingly, the dynamics of lysine ketoamide and Schiff base adducts on the same sites were highly correlated (Fig. 4B and 4C, Pearson coefficient = 0.835, p < 0.01), suggesting that these two types of lysine modifications might be removed by the same mechanism. This notion is also supported by the intrinsic relationship between these two types of modifications, in which the chemically labile Schiff base adducts transform to more stable ketoamide products via a carbinolamine intermediate (supplemental Scheme S1).
Fig. 4.

Dynamics of aONE-derived protein adduction in RKO cells. A, Box plots of ratios determined from four types of aONE-derived protein adducts in dynamic adduction analyses. RKO cells were first treated with 50 μm aONE. After 2 h incubation, cells were either harvested immediately and used as controls or placed in aONE-free medium for another 1 and 4 h recovery period. The aONE labeled proteomes were digested with trypsin and then biotinylated by click chemistry with the light (recovery) or heavy (control) labeled UV-cleavable azido biotin, followed by streptavidin enrichment, photorelease, and LC-MS/MS based identification and quantification. B, Heatmaps of ratios of changes of aONE-derived ketoamide and Schiff-base adducts to the same lysine sites, which show high correlation between these two types of adduction. Lower the measured ratio (L/H) indicates higher turnover rates. C, Representative XIC chromatograms with the profiles for light- and heavy- labeled peptides in red and blue, respectively, for aONE-derived adducts to ENO1 K80.
Interestingly, the adducts on three lysine residues in GAPDH exhibited dramatically different dynamics in cells. The ketoamide at K263 and the Schiff base adduct at K215 significantly decreased after 1–4 h recovery period, whereas the Schiff base adduct at K219 remained almost unchanged (supplemental Fig. S10). This finding suggested that the site-specific aONE adduction dynamics of GAPDH in cells was unlikely mediated by protein-level degradation. Strikingly, we found ∼32% of aONE adducts remained quite stable at 4h of recovery (RL/H>0.67), whereas such long-lived protein adducts of aHNE only accounted for ∼3% of all aHNE adducts identified previously in RKO cells (10). This finding indicates that the protein adducts of ONE are more difficult for biological systems to remove, compared with those derived from HNE, thereby leading to more potent and sustained cytotoxicity (15).
Comparison of the In Vitro and In Situ Stability of aONE-derived Adducts
To test the role of an intact cellular environment in the turnover of aONE-derived adducts, we first treated HeLa cells with 50 μm aONE. After 2 h incubation, we either replaced the labeling medium with aONE-free medium for another 4 h (in situ recovery) or lysed the cells and incubated the lysates at 37 °C for 4 h (in vitro recovery). Protein samples obtained from in situ and in vitro recovery experiments were processed to tryptic peptides, labeled with light and heavy isotope tags, respectively, and combined. After streptavidin enrichment and photorelease, the resulting peptides were subjected to LC-MS/MS analysis. We obtained quantitative data for 11 lysine ketoamide adducts, 7 lysine Schiff-base adducts, and 71 cysteine pyrrole adducts (supplemental Table S3–S4). Similar to the aforementioned finding, ketoamide and Schiff-base adducts on the same lysine sites (e.g. K205 of CH60 and K63 of ACTC) displayed nearly identical stability either in vitro or in situ. In addition, ∼68.5% of the aONE adduction events on cysteine and lysine exhibited similar turnover rates in vitro and in situ (0.67<RL/H<1.5, supplemental Fig. S11), whereas 31.5% of were found to be less stable in intact cells than in lysates (RL/H <0.67, supplemental Fig. S11). It appeared that the discrimination of in vitro and in situ stability of aONE adducts is independent of specific type of adduction. Notably, aONE adducts on several functionally important proteins exhibited much higher in situ turnover rates. For example, the pyrrole adduct of C179 of BAG3 (BCL-2-associated athanogene 3), a HSP70 co-chaperone that plays an essential role in cellular protein quality control, was more labile in cells than in lysates (RL/H = 0.14). Likewise, the pyrrole adduct of C342 of BCAT2 (Branched-chain-amino acid aminotransferase), which is associated with energy expenditure and protein turnover in cells under stress conditions (47, 48), was also reversed more rapidly in cells than in lysates (RL/H = 0.34). Together, these findings indicate that intact cellular environment contributes to the more rapid turnover of some aONE adducts.
Effects of Sirt2 Knockdown on the Turnover of aONE-derived Adductions
We next asked whether Sirt2 regulates ONE-derived lysine ketoamide adducts to nonhistone proteins. We first transfected HeLa cells with control and Sirt2 siRNA, and treated cells with aONE. After 2 h of incubation, we harvested cells immediately or placed cells in aONE-free medium for a 4 h recovery period. Protein lysates obtained from the aONE-treated cells with or without recovery were processed to tryptic peptides, labeled with light and heavy isotope tags, and combined. After streptavidin enrichment and photorelease, the resulting peptides were subjected to LC-MS/MS analysis. We obtained quantitative data for 18 histone lysine adducts, 37 nonhistone lysine adducts, and 64 cysteine adducts (supplemental Table S5–S6). Notably, Sirt2 knockdown significantly increased the global stability of lysine adducts on either histone or nonhistone proteins during the recovery period, whereas it did not change the stability of most aONE-derived cysteine adducts (Fig. 5A). For example, the in situ stability of lysine ketoamide adducts of a histone protein H3C (K24) and a nonhistone protein ACTBL (K329) increased by 2.6-fold and 8.6-fold, respectively, in Sirt2 knockdown cells, as compared with that determined in control cells (Fig. 5B). In contrast, the cysteine pyrrole adduct of UBA1 C632 exhibited similar turnover rates of in control and Sirt2 knockdown cells (Fig. 5B). Moreover, a few lysine adducts were unaffected by Sirt2 status, including ketoamide adducts of H2A2A K96, FLNB K681, NPM K239, and Schiff-base adducts on H3C K56, H4G K13, MVP K429, and S10A6 K47, which suggested the target selectivity of Sirt2 (supplemental Table S6). Surprisingly, we also observed that several aONE-derived adducts (18 cysteine pyrrole adducts and 4 lysine adducts) appeared to be more unstable in Sirt2 knockdown cells than control cells (RL/HKD/RL/HCrl < 0.67). Notably, ∼83% (15/18) of these pyrrole adducts also occurred at the S-sulfenylated cysteine sites identified in our previously published data set (25), including ACLY C20, C7orf50 C107, CAPZB C206, CSK C290, EEF2 C41, FUBP3 C460, HNRNPH1 C267, HUWE1 C1832, LMNA C522, MYH9 C988, PCBP1_C109, PRDX6 C91, QARS C657, SRRT C412, and VDAC3 C229. These changes may reflect a potential redox perturbation induced by Sirt2 knockdown (49, 50), such changes further highlight the potential crosstalk between ONE-derived cysteine adductions and redox thiol modifications. Moreover, RL/H values of ∼89% of lysine adducts were lower than 0.67, which means that a partial knockdown of Sirt2 could not completely reverse the turnover of aONE-derived lysine adducts in the recovery period. Nonetheless, we could not rule out the possibility that there might be other unknown chemical or enzymatic mechanisms, in addition to possible Sirt2-mediated deacylation, involved in regulation of the global turnover of this modification, which merits further exploration in the future.
Fig. 5.

Effects of Sirt2 knockdown on the turnover of aONE-derived adductions. A, HeLa cells were transfected with control and Sirt2 siRNA, and then treated with 50 μm aONE. After 2 h incubation, cells were either harvested immediately or subjected to 4h recovery in medium without aONE. Protein lysates obtained from the aONE-treated cells with or without recovery were processed to tryptic peptides, labeled with light and heavy isotope tags, and combined. After streptavidin enrichment and photorelease, the resulting peptides were subjected to LC-MS/MS analysis. The ratios between heavy (recovery) and light (without recovery) of aONE-derived modifications were measured. The ratios obtained from control and Sirt2 siRNA transfected HeLa cells were plotted into line series and compared with pair-sample t test. B–D, Extracted ion chromatograms of aONE-derived peptides from H3C K24 (Ketoamide adduct), ACTBL K329 (Ketoamide adduct), and UBA1 C632 (Pyrrole adduct) obtained from control (lower) and Sirt2 siRNA (upper) transfected cells. The experimental ratios are displayed below the individual chromatogram, respectively.
Bioinformatics Analysis of the aONE-derived Adductome
To gain more biological insights of aONE-derived adductions, we performed GO classification analysis of all the aONE-targeted proteins identified in this study. We found the aONE-adducted proteins presented in most major cellular compartments, including membrane, cytosol, and nucleus (supplemental Table S7). Of interest, these proteins were also enriched in extracellular exosome, which can protect cells against oxidative stress via removal and/or transport of damaged proteins (51). GO classification also revealed that the adducted proteins are involved in several important biological processes, such as cell-cell adhesion (p = 4.9 × 10−21), mRNA splicing (p = 1.1 × 10−7), and nucleosome assembly (p = 4.9 × 10−6) (supplemental Table S7). Notably, we and others have shown that mRNA splicing-related network is vulnerable under electrophilic stress (11, 52). Moreover, as aforementioned, a recent study has demonstrated that ONE can block nucleosome assembly and compromise canonical nucleosome formation (16). Nonetheless, how ONE affects cell-cell adhesion remained to be determined in the future, although it has been suggested that a low μm concentration of HNE can reduce HL-60 cell adhesion to human umbilical venous cells (53). Additionally, we found that aONE targets certain protein domains, such as importin-beta (p = 4.4 × 10−7), RNA recognition motif domain (p = 2.1 × 10−6), and histone core (p = 4.9 × 10−5) (supplemental Table S7).
DISCUSSION
Among all known LDEs, HNE is the most extensively studied, and hundreds of proteins have been identified as the targets of this electrophile by either traditional analytical approaches or state-of-the-art chemoproteomics (10–14, 54, 55). ONE, the 4-keto analog of HNE (supplemental Scheme S1), exhibits much higher intrinsic reactivity and cytotoxicity than those of HNE and has drawn much attention since its discovery approximately two decades ago (56). However, study of ONE has lagged far behind study of HNE. The potential target profile of ONE-derived adductions is far more complicated than that of HNE because of the distinct stereoelectronic properties of ONE. A recent proteomic analysis confirmed that HNE and ONE display significant differences in cellular protein targets and sites of reactivity (11). However, a site-specific target profile of ONE in cells has never been reported.
In this study, we present the first proteome-wide survey of ONE adduct chemistry, target preference, and site-specific selectivity in intact cells using a chemoproteomic approach. Our analysis reveals four types of ONE-derived modifications on 234 nucleophilic sites of 183 proteins in RKO and HeLa cells. Notably, we map ONE-derived lysine adductions (ketoamide or Schiff-base) onto many histone marks. We further demonstrate anti-synergistic crosstalk of ONE adduction with monoubiquitination of H2BK120, a key histone mark, which promotes IR-induced DNA damage (Fig. 2). We also uncovered potential crosstalk between ONE adduction and sulfinic acid formation of PARK7/DJ-1 C106 (supplemental Fig. S4). Considering the chemical diversity of ONE-derived adductions, the future exploration of crosstalk within the same protein or across a protein-protein network is warranted.
We also have identified and validated novel pyrrole adducts to cysteine residue, which are major products of ONE-proteome reactions. However, it is important to note that we do not yet know whether this novel modification exists in blood or tissues in vivo. This question merits future investigation. Moreover, the functional impact of ONE-derived adductions on individual protein targets or the global thiol proteome remains to be determined. Regardless, we foresee that our data set, together with proximity-directed chemical approaches described recently by Aye and coworkers (57–61), will greatly facilitate future exploration of the complex networks affected by this type of electrophilic stress.
Furthermore, our analysis and emerging evidence demonstrate that both ONE and HNE adduction reactions are highly dynamic cellular events (10, 62), although ONE-derived adducts exhibit relatively higher stability in biological systems. Interestingly, two independent groups have recently shown that ONE-derived lysine ketoamide adductions of histone proteins can be reversed by Sirt2-mediated deacylation (22, 23). Our analysis not only confirms this previous finding, but also reveals a broader role of Sirt2 in regulation of ONE-derived lysine adductions of nonhistones. In addition, our results suggest that the cellular turnover of ONE-derived adduction may also be controlled by yet unidentified mechanisms, especially for those adducts on cysteine residues. Our findings, therefore, provide much-needed mechanistic insights into the potential toxicities associated with ONE as well as the self-protection machinery against electrophilic stress in human cells.
It is important to note that endogenous LDE generation in specific cell and tissue locations and over a time course that tracks the onset of inflammation or toxicity. Thus, our experimental approach, in which we exogenously apply aONE (50 μm, 2 h) to cells, may not adequately reflect cellular microenvironmental factors that modulate native ONE formation and reactivity. Beavers et al. recently developed several metabolically competent surrogates including ω-alkynyl linoleic acid and ω-alkynyl arachidonic acid for tracking the fate and protein targets of LDEs derived from these polyunsaturated fatty acids in cells under physiological oxidative stress (63, 64). However, site-specific mapping of endogenous LDE-adducts generated from radical-mediated peroxidation of these alkynyl ω-lipid surrogates remains an unmet analytical challenge. We expect that the chemoproteomic workflow outlined here, which includes detection of unanticipated adducts by blind PTM search and isotope signature-based identification and quantification, can be applied to globally and site-specifically map and quantify known and unexpected chemical modifications derived from endogenously generated LDEs and to estimate their stoichiometry in situ.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (65) partner repository with the data set identifier PXD007149 and 10.6019/PXD007149.
Supplementary Material
Acknowledgments
We thank Quan Zhou and Dr. Wenchuan Leng from mass spectrometry facility of the National Center for Protein Science (Beijing) for expert technical assistance.
Footnotes
Author contributions: J.Y. and D.C.L. designed the research; R.S., L.F., and C.T. performed the research; K.A.T. and N.A.P. contributed new reagents or analytic tools; R.S., K.L., and Y.Y. analyzed the data; J.Y. and D.C.L. wrote the paper.
* This work was supported in part by the National Key R&D Program of China (No. 2016YFA0501303), the National Natural Science Foundation of China (No. 31500666 and No. 81573395), the Beijing Natural Science Foundation (No.5162009), and Beijing Nova Program (No. Z171100001117014) to J.Y. and the National Institutes of Health (U24CA159988) to D.C.L.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- HNE
- 4-hydroxy-2-nonenal
- ACN
- acetonitrile
- BCA
- bicinchoninic acid
- DMEM
- Dulbecco's modified eagle's medium
- DTT
- dithiothreitol
- FDR
- false discovery rate
- HCD
- high energy collisional dissociation
- LC-MS/MS
- liquid chromatography-tandem mass spectrometry
- LDE
- lipid derived electrophile
- IAM
- iodoacetamide
- IR
- ionizing radiation
- MeOH
- methanol
- ONE
- 4-oxo-2-nonenal
- PBS
- phosphate buffered saline
- PTM
- post-translational modification
- PVDF
- polyvinylidene difluoride
- PSM
- peptide-spectrum match
- RT
- room temperature
- RSA
- residue solvent accessibility
- SCX
- strong cation exchange
- S/N
- signal to noise
- TBST
- tris-buffered saline plus 0.05% Tween-20 (v/v)
- TBTA
- tris[(1-benzyl-1H-1,2,3-triazol-4-yl) methyl] amine.
REFERENCES
- 1. Ezraty B., Gennaris A., Barras F., and Collet J. F. (2017) Oxidative stress, protein damage and repair in bacteria. Nat. Rev. Microbiol. 15, 385–396 [DOI] [PubMed] [Google Scholar]
- 2. Sies H., Berndt C., and Jones D. P. (2017) Oxidative Stress. Annu. Rev. Biochem. 86, 715–748 [DOI] [PubMed] [Google Scholar]
- 3. Sayre L. M., Lin D., Yuan Q., Zhu X., and Tang X. (2006) Protein adducts generated from products of lipid oxidation: focus on HNE and one. Drug Metab. Rev. 38, 651–675 [DOI] [PubMed] [Google Scholar]
- 4. Schopfer F. J., Cipollina C., and Freeman B. A. (2011) Formation and signaling actions of electrophilic lipids. Chem. Rev. 111, 5997–6021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rudolph T. K., and Freeman B. A. (2009) Transduction of redox signaling by electrophile-protein reactions. Sci. Signal. 2, re7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Uchida K. (2017) HNE as an inducer of COX-2. Free Radic. Biol. Med. 111, 169–172 [DOI] [PubMed] [Google Scholar]
- 7. Cohen G., Riahi Y., Sunda V., Deplano S., Chatgilialoglu C., Ferreri C., Kaiser N., and Sasson S. (2013) Signaling properties of 4-hydroxyalkenals formed by lipid peroxidation in diabetes. Free Radic. Biol. Med. 65, 978–987 [DOI] [PubMed] [Google Scholar]
- 8. Di Domenico F., Tramutola A., and Butterfield D. A. (2017) Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic. Biol. Med. 111, 253–261 [DOI] [PubMed] [Google Scholar]
- 9. Koenitzer J. R., and Freeman B. A. (2010) Redox signaling in inflammation: interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Ann. N.Y. Acad. Sci. 1203, 45–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J., Tallman K. A., Porter N. A., and Liebler D. C. (2015) Quantitative chemoproteomics for site-specific analysis of protein alkylation by 4-hydroxy-2-nonenal in cells. Anal. Chem. 87, 2535–2541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Codreanu S. G., Ullery J. C., Zhu J., Tallman K. A., Beavers W. N., Porter N. A., Marnett L. J., Zhang B., and Liebler D. C. (2014) Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteomics 13, 849–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang C., Weerapana E., Blewett M. M., and Cravatt B. F. (2014) A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yuan W., Zhang Y., Xiong Y., Tao T., Wang Y., Yao J., Zhang L., Yan G., Bao H., and Lu H. (2017) Highly selective and large scale mass spectrometric analysis of 4-Hydroxynonenal modification via fluorous derivatization and fluorous solid-phase extraction. Anal. Chem. 89, 3093–3100 [DOI] [PubMed] [Google Scholar]
- 14. Chen Y., Cong Y., Quan B., Lan T., Chu X., Ye Z., Hou X., and Wang C. (2017) Chemoproteomic profiling of targets of lipid-derived electrophiles by bioorthogonal aminooxy probe. Redox Biol. 12, 712–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lin D., Lee H. G., Liu Q., Perry G., Smith M. A., and Sayre L. M. (2005) 4-Oxo-2-nonenal is both more neurotoxic and more protein reactive than 4-hydroxy-2-nonenal. Chem. Res. Toxicol. 18, 1219–1231 [DOI] [PubMed] [Google Scholar]
- 16. Galligan J. J., Rose K. L., Beavers W. N., Hill S., Tallman K. A., Tansey W. P., and Marnett L. J. (2014) Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics. J. Am. Chem. Soc. 136, 11864–11866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nasstrom T., Fagerqvist T., Barbu M., Karlsson M., Nikolajeff F., Kasrayan A., Ekberg M., Lannfelt L., Ingelsson M., and Bergstrom J. (2011) The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of alpha-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 50, 428–437 [DOI] [PubMed] [Google Scholar]
- 18. Camarillo J. M., Ullery J. C., Rose K. L., and Marnett L. J. (2017) Electrophilic modification of PKM2 by 4-hydroxynonenal and 4-oxononenal results in protein cross-linking and kinase inhibition. Chem. Res. Toxicol. 30, 635–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aluise C. D., Camarillo J. M., Shimozu Y., Galligan J. J., Rose K. L., Tallman K. A., and Marnett L. J. (2015) Site-specific, intramolecular cross-linking of Pin1 active site residues by the lipid electrophile 4-oxo-2-nonenal. Chem. Res. Toxicol. 28, 817–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang W. H., Liu J., Xu G., Yuan Q., and Sayre L. M. (2003) Model studies on protein side chain modification by 4-oxo-2-nonenal. Chem. Res. Toxicol. 16, 512–523 [DOI] [PubMed] [Google Scholar]
- 21. Doorn J. A., and Petersen D. R. (2002) Covalent modification of amino acid nucleophiles by the lipid peroxidation products 4-hydroxy-2-nonenal and 4-oxo-2-nonenal. Chem. Res. Toxicol. 15, 1445–1450 [DOI] [PubMed] [Google Scholar]
- 22. Cui Y., Li X., Lin J., Hao Q., and Li X. D. (2017) Histone ketoamide adduction by 4-oxo-2-nonenal is a reversible posttranslational modification regulated by Sirt2. ACS Chem. Biol. 12, 47–51 [DOI] [PubMed] [Google Scholar]
- 23. Jin J., He B., Zhang X., Lin H., and Wang Y. (2016) SIRT2 Reverses 4-oxononanoyl lysine modification on histones. J. Am. Chem. Soc. 138, 12304–12307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yang J., Gupta V., Tallman K. A., Porter N. A., Carroll K. S., and Liebler D. C. (2015) Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 10, 1022–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang J., Gupta V., Carroll K. S., and Liebler D. C. (2014) Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 5, 4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dasari S., Chambers M. C., Codreanu S. G., Liebler D. C., Collins B. C., Pennington S. R., Gallagher W. M., and Tabb D. L. (2011) Sequence tagging reveals unexpected modifications in toxicoproteomics. Chem. Res. Toxicol. 24, 204–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dasari S., Chambers M. C., Slebos R. J., Zimmerman L. J., Ham A. J., and Tabb D. L. (2010) TagRecon: high-throughput mutation identification through sequence tagging. J. Proteome Res. 9, 1716–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S., and Pevzner P. A. (2014) MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun. 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang da W, Sherman B. T., and Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 30. Zhu X., and Sayre L. M. (2007) Long-lived 4-oxo-2-enal-derived apparent lysine michael adducts are actually the isomeric 4-ketoamides. Chem. Res. Toxicol. 20, 165–170 [DOI] [PubMed] [Google Scholar]
- 31. Oe T., Arora J. S., Lee S. H., and Blair I. A. (2003) A novel lipid hydroperoxide-derived cyclic covalent modification to histone H4. J. Biol. Chem. 278, 42098–42105 [DOI] [PubMed] [Google Scholar]
- 32. Sun Z. W., and Allis C. D. (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418, 104–108 [DOI] [PubMed] [Google Scholar]
- 33. Sadeghi L., Siggens L., Svensson J. P., and Ekwall K. (2014) Centromeric histone H2B monoubiquitination promotes noncoding transcription and chromatin integrity. Nat. Struct. Mol. Biol. 21, 236–243 [DOI] [PubMed] [Google Scholar]
- 34. Trujillo K. M., and Osley M. A. (2012) A role for H2B ubiquitylation in DNA replication. Mol. Cell. 48, 734–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karpiuk O., Najafova Z., Kramer F., Hennion M., Galonska C., Konig A., Snaidero N., Vogel T., Shchebet A., Begus-Nahrmann Y., Kassem M., Simons M., Shcherbata H., Beissbarth T., and Johnsen S. A. (2012) The histone H2B monoubiquitination regulatory pathway is required for differentiation of multipotent stem cells. Mol. Cell 46, 705–713 [DOI] [PubMed] [Google Scholar]
- 36. Fuchs G., Shema E., Vesterman R., Kotler E., Wolchinsky Z., Wilder S., Golomb L., Pribluda A., Zhang F., Haj-Yahya M., Feldmesser E., Brik A., Yu X., Hanna J., Aberdam D., Domany E., and Oren M. (2012) RNF20 and USP44 regulate stem cell differentiation by modulating H2B monoubiquitylation. Mol. Cell 46, 662–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moyal L., Lerenthal Y., Gana-Weisz M., Mass G., So S., Wang S. Y., Eppink B., Chung Y. M., Shalev G., Shema E., Shkedy D., Smorodinsky N. I., van Vliet N., Kuster B., Mann M., Ciechanover A., Dahm-Daphi J., Kanaar R., Hu M. C., Chen D. J., Oren M., and Shiloh Y. (2011) Requirement of ATM-dependent monoubiquitylation of histone H2B for timely repair of DNA double-strand breaks. Mol. Cell 41, 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakamura K., Kato A., Kobayashi J., Yanagihara H., Sakamoto S., Oliveira D. V., Shimada M., Tauchi H., Suzuki H., Tashiro S., Zou L., and Komatsu K. (2011) Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol. Cell 41, 515–528 [DOI] [PubMed] [Google Scholar]
- 39. Bennaars-Eiden A., Higgins L., Hertzel A. V., Kapphahn R. J., Ferrington D. A., and Bernlohr D. A. (2002) Covalent modification of epithelial fatty acid-binding protein by 4-hydroxynonenal in vitro and in vivo. Evidence for a role in antioxidant biology. J. Biol. Chem. 277, 50693–50702 [DOI] [PubMed] [Google Scholar]
- 40. Doulias P. T., Greene J. L., Greco T. M., Tenopoulou M., Seeholzer S. H., Dunbrack R. L., and Ischiropoulos H. (2010) Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. U.S.A. 107, 16958–16963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Taira T., Saito Y., Niki T., Iguchi-Ariga S. M., Takahashi K., and Ariga H. (2004) DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 5, 213–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Klamt F., Zdanov S., Levine R. L., Pariser A., Zhang Y., Zhang B., Yu L. R., Veenstra T. D., and Shacter E. (2009) Oxidant-induced apoptosis is mediated by oxidation of the actin-regulatory protein cofilin. Nat. Cell Biol. 11, 1241–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cameron J. M., Gabrielsen M., Chim Y. H., Munro J., McGhee E. J., Sumpton D., Eaton P., Anderson K. I., Yin H., and Olson M. F. (2015) Polarized cell motility induces hydrogen peroxide to inhibit cofilin via cysteine oxidation. Curr. Biol. 25, 1520–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barglow K. T., Knutson C. G., Wishnok J. S., Tannenbaum S. R., and Marletta M. A. (2011) Site-specific and redox-controlled S-nitrosation of thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 108, E600–E606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Conway M. E., Poole L. B., and Hutson S. M. (2004) Roles for cysteine residues in the regulatory CXXC motif of human mitochondrial branched chain aminotransferase enzyme. Biochemistry 43, 7356–7364 [DOI] [PubMed] [Google Scholar]
- 46. Stradal T. B., Troxler H., Heizmann C. W., and Gimona M. (2000) Mapping the zinc ligands of S100A2 by site-directed mutagenesis. J. Biol. Chem. 275, 13219–13227 [DOI] [PubMed] [Google Scholar]
- 47. She P., Reid T. M., Bronson S. K., Vary T. C., Hajnal A., Lynch C. J., and Hutson S. M. (2007) Disruption of BCATm in mice leads to increased energy expenditure associated with the activation of a futile protein turnover cycle. Cell Metab. 6, 181–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vogel C., Silva G. M., and Marcotte E. M. (2011) Protein expression regulation under oxidative stress. Mol. Cell. Proteomics 10, M111 009217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu S. N., Wang T. S., Li X., and Wang Y. P. (2016) SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation. Sci. Rep. 6, 32734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fiskus W., Coothankandaswamy V., Chen J., Ma H., Ha K., Saenz D. T., Krieger S. S., Mill C. P., Sun B., Huang P., Mumm J. S., Melnick A. M., and Bhalla K. N. (2016) SIRT2 deacetylates and inhibits the peroxidase activity of peroxiredoxin-1 to sensitize breast cancer cells to oxidant stress-inducing agents. Cancer Res. 76, 5467–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Szabo-Taylor K., Ryan B., Osteikoetxea X., Szabo T. G., Sodar B., Holub M., Nemeth A., Paloczi K., Pallinger E., Winyard P., and Buzas E. I. (2015) Oxidative and other posttranslational modifications in extracellular vesicle biology. Semin. Cell Dev. Biol. 40, 8–16 [DOI] [PubMed] [Google Scholar]
- 52. Anavi S., Ni Z., Tirosh O., and Fedorova M. (2015) Steatosis-induced proteins adducts with lipid peroxidation products and nuclear electrophilic stress in hepatocytes. Redox Biol. 4, 158–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gentile F., Pizzimenti S., Arcaro A., Pettazzoni P., Minelli R., D'Angelo D., Mamone G., Ferranti P., Toaldo C., Cetrangolo G., Formisano S., Dianzani M. U., Uchida K., Dianzani C., and Barrera G. (2009) Exposure of HL-60 human leukaemic cells to 4-hydroxynonenal promotes the formation of adduct (s) with alpha-enolase devoid of plasminogen binding activity. Biochem. J. 422, 285–294 [DOI] [PubMed] [Google Scholar]
- 54. Carini M., Aldini G., and Facino R. M. (2004) Mass spectrometry for detection of 4-hydroxy-trans-2-nonenal (HNE) adducts with peptides and proteins. Mass Spectrom. Rev. 23, 281–305 [DOI] [PubMed] [Google Scholar]
- 55. Sousa B. C., Pitt A. R., and Spickett C. M. (2017) Chemistry and analysis of HNE and other prominent carbonyl-containing lipid oxidation compounds. Free Radic. Biol. Med. 111, 294–308 [DOI] [PubMed] [Google Scholar]
- 56. Lee S. H., and Blair I. A. (2000) Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem. Res. Toxicol. 13, 698–702 [DOI] [PubMed] [Google Scholar]
- 57. Fang X., Fu Y., Long M. J., Haegele J. A., Ge E. J., Parvez S., and Aye Y. (2013) Temporally controlled targeting of 4-hydroxynonenal to specific proteins in living cells. J. Am. Chem. Soc. 135, 14496–14499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Parvez S., Fu Y., Li J., Long M. J., Lin H. Y., Lee D. K., Hu G. S., and Aye Y. (2015) Substoichiometric hydroxynonenylation of a single protein recapitulates whole-cell-stimulated antioxidant response. J. Am. Chem. Soc. 137, 10–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Long M. J., Poganik J. R., and Aye Y. (2016) On-Demand Targeting: Investigating Biology with Proximity-Directed Chemistry. J. Am. Chem. Soc. 138, 3610–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Long M. J., Parvez S., Zhao Y., Surya S. L., Wang Y., Zhang S., and Aye Y. (2017) Akt3 is a privileged first responder in isozyme-specific electrophile response. Nat. Chem. Biol. 13, 333–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Parvez S., Long M. J., Lin H. Y., Zhao Y., Haegele J. A., Pham V. N., Lee D. K., and Aye Y. (2016) T-REX on-demand redox targeting in live cells. Nat. Protoc. 11, 2328–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Randall M. J., Hristova M., and van der Vliet A. (2013) Protein alkylation by the alpha, beta-unsaturated aldehyde acrolein. A reversible mechanism of electrophile signaling? FEBS Lett. 587, 3808–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Beavers W. N., Serwa R., Shimozu Y., Tallman K. A., Vaught M., Dalvie E. D., Marnett L. J., and Porter N. A. (2014) omega-Alkynyl lipid surrogates for polyunsaturated fatty acids: free radical and enzymatic oxidations. J. Am. Chem. Soc. 136, 11529–11539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Beavers W. N., Rose K. L., Galligan J. J., Mitchener M. M., Rouzer C. A., Tallman K. A., Lamberson C. R., Wang X., Hill S., Ivanova P. T., Brown H. A., Zhang B., Porter N. A., and Marnett L. J. (2017) Protein modification by endogenously generated lipid electrophiles: mitochondria as the source and target. ACS Chem. Biol. 12, 2062–2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vizcaino J. A., Csordas A., Del-Toro N., Dianes J. A., Griss J., Lavidas I., Mayer G., Perez-Riverol Y., Reisinger F., Ternent T., Xu Q. W., Wang R., and Hermjakob H. (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 44, 11033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (65) partner repository with the data set identifier PXD007149 and 10.6019/PXD007149.