

Abstract
Lung cancer is the leading cause of cancer mortality worldwide, with squamous cell carcinoma (SQCC) being the second most common form. SQCCs are thought to originate in bronchial basal cells through an injury response to smoking, which results in this stem cell population committing to hyperplastic squamous rather than mucinous and ciliated fates. Copy number gains in SOX2 in the region of 3q26–28 occur in 94% of SQCCs, and appear to act both early and late in disease progression by stabilizing the initial squamous injury response in stem cells and promoting growth of invasive carcinoma. Thus, anti-SOX2 targeting strategies could help treat early and/or advanced disease. Because SOX2 itself is not readily druggable, we sought to characterize SOX2 binding partners, with the hope of identifying new strategies to indirectly interfere with SOX2 activity. We now report the first use of proximity-dependent biotin labeling (BioID) to characterize the SOX2 interactome in vivo. We identified 82 high confidence SOX2-interacting partners. An interaction with the coactivator EP300 was subsequently validated in both basal cells and SQCCs, and we demonstrate that EP300 is necessary for SOX2 activity in basal cells, including for induction of the squamous fate. We also report that EP300 copy number gains are common in SQCCs and that growth of lung cancer cell lines with 3q gains, including SQCC cells, is dependent on EP300. Finally, we show that EP300 inhibitors can be combined with other targeted therapeutics to achieve more effective growth suppression. Our work supports the use of BioID to identify interacting protein partners of nondruggable oncoproteins such as SOX2, as an effective strategy to discover biologically relevant, druggable targets.
Lung cancer is the leading cause of cancer mortality worldwide (1). Squamous cell carcinoma (SQCC)1 is the second most common form of lung cancer and generally develops over many years through successive premalignant changes in the bronchial epithelium (2, 3). SQCCs have few effective treatment options with median survival times usually between 9–11 months (4). The poor survival rate is due in part to few targeted therapies and a limited understanding of SQCC pathogenesis. Mounting evidence suggests that SQCC is a stem cell disease. Most SQCCs express TP63 (5, 6), a hallmark of basal cells, stem cells of the tracheal and bronchial airways (7–11). In addition, SQCCs generally develop following smoking-induced squamous metaplasia, an injury response of basal cells that replaces the normal quiescent mucociliary epithelium with a hyperproliferative squamous epithelium (2, 12–16). With continued smoking, squamous metaplasia progresses to increasing grades of dysplasia, followed by invasive carcinoma (2), suggesting that SQCC pathogenesis originates from the squamous injury response of basal cells, which becomes progressively dysregulated.
Ninety-four percent of SQCCs harbor copy number gains in both SOX2 and PIK3CA at 3q26–28 (17, 18) (TCGA data, www.cbioportal.org), which are commonly selected during high grade dysplasia (19, 20). These gains appear to be selected to stabilize a normally self-limiting squamous injury response. Indeed, premalignant stages including squamous metaplasia and lower grade dysplasia, which generally do not have 3q copy number gains, frequently regress spontaneously (21–23), whereas high grade dysplasia, which commonly has 3q copy number gains, is less prone to regression and more likely to progress to invasive carcinoma than earlier stages (24–27). Notably, SOX2 and PI3K (PIK3CA), within the 3q amplicon, cooperate to induce a hyperplasic squamous-committed stem cell state in basal cells (28), providing a molecular and stem cell-based explanation for how the 3q amplicon stabilizes the squamous injury response. Because of the importance of SOX2 in this process, inhibition of its activity during early stages of SQCC pathogenesis could slow disease progression. In addition, because growth of many cancers is dependent on the transcription factors that specify their lineage from stem cells (29–35), targeting SOX2 later, in frank carcinoma, may also be beneficial. Accordingly, reduction in SOX2 activity inhibits growth of SQCC cell lines with 3q copy number gains (17, 36, 37). Furthermore, SOX2 also has been suggested to have roles affecting SQCC malignancy that do not necessarily arise from affecting stem cell fate along the squamous lineage (37, 38). Overall, SOX2 is thus a compelling target for treatment of premalignant and invasive disease.
Because SOX2 lacks an obvious small molecule binding domain, it is challenging to design an inhibitor, as compared with e.g. the estrogen and androgen receptors, where antagonists related to their natural ligands have been successfully developed to treat breast and prostate cancer, respectively (39, 40). Because PI3K cooperates with SOX2 to drive the squamous injury response in stem cells (28), PI3K inhibitors could theoretically be used in the treatment of SOX2-driven neoplasias. However, in a phase II SQCC clinical trial, the PI3K inhibitor BKM120 was ineffective at its maximum tolerated dose (41). Whereas PI3K inhibitors might be more effective during preneoplasia, their effectiveness in SQCC might be improved by combining them with drugs that more directly target SOX2 activity. Such drugs could inhibit protein-protein interactions or the activity of chromatin modifying enzymes that are essential for SOX2-dependent effects on transcription. For example, small molecules that inhibit the interaction between TP53 and MDM2, or interactions between BET domain-containing proteins and acetyl-lysine residues, are in clinical trials (42) (e.g. GSK525762, https://clinicaltrials.gov). In addition, romidepsin and vorinostat are histone deacetylase inhibitors that are approved for treatment of cutaneous T-cell lymphoma (43), whereas histone methyltransferase inhibitors are in clinical trials for different cancers (e.g. EPZ-5676, https://clinicaltrials.gov).
To rationally develop effective anti-SOX2 targeting strategies, a comprehensive understanding of the SOX2 interactome and its potentially diverse functions in SQCC pathogenesis is required. Although SOX2 interactomes have been described in several previous studies (37, 44–50), no interactors have been functionally characterized in basal cells, and only TP63 has been studied as a SOX2 interactor in SQCCs (37). Furthermore, these studies have utilized standard affinity purification combined with mass spectrometry (AP-MS), a technique that is prone to false negatives for chromatin-associated proteins because of difficulties of solubilizing such polypeptides, especially as part of intact complexes. A recently developed method that circumvents this limitation is the in vivo proximity-dependent biotinylation technique, BioID (51). In this method, the “bait” protein of interest is fused to a mutant E. coli biotin ligase (BirA R118G), which releases activated biotin to covalently label nearby lysine residues. The bait is expressed in living cells, where biotin labeling is performed before lysis. Because of the covalent nature of the biotin label, the integrity of protein complexes need not be maintained post-lysis. Thus, cells can be lysed under harsher buffer conditions to maximize solubilization of chromatin-associated proteins. Labeled proteins are then purified by high affinity streptavidin precipitation and identified by mass spectrometry. This method was originally developed to help identify poorly soluble proteins such as those found in the nuclear lamina (51). BioID has since been used for a wide range of bait proteins localized throughout the cell (52–58), and head to head comparisons with AP-MS for nearly 100 baits have revealed that BioiD and AP-MS generally detect complementary interactomes (53, 54, 56–58).
EXPERIMENTAL PROCEDURES
Ethics Approval
Human tracheobronchial tissue for the isolation of basal cells was obtained as surgical waste from lung transplant operations with approval of the University Health Network Research Ethics Board (08–0318-T). Primary patient lung SQCC tissue was also obtained with approval of the University Health Network Research Ethics Board (04–0557-T), and was implanted subcutaneously into NOD/SCID mice following Animal Usage Protocol 603. All animal work was carried out with the approval of the University Health Network Animal Care Committee and was registered and licensed under the province of Ontario's Animals for Research Act, and was compliant with the humane policies and guidelines of the Canadian Council on Animal Care.
Drugs
BKM120 and CBP30 were purchased from Selleckchem (Houston, TX) and Sigma (St. Louis, MO), respectively.
Plasmids
pcDNABirASOX2 encodes an N-terminal fusion of E. coli BirA (R118G) to SOX2. It was constructed by amplifying SOX2 from pOTB7-SOX2 (28) with SOX2–18 (5′-ATAAGAAT GCGGCCGCT ATG TAC AAC ATG ATG GAG ACG GAG-3′) and SOX2–19 (5′-ATACCG CTC GAG TCA CAT GTG TGA GAG GGG CAG TG-3′), digestion with NotI and XhoI, and ligation into NotI/XhoI-digested pcDNA5-FLAG-BirAR118G. pcDNA5-FLAG-BirAR118G encodes a FLAG-tagged R118G BirA mutant, which is under the control of a tet operator-regulated promoter, with the promoter and BirA gene flanked by FRT recombination sites (52). The cloning places SOX2 downstream and in-frame with BirA. pLenti-SOX2, the lentiviral construct that expresses SOX2, was constructed by cloning the human SOX2 cDNA into the bidirectional pMA1 lentiviral vector and has been previously described (28). In this vector, SOX2 expression is driven by the PGK promoter and GFP is coexpressed from a minimal CMV promoter. pLenti-TP63, which expresses TP63, was constructed by Gateway cloning of the human ΔNTP63α cDNA (GeneCopoeia, Rockville, MD) into pMA1. Lentiviral shRNA constructs included shEP300–1 (TRCN0000039882), shEP300–2 (TRCN0000039883), shEP300–3 (TRCN0000039884), shEP300–4 (TRCN0000039885), shEP300-5 (TRCN0000231136), and shluc (TRCN0000072246) (Open Biosystems/Dharmacon, Lafayette, CO).
BioID Sample Preparation
HEK293-TREx flp-in cells (Invitrogen/Thermofisher, Waltham, MA) were co-transfected with pOG44 and pcDNA5 FRT/TO FLAG-BirA-SOX2 using the PolyJet transfection reagent (SignaGen, Rockville, MD). Stable lines were selected in DMEM/10% FBS/200 μg/ml hygromycin B. After selection, cells were infected with a lentiviral TP63 construct, and were further selected by FACS purification using the GFP marker coexpressed by the TP63 lentiviral construct. For mass spectrometry experiments, cells were grown to 70% confluency on 5 × 150 mm2 dishes, then incubated for 24 h in complete media supplemented with 1 μg/ml tetracycline (Sigma) and 50 μm biotin (BioShop, Burlington, ON, Canada). Cells were washed twice with PBS, and dried pellets snap frozen. Pellets were subsequently lysed in 10 ml of modified RIPA buffer (50 mm Tris-HCl pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, 0.1% SDS, 1:500 protease inhibitor mixture (Sigma), 250U Turbonuclease (Accelagen, San Diego, CA) at 4°C for 1 h and sonicated (30 s, at 35% power, Sonic Dismembrator 500; Fisher Scientific, NH) to disrupt aggregates. Lysates were centrifuged at 35,000 × g for 30 min, and the supernatants incubated with 30 μl of packed, pre-equilibrated Streptavidin-Sepharose beads (GE, Pittsburgh, PA) at 4 °C for 3 h. Beads were collected by centrifugation (2000 rpm, 2 min), washed 6 times with 50 mm ammonium bicarbonate pH 8.2, and treated with TPCK-trypsin (Promega, Madison, WI) for 16 h at 37 °C. Supernatants containing tryptic peptides were then collected and lyophilized. Peptides were resuspended in 0.1% formic acid and 1/6th of the sample was analyzed per mass spectrometry run.
Liquid Chromatography-Mass Spectrometry
Liquid chromatography (LC) analytical columns (75 μm inner diameter) and precolumns (150 μm ID) were made in-house from fused silica capillary tubing from InnovaQuartz (Phoenix, AZ), and packed with 100Å C18-coated silica particles (Magic, Michrom Bioresources, Auburn, CA). LC-MS/MS was conducted using a 120-min reversed-phase buffer gradient running at 250 nl/min (column heated to 40 °C) on a Proxeon EASY-nLC pump in-line with a hybrid LTQ-Orbitrap velos mass spectrometer (ThermoFisher, Waltham, MA). A parent ion scan was performed in the Orbitrap, using a resolving power of 60,000. Simultaneously, up to the twenty most intense peaks were selected for MS/MS (minimum ion count of 1000 for activation) using standard CID fragmentation. Fragment ions were detected in the LTQ. Dynamic exclusion was activated such that MS/MS of the same m/z (within a 10 ppm window, exclusion list size 500) detected three times within 45 s were excluded from analysis for 30 s.
Experimental Design and Statistical Rationale
Four BioID runs were conducted on FlagBirA*-SOX2 expressing cells, consisting of two technical replicates (n = 2) from two biological replicates (n = 2; total n = 4). Ten control runs of a BioID analysis conducted on cells expressing the FlagBirA* tag alone were used for comparative purposes. For protein identification, raw files were converted to the .mzXML format using Proteowizard (v3.0.4468) (59), then searched using X!Tandem (v2013.06.15.1) (60) against Human RefSeq Version 45 (containing 36,113 entries). Search parameters specified a parent MS tolerance of 15 ppm and an MS/MS fragment ion tolerance of 0.4 Da, with up to two missed cleavages allowed for trypsin. No fixed modifications were set but oxidation of methionine was allowed as a variable modification. Data were analyzed using the trans-proteomic pipeline (61) via the ProHits 4.0.0 software suite (62). Proteins identified with a ProteinProphet cut-off of 0.85 (corresponding to ≤1% probabilistic FDR (63)) were analyzed with SAINT Express v. 3.3 (64, 65) to identify high-confidence interactors. The ten controls were collapsed to the highest three spectral counts for each hit. Proteins identified with two or more unique peptides and scoring above a Bayesian False Discovery Rate of 1% (corresponding to a SAINT score ≥ 0.75, see (65) for details on BFDR calculation) were high-confidence proximity interactors. All data are publicly available and have been uploaded to the MassIVE archive (www.massive.ucsd.edu) with ID: MSV000080175 (SOX2_BioID_Interactome).
Informatics and Genomics Data
Enrichment of Gene Ontology (GO) annotations was restricted to “biological processes” and was performed using PANTHER (www.pantherdb.org) (66, 67). Gene Set Enrichment Analysis (GSEA) (68, 69) was performed at http://software.broadinstitute.org/gsea/index.jsp, focusing on the Hallmark (H), Curated (C2), and Oncogenic signature (C6) gene sets. TCGA cancer genomics data were downloaded from www.cbioportal.org (70).
Primary Human Tracheal Basal Cells and Cell Lines
Normal basal cells were isolated from healthy adult human tracheobronchial tissue, and unless otherwise indicated, cultured on plastic dishes in LHC-9 medium, as previously described (28). Basal cells were used between passage 2 and 3, and included isolates from 3 different donors. For growth on Transwell filters, basal cells were cultured in LHC basal: DMEM (1:1) that was supplemented as described (71), which included 0.33 nm retinoic acid and 5 ng/ml EGF. Media was added to both chambers. ChaGoK1 and SW1573 were obtained from the ATCC, whereas MGH7 cells were isolated by our group (72).
Primary Patient Lung Squamous Cell Carcinoma (SQCC) Xenografts (PDXs)
Collection and description of lung squamous cell carcinoma (SQCC) tissue and its use for the generation of xenografts (PDXs) has been previously described (28, 73, 74).
Immunostaining and Proximity Ligation Assay (PLA)
Immunostaining of cultured basal cells and SQCC xenograft tissue was performed using standard methods, as described (28). PDX#77 tissue was cryopreserved in OCT and was sectioned for staining without fixation. PDX#188 tissue was fixed in formalin and paraffin-embedded. Cultured cells were fixed in 4% paraformaldehyde in PBS. PLA was done with the Duolink In Situ Red Starter Kit (Sigma). Antibodies were α-ARID1B (1:200, A301–046A, Bethyl Laboratories, Montgomery, TX), α-EP300 (1:100, #sc-584, Santa Cruz, Dallas, TX), α-SOX2 (1:100, MAB2018, R&D Systems, Minneapolis, MN), and α-TP63 (1:100, GTX102425, Genetex, Irvine, CA). Nuclei were visualized by DAPI staining and were scored for presence of foci.
Transient Transfection and PLA of MGH7 Cells
MGH7 cells were seeded onto glass coverslips coated with 48 μg/ml PureCol (Advanced BioMatrix, Carlsbad, CA) in 12-well dishes and transfected with the PolyJet transfection reagent (SignaGen). Cells were transfected with 1 μg of the GFP-expressing lentiviral vector pMA1 as a marker for transfected cells and 150 ng of adenoviral E1A expression vector (gifts from Ed Harlow). Three days following transfection, cells were fixed in 4% paraformaldehyde and subjected to PLA.
Human Protein Atlas Images
α-EP300 (HPA003128, Sigma) stained images from the Human Protein Atlas (www.proteinatlas.org) (75–77) included normal nasopharyngeal epithelium (8834_B_7_2) and lung SQCC (8900_B_1_7, 8900_B_2_2, 8900_B_2_8).
Lentiviral Infections
Lentiviruses were generated and used to infect cells as previously described (28), with transduction efficiencies typically in the range of 80–90%. To select for shRNA lentiviral infection, cells were treated with puromycin (3 μg/ml) 24 h after infection for 24–48 h.
RNA Isolation and qRT-PCR
RNA isolation, reverse transcription, qRT-PCR, and some primers are described in (28). Briefly, qRT-PCR was performed using SYBR green (Bio-Rad, Hercules, CA), and target gene expression quantified by the ΔΔCt method using TBP expression for normalization. Additional primers for this study include: EP300 FWD-5′-GCA GGC ATG GTT CCA GTT TC-3′, EP300-RVS-5′-GGT CAG GTA GAG GGC CAT AGA-3′; CREBBP-3 5′-CGT GTC ACA GGG ACA GGT G-3′, CREBBP-4 5′-GTG ACT GTG TCA CTG GAG GG-3′; ADH7-F 5′-TCC ACT GGA TAT GGC GCT GC-3′, ADH7-R 5′-TCC CAG GCC AAA GAC GAC GC-3′; KIAA1199–3 5′-GAT ACG TCT CCA GAG GCC CAT G-3′, KIAA1199–4 5′-C TCA GTG TCC AGT GTC ACC CAG-3′; EDN1–3 5′-GAG AAA CCC ACT CCC AGT CC-3′, EDN1–4 5′-GAT GTC CAG GTG GCA GAA GT-3′; SMARCA4-F 5′-GGC TCA TGG CTG AAG ATG AGG AG-3′, SMARCA4-R 5′-CTC CGT GAG GTT AGC CAC GTA CTC-3′
siRNA Experiments
siRNAs were purchased from Dharmacon (Lafayette, CO) and included the ON-TARGETplus Human EP300 (2033) SMART pool (#l-003486–00-0005) and the ON-TARGETplus Non-targeting pool (#d-001810–10−05) (control, SiCon). siRNAs were transfected overnight at 20–50 nm using RNAiMAX (Invitrogen/Thermofisher) and following the manufacturers' instructions.
SDS-PAGE and Western Blotting
Cell lysis, SDS-PAGE, and Western blotting were performed as previously described (28), with the exception that a wet tank blotting system was used (Bio-Rad). Proteins were transferred in 25 mm Tris base, 190 mm glycine, and 10% methanol at 80 volts for 2.5 h. Primary antibodies included α-EP300 (sc-584, 1:500, Santa Cruz), α-CREBBP (sc-7300, 1:500, Santa Cruz), and α-PTPN11 (C-18, 1:2000, Santa Cruz).
AlamarBlue Growth Assays
AlamarBlue was purchased from Thermofisher and used according to the manufacturer's instructions.
Statistical Analyses for Biological Data
Statistical significance was calculated using either 2-tailed t or Fisher's exact tests, as indicated in the figure legends.
RESULTS
A SOX2 BioID-based Interactome
SOX2 BioID was conducted in 293 T-REx cells, a model system that we and others have previously used to identify proximal interactors for a large number of other bait proteins, including transcription factors (52–56, 78). SOX family proteins generally dimerize with other DNA-binding transcription factors to perform cell-specific functions (79). Because 293 T-REx cells are derived from HEK293 cells, which appear to have an embryonic neuroendocrine (adrenal gland) origin that lacks TP63 (80, 81), the major SOX2 partner in SQCCs (37), we stably expressed TP63 in these cells from a lentiviral vector. Altogether, 82 high-confidence SOX2-interactors were identified in this analysis (1% FDR; Fig. 1, Table I, supplemental Table S1, S2).
Fig. 1.

The SOX2 BioID interactome. SOX2 interactors were classified according to information compiled at GeneCards (www.genecards.org) and literature searches. Interactors uniquely identified in our study are depicted in black, while those identified in at least one previous AP-MS study are shown in red (see supplemental Table S5 for details). CPC = chromosomal passenger complex. Node size is proportional to total number of peptides identified in four mass spectrometry runs.
Table I. High-confidence SOX2 interactors identified using BioID.
BioID was performed on two SOX2-BirA* biological replicates, each divided into two technical replicates for mass spectrometry. Spectral counts from each technical replicate are shown. Proteins scoring above a Bayesian False Discovery Rate of 1% (in this analysis corresponding to a SAINT score > 0.75) were considered high-confidence proximity interactors. Novel interactions (not reported in previous SOX2 AP-MS studies) are highlighted in blue.
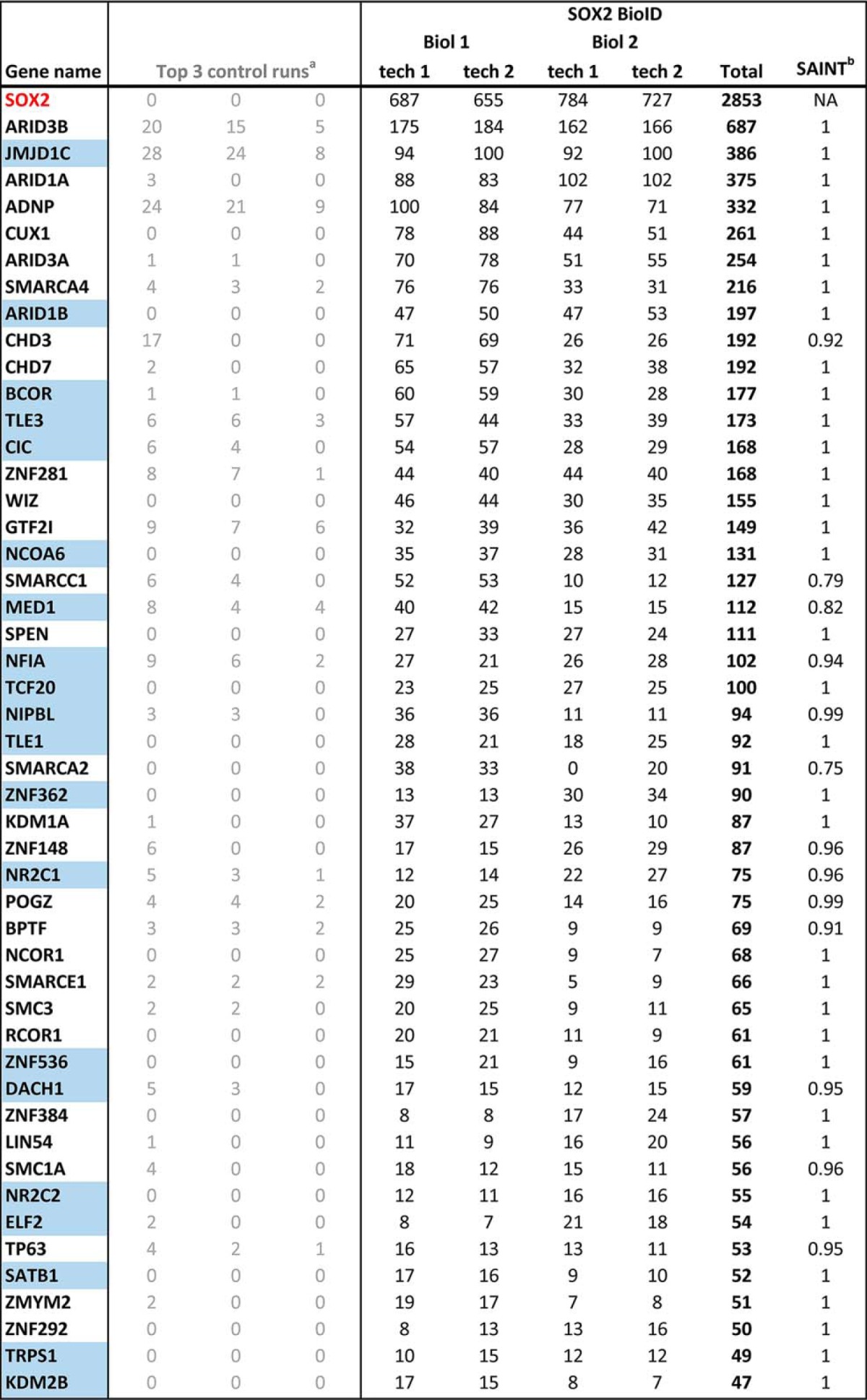
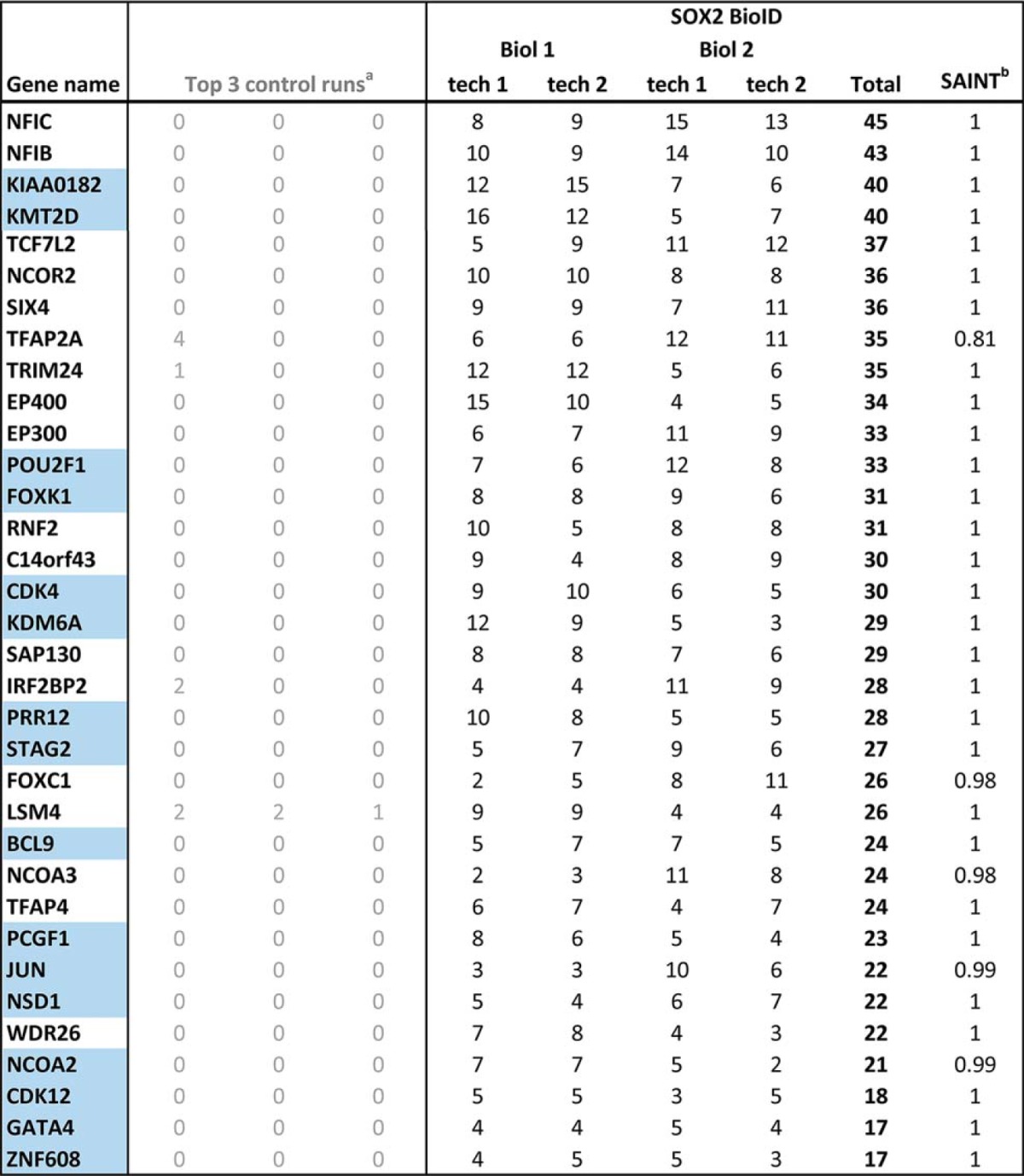
aControl runs consisted of five biological replicates of Flag-BirA*only that were each divided into two technical replicates. These 10 control runs were then collapsed to the three highest spectral counts for each hit.
bSAINT = Significance Analysis of INTeractome confidence score. NA = not applicable.
Gene Ontology (GO) analysis revealed that 87% of the BioID hits could be classified as transcriptional regulators (Fig. 2, supplemental Table S3), consistent with BirA labeling proteins close to the transcription factor bait in vivo. The interactome largely comprised sequence-specific transcription factors, transcriptional corepressors and coactivators, chromatin modifiers and remodeling proteins, and members of the cohesin complex (Fig. 1). As expected, given the central roles of many of the SOX2-interactors in transcriptional regulation, Gene Set Enrichment Analysis (GSEA) showed enrichment of gene sets associated with multiple transcription factors (supplemental Table S4). However, some gene sets annotated as responses to specific stimuli were also enriched (Fig. 2, supplemental Table S4). These stimuli included anticancer treatments and signaling pathway agonists and receptors, suggesting potential ways of modulating SOX2 activity through extracellular treatments.
Fig. 2.

Summary of select characteristics that are significantly enriched in the SOX2 BioID interactome. The number of transcriptional regulators was determined by Gene Ontology (GO) analysis (supplemental Table S3), whereas other characteristics were evaluated by Gene Set Enrichment Analysis (GSEA) (supplemental Table S4).
When compared with eight other SOX2 interactome studies conducted by AP-MS in neuronal, embryonic stem (ES), and TP63-expressing SQCC cell types (37, 44–50), 46 (56%) of the BioID interactors were identified in at least one other study (Fig. 1, 2, Table I, supplemental Table S5), supporting the ability of BioID to detect genuine SOX2 interactors. Indeed, TP63 was included among these interactors, which associates with SOX2 and coregulates gene expression in SQCC (37), as well as CHD7, a helicase that coregulates a subset of SOX2 targets in neural stem cells (46). In addition, several of the common interactors have been linked to SOX2 function in ES cells and in the reprogramming of somatic cells into ES cell-like stem cells (iPS cells). These interactors include SMC1A and SMC3, which colocalize with SOX2 at some sites in ES cells while helping maintain pluripotency (82, 83); EP300, which colocalizes with SOX2, OCT4, and NANOG in many enhancer regions of ES cells (84, 85); and SMARCA4, a core component of the SWI/SNF complex that enhances generation of iPS cells by SOX2 (86).
Although 36 (44%) of the interactors were not previously reported in other studies, several lines of evidence support these interactions as also being genuine and relevant to SOX2 function. For example, JMJD1C, a histone demethylase, coregulates the miR-302 promoter with SOX2 in ES cells and promotes pluripotency (87, 88). BCOR, a corepressor found in some Polycomb complexes (89), was also detected. Its mutation is linked to anophthalmia/microphthalmia syndromes that are also caused by SOX2 mutation (90, 91), supporting it acting with SOX2 in eye development. In addition, through GSEA analysis we found that 7 novel interactors belong to a larger group of 11 that are direct SOX2 transcriptional targets in ES cells (Fig. 2, supplemental Table S4). Although such targets do not necessarily physically interact with SOX2 to promote SOX2 function, we corroborated this possibility for at least one of the targets using a different method.
We used the proximity ligation assay (PLA) to validate the SOX2 interaction with ARID1B, a SWI/SNF complex component that is both a novel interactor and a direct SOX2 target in ES cells (92). With this assay, proximity of proteins within 30 nm of each other can be visualized in situ (93). Typically, standard primary antibodies to the proteins of interest are first incubated with fixed cells, followed by binding of specialized secondary antibodies with unique covalently attached DNA strands. Connector oligonucleotides are then ligated to the DNA strands of the secondary antibodies to create a circularized template, which is amplified with a polymerase. Amplified circles are then detected with complementary fluorescent oligonucleotide probes and visualized as discrete foci. We performed PLA on endogenous SOX2 and ARID1B in MGH7 cells, a lung SQCC cell line that has high copy 3q amplification and expresses high levels of SOX2 (72, 94) (supplemental Fig. S1). Nuclear fluorescent foci were observed in many cells and were dependent on the presence of both primary antibodies (Fig. 3). These data support the PLA reaction being dependent on both SOX2 and ARID1B proteins, and are consistent with these proteins being close to each other in vivo. Overall, both the PLA data, as well as the identities of some of the common and novel SOX2 interactors, support BioID as a valid method to detect proteins relevant to SOX2 function.
Fig. 3.

Detection of proximity between SOX2 and ARID1B in SQCC cells with 3q amplification. Cytospun MGH7 cells were fixed and subjected to the proximity ligation assay (PLA). Cells were stained with either both α-SOX2 and α-ARID1B primary antibodies, or single antibody controls. Nuclei were visualized with DAPI staining. Scale is 10 μm. Mean percentages of foci-positive nuclei ± S.E. are shown, and were calculated by scoring five fields comprising 239–582 nuclei per field. Significance was calculated using a 2-tailed t test. ***p = 8.9 × 10−9 and 8.3 × 10−9 relative to α-SOX2 alone and α-ARID1B alone, respectively, primary antibody controls.
EP300 is Expressed In Normal Respiratory Basal Cells and In SQCCs
To prioritize BioID hits for study in basal cells and SQCCs, we considered the presence of potentially druggable domains and previous studies with the interactor. EP300 is a BioID SOX2-interactor that acetylates histones and nonhistone proteins and promotes transcriptional activation by a number of transcription factors (95). It has several regions that are involved in protein-protein interactions (Fig. 4A), which include KIX, zinc finger (TAZ, ZZ), and LXXLL (L) domains, as well as a bromodomain (Br) that binds acetyl-lysine residues in other transcription factors and histones. EP300 also has a RING finger that autoregulates acetyltransferase activity (96). This architecture allows EP300 to be sensitive to different types of modulatory compounds including distinct research tools, natural products, and FDA-approved drugs (97–105). Although some previous work has linked EP300 to regulation of SOX2 activity, its inconsistent detection in AP-MS studies and opposing models for regulation of SOX2 did not cause us to consider it prior to our work. EP300 acetylates SOX2 in vitro (106), but it has only been detected as a SOX2-interactor in a subset of AP-MS studies in ES cells (48, 49). In ES cells, EP300 has been proposed to be both an inhibitor (106), as well as a coactivator, with the coactivator function depending on other pluripotency factors including OCT4 (84, 107, 108). Because the parental HEK293 cells used in our screen do not express OCT4 under standard culture conditions (109), our reproducible identification of EP300 as a SOX2-interactor prompted us to consider that EP300 might regulate SOX2 activity outside of ES cells, in contexts relevant to SQCC pathogenesis. Indeed, such a role for EP300 could relate to its mutation in cervical SQCCs (110) and why its expression is associated with poor prognosis in cutaneous, laryngeal, and esophageal SQCCs (111–113), all of which are characterized by varying frequencies of 3q copy number gains (17, 114, 115). We thus, focused on EP300 for further study.
Fig. 4.

Expression of EP300 in normal respiratory basal cells and in SQCCs. A, Schematic of EP300 architecture. Domains include L (LXXLL motif-containing), TAZ (TAZ Zinc finger), KIX (kinase domain inducible interacting), Br (bromodomain), RING (really interesting new gene), PHD (plant homeodomain), HAT (acetyltransferase catalytic domain), ZZ (ZZ Zinc finger), and NCBD (nuclear receptor coactivator binding domain). B, Immunohistochemistry of EP300 in normal human nasopharyngeal epithelia. Arrows in inset point to basal cells. Scale is 20 μm. Images are from The Human Protein Atlas (www.proteinatlas.org). C, EP300 immunofluorescence staining of second passage primary human tracheobronchial basal cells. Scale is 10 μm. D, SOX2 and EP300 immunofluorescence staining of two different primary patient-derived SQCC xenografts (PDXs). Scale is 20 μm. E, Immunohistochemistry of EP300 in three different primary patient SQCCs. Scale is 20 μm. Images are from The Human Protein Atlas.
We first examined EP300 expression in cell types relevant to lung SQCC pathogenesis. EP300 was readily detected by immunohistochemistry in all cells of normal respiratory epithelia, including basal cells (Fig. 4B), which also express SOX2 (28). EP300 was also expressed in ex vivo cultures of purified tracheobronchial basal cells that retain stem cell activity (Fig. 4C) (28, 116). In primary patient SQCCs harboring SOX2 amplification that were grown as xenografts (PDXs), EP300 was expressed in most tumor cells, along with SOX2 (Fig. 4D). This expression pattern was similar to that in nonxenografted SQCC tissue, as reported by the Human Protein Atlas, which found EP300 to be expressed at moderate/high levels in five of five primary patient SQCCs (Fig. 4E, three representative images are shown). Thus, both EP300 and SOX2 are expressed in the putative normal stem cells of origin and invasive SQCC disease.
EP300 Is Proximal to SOX2 In Normal Basal Cells and SQCCs
We next used PLA to assess the proximity of SOX2 and EP300 in basal cells and SQCCs. When cultured ex vivo, basal cells enter a SOX2Lo state that is transiently observed in vivo during recovery from injury (28). Re-expression of physiologic high levels of SOX2 in these cells drives different types of differentiation, depending on the signaling context (28). For example, when subconfluent basal cells are cultured in normal growth media, PI3K cooperates with lentivirally expressed SOX2 to induce a squamous fate (28). Under these conditions, PLA with both α-SOX2 and α-EP300 primary antibodies resulted in more fluorescent foci than single primary antibody controls (Fig. 5), supporting their proximity at a time when basal cells functionally respond to SOX2. Further evidence for the PLA-generated foci reflecting proximity of SOX2 and EP300 was obtained by using basal cells that had been transduced with empty vector instead of Lenti-SOX2. Omission of exogenous SOX2 expression in these cells significantly reduced the number of foci generated by simultaneous presence of both α-SOX2 and α-EP300 primary antibodies (Fig. 5).
Fig. 5.

Detection of proximity between SOX2 and EP300 in primary human tracheobronchial basal cells. Second passage basal cells were transduced with Lenti-SOX2 and after 2 days, cytospun, fixed, and subjected to the proximity ligation assay (PLA). Cytospins were stained with either both α-SOX2 and α-EP300 primary antibodies, or single antibody controls. Nuclei were visualized with DAPI staining. Scale is 10 μm. Mean percentages of foci-positive nuclei ± S.E. are shown, and were calculated by scoring five fields and counting 50–100 nuclei per field. Significance was calculated using a 2-tailed t test. **p = 0.00003 and 0.0007 relative to α-SOX2 alone and α-EP300 alone, respectively (in Lenti-SOX2-transduced basal cells), and 0.0009 relative to the combination of α-SOX2 and α-EP300 in empty vector control-transduced cells.
We also used PLA to examine the proximity of SOX2 and EP300 in lung cancer cell lines with 3q copy number gains, as well as a primary patient SQCC PDX tissue that we had previously determined to have SOX2 amplification (28). The MGH7 SQCC cell line was described earlier and ChaGoK1 was derived from an undifferentiated bronchogenic carcinoma (117). ChaGoK1 has low level gain at 3q and expresses TP63 and SOX2 (supplemental Fig. S1), common SQCC markers located within the 3q amplicon (5, 6, 17, 18, 28, 118). In this assay >50% of cells in culture and in SQCC tissue displayed nuclear foci that were strongly dependent on the presence of both α-SOX2 and α-EP300 primary antibodies (Fig. 6). In addition, the number of cells showing nuclear foci, as well as the number of foci per cell, were reduced when MGH7 cells (the SQCC cell line that had the highest levels of SOX2 expression) were transfected with expression vectors for adenoviral E1A oncoproteins (supplemental Fig. S2). Both the 13S and 12S E1A isoforms are well-established to bind EP300 through their common amino termini (119, 120), with sequestration of EP300 thought to be a major component of their transforming properties (121). In addition, E1A has been shown to be able to sequester EP300 away from a GAL4-SOX2 chimeric transcription factor (108). Thus, the ability of both the 13S and 12S E1A isoforms to reduce the number of SOX2-EP300 PLA foci provide further evidence that these foci signify a bona fide SOX2-EP300 interaction. Overall, the PLA data are consistent with the proximity between SOX2 and EP300 observed by BioID in HEK293 cells, and by AP-MS and ChIP in ES cells (48, 49, 84), but provide evidence that their interaction is not limited to ES cell-like contexts and occurs in normal basal and SQCC cells.
Fig. 6.

Detection of proximity between SOX2 and EP300 in lung cancers with 3q gains. A primary patient-derived SQCC xenograft (PDX) and two lung cancer cell lines (1 definitive SQCC), all harboring SOX2 gains, were subjected to the proximity ligation assay (PLA). Slides were stained with either both α-SOX2 and α-EP300 primary antibodies, or single antibody controls. Nuclei were visualized with DAPI staining. Scale is 10 μm. Mean percentages of foci-positive nuclei ± S.E. are shown, and were calculated by scoring five fields and counting 60–200 nuclei per field. Significance was calculated using a 2-tailed t test. ***p = 0.00007 and 0.00007 relative to α-SOX2 alone and α-EP300 alone, respectively (SQCC PDX); 5.7 × 10−9 and 7.4 × 10−11 relative to α-SOX2 alone and α-EP300 alone, respectively (MGH7); 6.2 × 10−9 and 9.8 × 10−9 relative to α-SOX2 alone and α-EP300 alone, respectively (ChaGoK1).
EP300 Is Required for SOX2 Activity in Basal Cells
To determine if EP300 affects SOX2 activity in an SQCC-relevant context, we first asked whether an EP300 chemical inhibitor interferes with SOX2's ability to induce a squamous fate in basal cells. The induction of the squamous fate by SOX2 reflects a core SOX2 activity in basal cells and appears to be one of the earliest points of genetic dysregulation in SQCC pathogenesis (28). Basal cells were infected with either control vector or Lenti-SOX2 in the presence or absence of CBP30, a compound that preferentially binds to the bromodomains of EP300 and related CREBBP to inhibit their interactions with acetyl-lysine residues (100–102). After 5 days, squamous lineage marker expression was quantified (Fig. 7). CBP30 strongly suppressed induction of squamous lineage markers by Lenti-SOX2. To determine if the inhibitory effect of CBP30 on SOX2 activity was also manifested on earlier transcriptional responses to SOX2, we examined expression levels of a panel of SOX2 early response genes. After addition of Lenti-SOX2, most basal cells do not express SOX2 protein until ∼36 h post-infection (28). At this time point, we previously identified a set of genes that were induced by SOX2 (28). CBP30 strongly suppressed induction of all three tested early response genes (ADH7, KIAA1199, EDN1), as well as a gene whose expression is correlated with SOX2 in SQCC that we also found is a target of SOX2 in basal cells (FOXE1) (17, 28) (Fig. 7). By contrast, PIK3CA and SMARCA4 were not induced by SOX2 and were insensitive to CBP30 (Fig. 7).
Fig. 7.

Chemical inhibition of EP300 suppresses SOX2 activity in tracheobronchial basal cells. Primary tracheobronchial basal cells were infected with control vector or Lenti-SOX2 ± co-treatment with 12.5 μm CBP30 (EP300 inhibitor) or DMSO (inhibitor control). Media and inhibitor were replaced every other day, and after 5 days, gene expression quantified by qRT-PCR. Data are normalized to TBP expression and plotted as fold induction relative to vector control. Means ± S.E. of 3 replicates are shown. Significance of potential gene expression induction by SOX2 was calculated by comparing values between vector and Lenti-SOX2/DMSO using paired 2-tailed t tests. *p = 0.006 (TMPRSS11B), 0.05 (SPRR3), 0.04 (ADH7), 0.01 (KIAA1199), 0.01 (FOXE1), 0.02 (EDN1). ns = not significant. ns, p = 0.16 (PIK3CA), 0.33 (SMARCA4). Significance of potential inhibition of SOX2 activity was calculated by comparing values between Lenti-SOX2/DMSO and Lenti-SOX2/CBP30 using 2-tailed t tests. *p = 0.01 (SPRR3), 0.01 (ADH7); **p = 0.001 (KIAA1199), 0.003 (FOXE1), 0.002 (EDN1); ***p = 0.0001 (TMPRSS11B); ns, p = 0.28 (PIK3CA), 0.53 (SMARCA4).
To more specifically interfere with EP300 activity, we used lentiviral shRNA constructs that target EP300, but not the related gene CREBBP. In our first screen of shEP300 constructs, we identified shEP300 #3, which reduced EP300, but not CREBBP protein levels in basal cells (Fig. 8). We later identified another shRNA construct, shEP300 #5, that also reduced EP300, but not CREBBP protein levels in basal cells (Fig. 8). To test the effects of EP300 knockdown on SOX2 activity, the shEP300 constructs or control shluciferase (shluc) were coinfected with Lenti-SOX2 in basal cells (Fig. 9). In perfect agreement with the CBP30 data, both shEP300 constructs suppressed induction of squamous lineage marker and early response genes by SOX2 (Fig. 9A, 9B). Also in accord with the CBP30 results, SOX2-nonresponsive PIK3CA and SMARCA4 expression were not reduced by the shEP300 constructs (Fig. 9A, 9B). As expected, both shEP300 constructs reduced EP300, but not CREBBP mRNA expression (Fig. 9A, 9B). Altogether, these data support EP300 being necessary for several basal cell transcriptional responses to SOX2, as well as their biological differentiation into the squamous lineage.
Fig. 8.

Identification of lentiviral shRNA constructs that reduce EP300, but not CREBBP expression in basal cells. Primary tracheobronchial basal cells were infected with lentivirus overnight. After 24 h, infected cells were selected with puromycin and protein levels quantified by Western blotting between 48–72 h. Left panel, four different shEP300 constructs were initially tested, which led to identification of shEP300 #3. Right panel, characterization of shEP300 #5. Protein levels were quantified by densitometry, with EP300 and CREBBP expression normalized to PTPN11 (loading control) levels. Normalized EP300 and CREBBP expression was then adjusted relative to control shluciferase (shluc)-transduced cells, which was assigned a value of 100. NS = nonspecific band.
Fig. 9.

shRNA-mediated knockdown of EP300 suppresses SOX2 activity in tracheobronchial basal cells. Primary tracheobronchial basal cells were infected with control vector or Lenti-SOX2 ± shEP300 or shluc control, and after 5 days, gene expression quantified by qRT-PCR. Data are normalized to TBP expression and plotted as fold induction relative to vector control. Means ± S.E. of 3 replicates are shown. A, Cells infected with shEP300 #3. Significance of potential gene expression induction by SOX2 was calculated by comparing values between vector and Lenti-SOX2/DMSO using paired 2-tailed t tests. *p = 0.01 (TMPRSS11B), 0.02 (SPRR3), 0.04 (ADH7), 0.02 (FOXE1); **p = 0.0008 (KIAA1199), 0.003 (EDN1). ns = not significant. ns, p = 0.47 (PIK3CA), 0.16 (SMARCA4), 0.73 (EP300), 0.19 (CREBBP). Significance of potential inhibition of SOX2 activity was calculated by comparing values between Lenti-SOX2/DMSO and Lenti-SOX2/CBP30 using 2-tailed t tests. *p = 0.009 (ADH7); **p = 0.002 (SPRR3), 0.003 (FOXE1), 0.003 (EP300); ***p = 0.0005 (TMPRSS11B), 0.000003 (KIAA1199), 0.00006 (EDN1); ns, p = 0.17 (PIK3CA), 0.39 (SMARCA4), 0.99 (CREBBP). B, Cells infected with shEP300 #5. Significance of potential gene expression induction by SOX2 was calculated by comparing values between vector and Lenti-SOX2/DMSO using paired 2-tailed t tests. *p = 0.008 (TMPRSS11B), 0.05 (SPRR3), 0.009 (ADH7), 0.03 (KIAA1199), 0.02 (FOXE1), 0.05 (EDN1), 0.008 (EP300). ns = not significant. ns, p = 0.16 (PIK3CA), 0.67 (SMARCA4), 0.28 (CREBBP). Significance of potential inhibition of SOX2 activity was calculated by comparing values between Lenti-SOX2/DMSO and Lenti-SOX2/CBP30 using 2-tailed t tests. *p = 0.02 (SPRR3), 0.01 (KIAA1199), 0.01 (FOXE1), 0.02 (EDN1); **p = 0.004 (TMPRSS11B), 0.0006 (ADH7), 0.002 (PIK3CA); ***p = 0.00005 (EP300); ns, p = 0.20 (SMARCA4), 0.34 (CREBBP).
EP300 Copy Number Gains Are Commonly Selected in Lung SQCCs
Copy number gains in SOX2 at 3q26–28 occur in 94% of lung SQCCs (17, 18), the highest frequency of all studied cancers (supplemental Fig. S3). Notably, EP300 gains at 22q13 are also common in SQCCs (45% of cases), whereas only 20% of SQCCs display heterozygous loss, and no samples appear to have biallelic inactivation (Fig. 10A) (18). In SQCCs, EP300 copy number gains are correlated with increased EP300 mRNA expression (Fig. 10B), supporting the gains affecting EP300 expression. The pattern of EP300 copy number variation favoring gains over losses was specific to SQCCs as compared with lung adenocarcinomas (ADCs) (18, 122). In ADCs, the opposite pattern was observed, with only 13% of samples displaying EP300 gains and 44% of cases showing heterozygous loss (Fig. 10A, 10C). When compared with 19 other major cancers, EP300 copy number gains ranked highest in lung SQCCs and were significantly more enriched than in 18 of the other cancers (supplemental Fig. S4). By contrast, heterozygous EP300 losses were not commonly selected in lung SQCCs, with the SQCC frequency being lower than at least 8 other cancers (supplemental Fig. S4). Similarly, EP300 mutations were observed in only 4.5% of lung SQCCs, a frequency that was statistically like 10 other cancers (supplemental Fig. S5). Together these data suggest that both increased EP300 and SOX2 activity are genetically coselected in SQCCs.
Fig. 10.

EP300 copy number gains are enriched in lung SQCCs over ADCs. Data were obtained from the TCGA. A, Distribution of EP300 DNA alterations in lung SQCC and ADC cohorts. B, Quantification of EP300 mRNA levels relative to EP300 copy number in the SQCC cohort. RPKM = Reads Per Kilobase of transcript per Million mapped reads. Means ± S.E. are shown. Significance was calculated relative to diploid expression using a 2-tailed t test. *p = 0.04 and **p = 0.004. C, Comparison of EP300 copy number variations between SQCC and ADC cohorts. Significance was calculated using a 2-tailed Fisher's exact test. ***p = 3 × 10−17 (copy number gains) and 6 × 10−7 (het loss).
EP300 Promotes Growth Of Lung Cancer Cells With 3q Copy Number Gains
The growth of lung and esophageal SQCC cell lines with 3q copy number gains depends on SOX2 expression (17, 36, 37). To investigate if 3q gains in lung cancer might also be associated with sensitivity to EP300 levels, we examined the effect of EP300 knockdown on cell growth. To this end, we first reduced EP300 expression with a pool of 4 different EP300 siRNAs in MGH7 SQCC cells, which have high level 3q amplification (94). As a comparator, we also reduced EP300 expression in SW1573 lung cancer cells. SW1573 was derived from an alveolar lung cancer (ATCC), and harbors an activating KRAS mutation, but does not have 3q copy number gains (supplemental Fig. S1A). This cell line also expresses significantly lower levels of TP63 and SOX2 mRNA than MGH7 and ChaGoK1 (supplemental Fig. S1B), and does not express detectable TP63 and SOX2 proteins (supplemental Fig. S1C). Relative to a control non-targeting pool of siRNAs, the siEP300 pool reduced EP300 (but not CREBBP) protein and mRNA expression in both MGH7 and SW1573 cells (Fig. 11A, 11B). However, whereas MGH7 growth was severely impaired by siEP300 treatment, SW1573 growth was not affected (Fig. 11C). To further verify dependence of MGH7 growth on EP300, we also infected MGH7 cells with two shEP300 lentiviruses. Both shEP300 #5 and #3 reduced EP300 (but not CREBBP) protein and mRNA expression in MGH7 cells (Fig. 11D, 11E), and inhibited growth of this cell line (Fig. 11F). We then tested the effect of reducing EP300 expression in ChaGoK1 cells, which have low level 3q gain (supplemental Fig. S1A). Using shEP300 #3, we were able to achieve specific knockdown of EP300 protein and mRNA levels over CREBBP (Fig. 12A). As with MGH7, knockdown of EP300 in ChaGoK1 was associated with growth inhibition (Fig. 12B). Thus, growth of at least some lung cancers with 3q copy number gains, including SQCCs, is dependent on EP300.
Fig. 11.

EP300 promotes growth of 3q-amplified MGH7 SQCC cells, but not non-3q-amplified SW1573 lung cancer cells. A, B, Cell lines were transfected overnight with the indicated siRNA pools (siCon = control non-EP300 targeting pool of siRNAs) and at 48 h, EP300 and CREBBP expression was quantified by Western blotting (A) and qRT-PCR (B). A, For Western blotting, protein levels were quantified by densitometry, with EP300 and CREBBP expression normalized to PTPN11 levels (loading control). Normalized EP300 and CREBBP expression was then adjusted relative to siCon-transfected cells, which was assigned a value of 100. NS = nonspecific band. B, For qRT-PCR, EP300 and CREBBP expression was normalized to levels of TBP and plotted relative to siCon, which was assigned a value of 1.0. Means ± S.E. of 3 replicates are shown. Significance was calculated using paired 2-tailed t tests between siCon and siEP300-transfected populations. Only p values ≤ 0.05 are indicated. For MGH7, ***p = 0.0005 and *p = 0.01. For SW1573, **p = 0.001. C, siEP300 inhibits growth of MGH7 cells. Cells were transfected with either an siEP300 or siCon pool in triplicate and growth quantified by alamarBlue after 5–6 days. Data are plotted relative to siCon transfected cells, which was assigned a value of 100. Means ± S.E. are shown. Significance was calculated using a 2-tailed t test between siEP300 and siCon cells. Only p values ≤ 0.05 are indicated. ***p = 4 × 10−6. D, E, shEP300 lentiviruses reduce EP300, but not CREBBP expression in MGH7 cells. Cells were infected overnight with shEP300 or shluciferase (shluc) control lentiviruses and at 24 h, selected in puromycin. At 48 h post-virus addition, EP300 and CREBBP expression was quantified by Western blotting (D) and qRT-PCR (E). D, For Western blotting, protein levels were quantified by densitometry, as described in (A). NS = nonspecific band. E, For qRT-PCR, EP300 and CREBBP expression was quantified as described in (B). Means ± S.E. of 3 replicates are shown. Significance was calculated using paired 2-tailed t tests between shluc and shEP300-infected populations. Only p values ≤ 0.05 are indicated. ***p = 0.0004 (shEP300 #5), 0.0001 (shEP300 #3). F, shEP300 lentiviruses inhibit growth of MGH7 cells. Cells were infected overnight with shEP300 and shluc control lentiviruses and selected in puromycin after 24 h for an additional 24 h. Selected cells were then seeded in replicate for growth assays. Following 7–10 days of culture, cell growth was quantified by alamarBlue. Data are plotted relative to control shluc-infected cells, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. ***p = 0.0004 (shEP300 #5), 5 × 10−8 (shEP300 #3).
Fig. 12.

EP300 promotes growth of ChaGoK1 lung cancer cells with 3q gains. A, An shEP300 lentivirus reduces EP300, but not CREBBP expression in ChaGoK1 cells. Cells were infected overnight with shEP300 #3 or shluciferase (shluc) control lentiviruses and at 24 h, selected in puromycin. At 48 h post-virus addition, EP300 and CREBBP expression was quantified by Western blotting and qRT-PCR. For Western blotting, protein levels were quantified by densitometry, with EP300 and CREBBP expression normalized to PTPN11 levels (loading control). Normalized EP300 and CREBBP expression was then adjusted relative to shluc-transfected cells, which was assigned a value of 100. NS = nonspecific band. For qRT-PCR, EP300 and CREBBP expression was normalized to levels of TBP and plotted relative to shluc, which was assigned a value of 1.0. Means ± S.E. of 3 replicates are shown. Significance was calculated using paired 2-tailed t tests between shluc and shEP300-infected populations. **p = 0.005 and *p = 0.03. B, An shEP300 lentivirus inhibits growth of ChaGoK1 cells. Cells were infected overnight with shEP300 #3 and shluc control lentiviruses and selected in puromycin after 24 h for an additional 24 h. Selected cells were then seeded in replicate for growth assays. Following 7–10 days of culture, cell growth was quantified by alamarBlue. Data are plotted relative to control shluc-infected cells, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. ***p = 1 × 10−6.
An EP300 Chemical Inhibitor Suppresses Growth of Lung Cancer Cell Lines With 3q Gains and Increases Sensitivity to a PI3K Inhibitor
We next explored whether EP300 chemical inhibitors suppress growth of lung cancer cells with 3q copy number gains. At 20 μm, CBP30 impaired growth of both lung cancer cell lines with 3q gains by almost 60% (MGH7, ChaGoK1), whereas growth of SW1573 cells, which do not have 3q gains, was only reduced by 14% (Fig. 13A). Because of the recent failure of the PI3K inhibitor BKM120 in a lung SQCC clinical trial, it was proposed that PI3K inhibitors might be more effective if combined with other targeted therapeutics (41). We therefore, tested if growth suppression by a low dose of BKM120 could be enhanced by simultaneous treatment with CBP30. Indeed, combination of 0.5 μm BKM120 with 20 μm CBP30 impaired growth of the two cell lines with 3q copy number gains more strongly than single agent treatment (Fig. 13B). The combination treatment strategy was not effective in SW1573 cells (Fig. 13B). Thus, CBP30 recapitulates the sensitivity of lung cancer cells with 3q copy number gains to EP300 genetic inhibition, and enhances sensitivity of these cells to low doses of a PI3K inhibitor.
Fig. 13.

An EP300 chemical inhibitor suppresses growth of lung cancer cell lines with 3q gains and increases sensitivity to a PI3K inhibitor. A, Single agent EP300 chemical inhibitor treatment suppresses growth of lung cancer cell lines with 3q gains. Cells were fed every other day with the indicated CBP30 concentration and growth quantified after 7–10 days by alamarBlue. Data are plotted relative to control vehicle-treated cells, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. Unless otherwise indicated, p values are relative to respective DMSO-treated control cultures. MGH7, ***p = 0.0001 (10 μm CBP30), 1.9 × 10−6 (20 μm CBP30); ChaGoK1, ***p = 3.5 × 10−6 (10 μm CBP30), 1.4 × 10−5 (20 μm CBP30); SW1573, **p = 0.0007 (10 μm CBP30), 0.001 (20 μm CBP30); ***p = 7.1 × 10−6 (20 μm CBP30/MGH7 versus 20 μm CBP30/SW1573), 6.7 × 10−5 (20 μm CBP30/ChaGoK1 versus 20 μm CBP30/SW1573). B, At low doses, combination of PI3K and EP300 chemical inhibitors results in more growth inhibition than either single agent alone. Growth assays were performed as described in (A). Significance was calculated using a 2-tailed t test. ns = not significant. MGH7, **p = 0.001, ***p = 5 × 10−5; ChaGoK1, *p = 0.009, **p = 0.005, SW1573, ns, p = 0.40 (to CBP30 alone), 0.17 (to BKM120 alone).
Tracheobronchial Basal Cell Growth is Dependent on EP300, but Quiescence Reduces Toxicity of EP300 Inhibitors
Because many SQCCs are likely to originate from TP63-expressing basal cells, some of the sensitivity of SQCCs to EP300 inhibition may reflect an inherent dependence of the putative cell of origin on EP300 levels. To test this possibility, we measured long-term growth in tracheobronchial basal cell cultures that had been infected with one of the shEP300 lentiviruses. After 7–10 days of shEP300 infection, all basal cells had died (Fig. 14A). This extreme sensitivity to EP300 inhibition was recapitulated by CBP30 treatment, with basal cell growth reduced to 38% when exposed to 10 μm CBP30 (Fig. 14B). By contrast, it has been reported that 10 μm CBP30 is not profoundly toxic in 12 different normal primary cell types (101). However, in the healthy native tracheobronchial epithelium, most basal cells are quiescent, with only 1.7% expressing Ki-67, a marker of cycling cells (123). We therefore tested whether EP300 inhibition would be equally toxic in the largely quiescent cell populations that are found under normal homeostatic conditions. To recapitulate these conditions, we grew basal cells on Transwell filters, where quiescence is induced at confluence (28). We then compared the effects of exposure to the combination of CBP30 and BKM120 between proliferating (plastic) and quiescent (Transwell) cultures. Over an 8-day treatment period, the drug combination reduced cell growth on plastic by 97%, but on Transwell filters, it reduced cell number by only 20% (Fig. 14C). Although these data are supportive, further studies will be required to determine if an appropriate therapeutic index might be attainable for this class or other classes of EP300 chemical inhibitors.
Fig. 14.

Quiescence reduces the toxicity of EP300 and PI3K chemical inhibitors in tracheobronchial basal cells. A, Knockdown of EP300 inhibits basal cell growth. Basal cells proliferating on plastic were infected overnight with shEP300 #3 and shluc control lentiviruses and selected in puromycin after 24 h for an additional 24 h. Selected cells were then seeded in replicate for growth assays. Following 7–10 days of culture, cell growth was quantified by alamarBlue. Data are plotted relative to control shluc-infected cells, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. ***p = 1 × 10−8. B, Single agent EP300 chemical inhibitor treatment suppresses basal cell growth. Cells were fed every other day with the indicated CBP30 concentration and growth quantified after 7–10 days by alamarBlue. Data are plotted relative to control vehicle-treated cells, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. ***p = 0.0002 (10 μm CBP30), 0.00004 (20 μm CBP30). C, Quiescence induced by growth on Transwell filters reduces toxicity of PI3K and EP300 chemical inhibitors on basal cells. Basal cells were cultured either subconfluently on plastic dishes or at confluence on Transwell filters, and were treated with fresh drugs every other day. After 8 days, growth was measured be either alamarBlue (plastic) or counting total cell number (Transwell filters). Data are plotted relative to cultures treated with control vehicle, which was assigned a value of 100. Means ± S.E. from triplicate cultures are shown. Significance was calculated using a 2-tailed t test. ***p = 9.1 × 10–7, *p = 0.03.
DISCUSSION
The major goals of this study were to determine if BioID could be used to characterize the SOX2 interactome and to gain insight into the mechanism and treatment of SOX2-driven SQCC pathogenesis. Here we report the first SOX2 interactome characterized by BioID. This interactome has some overlap with previously reported SOX2 AP-MS studies (37, 44–50), but also identifies 36 novel high-confidence interactions. An additional 15 interactions were largely unique to our data set as they were reported in only one other study where the maximum number of spectral counts for these interactors ranged between 1 and 2 (37). The additional interactors identified in our study likely reflect bona fide SOX2-interactions because a number of both common and novel interactors have known connections to SOX2 activity, including EP300, which we functionally validated outside of an ES cell context for the first time. In addition, we found that the novel interaction with ARID1B could be recapitulated by PLA. Our data thus support BioID as an effective method to interrogate SOX2 interactomes.
We also characterized roles of EP300 in early SQCC pathogenesis and growth of invasive carcinoma, processes that are regulated by SOX2. Previous work proposed contrasting functions for EP300 on SOX2 activity. Recombinant EP300 protein has been shown to acetylate SOX2 on many lysine residues in vitro, with an alanine substitution mutation at one of the lysine residues, K75A, inhibiting cytoplasmic accumulation of SOX2 in ES cells (106). Based on these findings, the authors of that study proposed that EP300 inhibits SOX2 activity through acetylation-dependent nuclear export. However, lysines can also acquire other modifications (e.g. ubiquitin), making it difficult to ascribe the effect of the K75A mutation solely to defective acetylation. Furthermore, it is not clear to what extent EP300 acetylates SOX2 in vivo, with one study suggesting that EP300 might only be a minor contributor (124). Thus, the extent to which this EP300 mechanism operates in distinct cell types is still unknown. Contrary to the inhibitor model, other studies support EP300 being a SOX2 coactivator. In ES cells, EP300 is colocalized with SOX2 at many enhancer regions (84), and it cooperates with SOX2 and OCT4 to promote transcriptional activation from an FGF4 enhancer that is active in these cells (107, 108). Our findings also support a coactivator over inhibitor role for EP300 in regulating SOX2 activity in basal cells. Inhibition of EP300 activity through both chemical and genetic methods suppresses SOX2-dependent transcriptional changes and squamous differentiation of basal cells. However, there appears to be mechanistic differences between ES and basal cells in how EP300 functions with SOX2. In ES cells, both OCT4 and NANOG are necessary for global recruitment of EP300 to SOX2 sites (84), and at least at the FGF4 enhancer, OCT4 is necessary for EP300 to promote transcriptional activation by SOX2 (108). By contrast, in SQCCs, the OCT4 binding motif is not enriched at SOX2 binding regions (37), and most SQCCs do not express OCT4 protein (125, 126). Thus, in basal cells and SQCCs, other factors such as TP63 may be involved in EP300 recruitment or SOX2 may be able to directly recruit EP300.
Although EP300 has been suggested to have both tumor suppressor and oncogenic activities (95), such putative functions have not been clearly established for most cancers. In the TCGA somatic cancer cohorts, EP300 is mutated at <10% frequency in most types of cancer, including lung SQCCs. At least one missense mutation affects an amino acid in the catalytic HAT domain (D1399N), that when altered to D1399Y (or D1435E in CREBBP), strongly reduces acetyl transferase activity (96, 127). However, in murine hematopoietic stem and progenitor cells, similar HAT inactivating mutations in EP300 are biological gain-of-functions with regards to growth of these cells (128). Thus, it remains to be seen whether in SQCCs, the EP300 mutations are passenger or oncogenic, or if they define a rare disease subclass where EP300 is a tumor suppressor. By contrast, EP300 gains are common in SQCCs, occurring in 45% of cases, which is the highest frequency among surveyed TCGA cohorts. SQCCs also have the highest frequency of SOX2 copy number gains, supporting co-selection of increases in both SOX2 and EP300 activities during SQCC pathogenesis. Consistent with this possibility, we find evidence for SOX2 and EP300 being in close physical proximity in basal cells and SQCCs. Furthermore, we find that EP300 is necessary for SOX2 activity in basal cells, including for the induction of the squamous injury response by SOX2, a key initiating event in SQCC pathogenesis. Also, we find that like SOX2 (17, 36, 37), EP300 promotes growth of at least some lung cancer cell lines with 3q gains.
EP300 is not generally required for all cell growth, as normal ES cells and some cancer cell lines still grow when EP300 is homozygously inactivated (129, 130). However, certain cancers are very dependent on EP300 for their growth. For example, EP300 chemical inhibitors that target either the bromodomain or the catalytic HAT domain suppress growth of AML1-ETO-driven AMLs (103, 105, 131). They also synergistically inhibit growth of some AML cell lines when combined with the chemotherapeutic doxorubicin (103). The growth inhibitory effect of these drugs is thought to arise from EP300 inhibition because EP300 acetylates AML1-ETO and is necessary for its leukemogenic activity (132). Our studies suggest that SOX2-driven SQCCs could be another EP300-dependent cancer. This dependence could arise through both the developmental origin of the cancer from basal cells, which are also very dependent on EP300 for growth, as well as the requirement of SOX2 for EP300 for some of its major functions during SQCC pathogenesis. Accordingly, it would be worthwhile to develop EP300 inhibitors for clinical trials involving SQCCs. Notably, some FDA-approved drugs that contain salicylate moieties inhibit EP300 catalytic activity, including the anti-inflammatory diflunisal, at concentrations that can be achieved in patients (105, 133). In addition, natural products including curcumin, anacardic acid, and luteolin, inhibit EP300 catalytic activity (97, 98, 104), with luteolin also being able to suppress growth of a head and neck SQCC xenograft in mice (104). As suggested by our work, compounds that target EP300 could have therapeutic benefit in lung SQCCs on their own or in conjunction with other treatments. Other treatments could include drugs, radiation, or agonists/antagonists of signaling receptors that we found affect expression of components of the SOX2 interactome, or specific targeted therapeutics such as PI3K inhibitors. Indeed, we found that an EP300 inhibitor increases the effectiveness of low dose of the PI3K inhibitor, BKM120, in suppressing growth of lung cancer cell lines with 3q gains. Although normal basal cell proliferation is also very sensitive to this drug combination, toxicity is significantly reduced under physiologic conditions where they are largely quiescent. Further work will be required to determine if a tolerable therapeutic index may be attainable for clinical grade EP300 inhibitors. Additionally, the growing number of natural products with EP300 inhibitory activity suggests potential chemoprevention strategies, which potentially can be combined with myo-inositol, a natural PI3K inhibitor that promoted regression of premalignant squamous lesions in a phase I clinical trial (134–136). In conclusion, our work supports the continued use of BioID to study SOX2, and suggests that it may be effective to identify druggable targets of a wide range of “nondruggable” oncoproteins.
DATA AVAILABILITY
All mass spectrometry data have been uploaded to the public MassIVE archive (www.massive.ucsd.edu) with ID: MSV000080175 (SOX2_BioID_Interactome).
Supplementary Material
Footnotes
Author contributions: B.K., E.C., B.R., and N.M. designed research; B.K., E.C., E.L., E.V., and N.M. performed research; M.S.T. contributed new reagents or analytic tools; B.K., E.C., J.S., B.R., and N.M. analyzed data; B.K., J.S., B.R., and N.M. wrote the paper.
* This research was supported by an Investigator Award (IA-006) from the Ontario Institute for Cancer Research (http://oicr.on.ca/) to NM, ORF-RE-03-020 from the Ontario Ministry of Research and Innovation (https://www.ontario.ca/ministry-research-and-innovation), Canadian Cancer Society Research Institute grant #701595 (CCSRI, http://www.cancer.ca/research), and the Canadian Institutes of Health Research (CIHR, http://www.cihr-irsc.gc.ca/e/193.html) grant MOP137136 to MST. Work in the BR lab was supported by CIHR operating grants MOP130340 and MOP119289. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SQCC
- squamous cell carcinoma
- ADC
- adenocarcinoma
- AP-MS
- affinity purification-mass spectrometry
- SAINT
- significance analysis of the interactome
- FDR
- false discovery rate
- TCGA
- The Cancer Genome Atlas
- PI3K
- phosphatidylinositol-4,5-bisphosphate 3-kinase
- PDX
- primary patient-derived tumor xenograft
- PLA
- proximity ligation assay
- AML
- acute myeloid leukemia
- HAT
- histone acetyltransferase
- CCLE
- Cancer Cell Line Encyclopedia
- ATCC
- American Type Culture Collection
- ES
- embryonic stem.
REFERENCES
- 1. Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., and Jemal A. (2015) Global cancer statistics, 2012. CA. Cancer J. Clin. 65, 87–108 [DOI] [PubMed] [Google Scholar]
- 2. Ishizumi T., McWilliams A., MacAulay C., Gazdar A., and Lam S. (2010) Natural history of bronchial preinvasive lesions. Cancer Metastasis Rev. 29, 5–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Meza R., Meernik C., Jeon J., and Cote M. L. (2015) Lung cancer incidence trends by gender, race and histology in the United States, 1973–2010. PLoS ONE 10, e0121323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Langer C. J., Obasaju C., Bunn P., Bonomi P., Gandara D., Hirsch F. R., Kim E. S., Natale R. B., Novello S., Paz-Ares L., Perol M., Reck M., Ramalingam S. S., Reynolds C. H., Socinski M. A., Spigel D. R., Wakelee H., Mayo C., and Thatcher N. (2016) Incremental innovation and progress in advanced squamous cell lung cancer: current status and future impact of treatment. J. Thoracic Oncol. 11, 2066–2081 [DOI] [PubMed] [Google Scholar]
- 5. Au N. H., Gown A. M., Cheang M., Huntsman D., Yorida E., Elliott W. M., Flint J., English J., Gilks C. B., and Grimes H. L. (2004) P63 expression in lung carcinoma: a tissue microarray study of 408 cases. Appl. Immunohistochem. Mol. Morphol. 12, 240–247 [DOI] [PubMed] [Google Scholar]
- 6. Ocque R., Tochigi N., Ohori N. P., and Dacic S. (2011) Usefulness of immunohistochemical and histochemical studies in the classification of lung adenocarcinoma and squamous cell carcinoma in cytologic specimens. Am. J. Clin. Pathol. 136, 81–87 [DOI] [PubMed] [Google Scholar]
- 7. Engelhardt J. F., Schlossberg H., Yankaskas J. R., and Dudus L. (1995) Progenitor cells of the adult human airway involved in submucosal gland development. Development 121, 2031–2046 [DOI] [PubMed] [Google Scholar]
- 8. Hajj R., Baranek T., Le Naour R., Lesimple P., Puchelle E., and Coraux C. (2007) Basal cells of the human adult airway surface epithelium retain transit-amplifying cell properties. Stem Cells 25, 139–148 [DOI] [PubMed] [Google Scholar]
- 9. Kumar P. A., Hu Y., Yamamoto Y., Hoe N. B., Wei T. S., Mu D., Sun Y., Joo L. S., Dagher R., Zielonka E. M., Wang de Y., Lim B., Chow V. T., Crum C. P., Xian W., and McKeon F. (2011) Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rock J. R., Onaitis M. W., Rawlins E. L., Lu Y., Clark C. P., Xue Y., Randell S. H., and Hogan B. L. (2009) Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. U.S.A. 106, 12771–12775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Watson J. K., Rulands S., Wilkinson A. C., Wuidart A., Ousset M., Van Keymeulen A., Gottgens B., Blanpain C., Simons B. D., and Rawlins E. L. (2015) Clonal Dynamics Reveal Two Distinct Populations of Basal Cells in Slow-Turnover Airway Epithelium. Cell Reports 12, 90–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dupuit F., Gaillard D., Hinnrasky J., Mongodin E., de Bentzmann S., Copreni E., and Puchelle E. (2000) Differentiated and functional human airway epithelium regeneration in tracheal xenografts. Am. J. Physiol. Lung Cell Mol Physiol 278, L165–L176 [DOI] [PubMed] [Google Scholar]
- 13. Lee J. J., Liu D., Lee J. S., Kurie J. M., Khuri F. R., Ibarguen H., Morice R. C., Walsh G., Ro J. Y., Broxson A., Hong W. K., and Hittelman W. N. (2001) Long-term impact of smoking on lung epithelial proliferation in current and former smokers. J. Natl. Cancer Inst. 93, 1081–1088 [DOI] [PubMed] [Google Scholar]
- 14. Peters E. J., Morice R., Benner S. E., Lippman S., Lukeman J., Lee J. S., Ro J. Y., and Hong W. K. (1993) Squamous metaplasia of the bronchial mucosa and its relationship to smoking. Chest 103, 1429–1432 [DOI] [PubMed] [Google Scholar]
- 15. Tipton D. L., and Crocker T. T. (1964) Duration of bronchial squamous metaplasia produced in dogs by cigarette smoke condensate. J. Natl. Cancer Inst. 33, 487–495 [PubMed] [Google Scholar]
- 16. Willey J. C., Grafstrom R. C., Moser C. E. Jr, Ozanne C., Sundquvist K., and Harris C. C. (1987) Biochemical and morphological effects of cigarette smoke condensate and its fractions on normal human bronchial epithelial cells in vitro. Cancer Res. 47, 2045–2049 [PubMed] [Google Scholar]
- 17. Bass A. J., Watanabe H., Mermel C. H., Yu S., Perner S., Verhaak R. G., Kim S. Y., Wardwell L., Tamayo P., Gat-Viks I., Ramos A. H., Woo M. S., Weir B. A., Getz G., Beroukhim R., O'Kelly M., Dutt A., Rozenblatt-Rosen O., Dziunycz P., Komisarof J., Chirieac L. R., Lafargue C. J., Scheble V., Wilbertz T., Ma C., Rao S., Nakagawa H., Stairs D. B., Lin L., Giordano T. J., Wagner P., Minna J. D., Gazdar A. F., Zhu C. Q., Brose M. S., Cecconello I., Jr U. R., Marie S. K., Dahl O., Shivdasani R. A., Tsao M. S., Rubin M. A., Wong K. K., Regev A., Hahn W. C., Beer D. G., Rustgi A. K., and Meyerson M. (2009) SOX2 is an amplified lineage-survival oncogene in lung and esophageal squamous cell carcinomas. Nat. Genet. 41, 1238–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hammerman P. S., Lawrence M. S., Voet D., Jing R., Cibulskis K., Sivachenko A., Stojanov P., McKenna A., Lander E. S., Gabriel S., Getz G., Sougnez C., Imielinski M., Helman E., Hernandez B., Pho N. H., Meyerson M., Chu A., Chun H. J., Mungall A. J., Pleasance E., Gordon Robertson A., Sipahimalani P., Stoll D., Balasundaram M., Birol I., Butterfield Y. S., Chuah E., Coope R. J., Corbett R., Dhalla N., Guin R., He A., Hirst C., Hirst M., Holt R. A., Lee D., Li H. I., Mayo M., Moore R. A., Mungall K., Ming Nip K., Olshen A., Schein J. E., Slobodan J. R., Tam A., Thiessen N., Varhol R., Zeng T., Zhao Y., Jones S. J., Marra M. A., Saksena G., Cherniack A. D., Schumacher S. E., Tabak B., Carter S. L., Nguyen H., Onofrio R. C., Crenshaw A., Ardlie K., Beroukhim R., Winckler W., Protopopov A., Zhang J., Hadjipanayis A., Lee S., Xi R., Yang L., Ren X., Zhang H., Shukla S., Chen P. C., Haseley P., Lee E., Chin L., Park P. J., Kucherlapati R., Socci N. D., Liang Y., Schultz N., Borsu L., Lash A. E., Viale A., Sander C., Ladanyi M., Todd Auman J., Hoadley K. A., Wilkerson M. D., Shi Y., Liquori C., Meng S., Li L., Turman Y. J., Topal M. D., Tan D., Waring S., Buda E., Walsh J., Jones C. D., Mieczkowski P. A., Singh D., Wu J., Gulabani A., Dolina P., Bodenheimer T., Hoyle A. P., Simons J. V., Soloway M. G., Mose L. E., Jefferys S. R., Balu S., O'Connor B. D., Prins J. F., Liu J., Chiang D. Y., Neil Hayes D., Perou C. M., Cope L., Danilova L., Weisenberger D. J., Maglinte D. T., Pan F., Van Den Berg D. J., Triche T. Jr., Herman J. G., Baylin S. B., Laird P. W., Noble M., Gehlenborg N., Dicara D., Wu C. J., Yingchun Liu S., Zou L., Lin P., Cho J., Nazaire M. D., Robinson J., Thorvaldsdottir H., Mesirov J., Sinha R., Ciriello G., Cerami E., Gross B., Jacobsen A., Gao J., Arman Aksoy B., Weinhold N., Ramirez R., Taylor B. S., Antipin Y., Reva B., Shen R., Mo Q., Seshan V., Paik P. K., Akbani R., Zhang N., Broom B. M., Casasent T., Unruh A., Wakefield C., Craig Cason R., Baggerly K. A., Weinstein J. N., Haussler D., Benz C. C., Stuart J. M., Zhu J., Szeto C., Scott G. K., Yau C., Ng S., Goldstein T., Waltman P., Sokolov A., Ellrott K., Collisson E. A., Zerbino D., Wilks C., Ma S., Craft B., Du Y., Cabanski C., Walter V., Marron J. S., Liu Y., Wang K., Creighton C. J., Zhang Y., Travis W. D., Rekhtman N., Yi J., Aubry M. C., Cheney R., Dacic S., Flieder D., Funkhouser W., Illei P., Myers J., Tsao M. S., Penny R., Mallery D., Shelton T., Hatfield M., Morris S., Yena P., Shelton C., Sherman M., Paulauskis J., Govindan R., Azodo I., Beer D., Bose R., Byers L. A., Carbone D., Chang L. W., Chiang D., Chun E., Collisson E., Ding L., Heymach J., Ida C., Johnson B., Jurisica I., Kaufman J., Kosari F., Kwiatkowski D., Maher C. A., Mungall A., Pao W., Peifer M., Robertson G., Rusch V., Siegfried J., Stuart J., Thomas R. K., Tomaszek S., Vaske C., Weisenberger D., Wigle D. A., Yang P., John Zhang J., Jensen M. A., Sfeir R., Kahn A. B., Chu A. L., Kothiyal P., Wang Z., Snyder E. E., Pontius J., Pihl T. D., Ayala B., Backus M., Walton J., Baboud J., Berton D. L., Nicholls M. C., Srinivasan D., Raman R., Girshik S., Kigonya P. A., Alonso S., Sanbhadti R. N., Barletta S. P., Greene J. M., Pot D. A., Bandarchi-Chamkhaleh B., Boyd J., Weaver J., Azodo I. A., Tomaszek S. C., Christine Aubry M., Ida C. M., Brock M. V., Rogers K., Rutledge M., Brown T., Lee B., Shin J., Trusty D., Dhir R., Siegfried J. M., Potapova O., Fedosenko K. V., Nemirovich-Danchenko E., Zakowski M., Iacocca M. V., Brown J., Rabeno B., Czerwinski C., Petrelli N., Fan Z., Todaro N., Eckman J., Kimryn Rathmell W., Thorne L. B., Huang M., Boice L., Hill A., Curley E., Morrison C., Gaudioso C., Bartlett J. M., Kodeeswaran S., Zanke B., Sekhon H., David K., Juhl H., Van Le X., Kohl B., Thorp R., Viet Tien N., Van Bang N., Sussman H., Duc Phu B., Hajek R., Phi Hung N., Khan K. Z., Muley T., Mills Shaw K. R., Sheth M., Buetow K., Davidsen T., Demchok J. A., Eley G., Ferguson M., Dillon L. A., Schaefer C., Guyer M. S., Ozenberger B. A., Palchik J. D., Peterson J., Sofia H. J., Thomson E., and Johnson B. E. (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McCaughan F., Pole J. C., Bankier A. T., Konfortov B. A., Carroll B., Falzon M., Rabbitts T. H., George P. J., Dear P. H., and Rabbitts P. H. (2010) Progressive 3q amplification consistently targets SOX2 in preinvasive squamous lung cancer. Am. J. Respir. Crit. Care Med. 182, 83–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schneider F., Luvison A., Cieply K., and Dacic S. (2013) Sex-determining region Y-box 2 amplification in preneoplastic squamous lesions of the lung. Hum. Pathol. 44, 706–711 [DOI] [PubMed] [Google Scholar]
- 21. Bota S., Auliac J. B., Paris C., Metayer J., Sesboue R., Nouvet G., and Thiberville L. (2001) Follow-up of bronchial precancerous lesions and carcinoma in situ using fluorescence endoscopy. Am. J. Respir. Crit. Care Med. 164, 1688–1693 [DOI] [PubMed] [Google Scholar]
- 22. Hoshino H., Shibuya K., Chiyo M., Iyoda A., Yoshida S., Sekine Y., Iizasa T., Saitoh Y., Baba M., Hiroshima K., Ohwada H., and Fujisawa T. (2004) Biological features of bronchial squamous dysplasia followed up by autofluorescence bronchoscopy. Lung Cancer 46, 187–196 [DOI] [PubMed] [Google Scholar]
- 23. Pasic A., van Vliet E., Breuer R. H., Risse E. J., Snijders P. J., Postmus P. E., and Sutedja T. G. (2004) Smoking behavior does not influence the natural course of pre-invasive lesions in bronchial mucosa. Lung Cancer 45, 153–154 [DOI] [PubMed] [Google Scholar]
- 24. Breuer R. H., Pasic A., Smit E. F., van Vliet E., Vonk Noordegraaf A., Risse E. J., Postmus P. E., and Sutedja T. G. (2005) The natural course of preneoplastic lesions in bronchial epithelium. Clin. Cancer Res. 11, 537–543 [PubMed] [Google Scholar]
- 25. Jeremy George P., Banerjee A. K., Read C. A., O'Sullivan C., Falzon M., Pezzella F., Nicholson A. G., Shaw P., Laurent G., and Rabbitts P. H. (2007) Surveillance for the detection of early lung cancer in patients with bronchial dysplasia. Thorax 62, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lam S., leRiche J. C., Zheng Y., Coldman A., MacAulay C., Hawk E., Kelloff G., and Gazdar A. F. (1999) Sex-related differences in bronchial epithelial changes associated with tobacco smoking. J. Natl. Cancer Inst. 91, 691–696 [DOI] [PubMed] [Google Scholar]
- 27. van Boerdonk R. A., Smesseim I., Heideman D. A., Coupe V. M., Tio D., Grunberg K., Thunnissen E., Snijders P. J., Postmus P. E., Smit E. F., Daniels J. M., and Sutedja T. G. (2015) Close Surveillance with Long-Term Follow-up of Subjects with Preinvasive Endobronchial Lesions. Am. J. Respir. Crit. Care Med. 192, 1483–1489 [DOI] [PubMed] [Google Scholar]
- 28. Kim B. R., Van de Laar E., Cabanero M., Tarumi S., Hasenoeder S., Wang D., Virtanen C., Suzuki T., Bandarchi B., Sakashita S., Pham N. A., Lee S., Keshavjee S., Waddell T. K., Tsao M. S., and Moghal N. (2016) SOX2 and PI3K Cooperate to Induce and Stabilize a Squamous-Committed Stem Cell Injury State during Lung Squamous Cell Carcinoma Pathogenesis. PLos Biol. 14, e1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buchwalter G., Hickey M. M., Cromer A., Selfors L. M., Gunawardane R. N., Frishman J., Jeselsohn R., Lim E., Chi D., Fu X., Schiff R., Brown M., and Brugge J. S. (2013) PDEF promotes luminal differentiation and acts as a survival factor for ER-positive breast cancer cells. Cancer Cell 23, 753–767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chi P., Chen Y., Zhang L., Guo X., Wongvipat J., Shamu T., Fletcher J. A., Dewell S., Maki R. G., Zheng D., Antonescu C. R., Allis C. D., and Sawyers C. L. (2010) ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature 467, 849–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin L., Bass A. J., Lockwood W. W., Wang Z., Silvers A. L., Thomas D. G., Chang A. C., Lin J., Orringer M. B., Li W., Glover T. W., Giordano T. J., Lam W. L., Meyerson M., and Beer D. G. (2012) Activation of GATA binding protein 6 (GATA6) sustains oncogenic lineage-survival in esophageal adenocarcinoma. Proc. Natl. Acad. Sci. U.S.A. 109, 4251–4256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. McGill G. G., Horstmann M., Widlund H. R., Du J., Motyckova G., Nishimura E. K., Lin Y. L., Ramaswamy S., Avery W., Ding H. F., Jordan S. A., Jackson I. J., Korsmeyer S. J., Golub T. R., and Fisher D. E. (2002) Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell 109, 707–718 [DOI] [PubMed] [Google Scholar]
- 33. Salari K., Spulak M. E., Cuff J., Forster A. D., Giacomini C. P., Huang S., Ko M. E., Lin A. Y., van de Rijn M., and Pollack J. R. (2012) CDX2 is an amplified lineage-survival oncogene in colorectal cancer. Proc. Natl. Acad. Sci. U.S.A. 109, E3196–E3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanaka H., Yanagisawa K., Shinjo K., Taguchi A., Maeno K., Tomida S., Shimada Y., Osada H., Kosaka T., Matsubara H., Mitsudomi T., Sekido Y., Tanimoto M., Yatabe Y., and Takahashi T. (2007) Lineage-specific dependency of lung adenocarcinomas on the lung development regulator TTF-1. Cancer Res. 67, 6007–6011 [DOI] [PubMed] [Google Scholar]
- 35. Weir B. A., Woo M. S., Getz G., Perner S., Ding L., Beroukhim R., Lin W. M., Province M. A., Kraja A., Johnson L. A., Shah K., Sato M., Thomas R. K., Barletta J. A., Borecki I. B., Broderick S., Chang A. C., Chiang D. Y., Chirieac L. R., Cho J., Fujii Y., Gazdar A. F., Giordano T., Greulich H., Hanna M., Johnson B. E., Kris M. G., Lash A., Lin L., Lindeman N., Mardis E. R., McPherson J. D., Minna J. D., Morgan M. B., Nadel M., Orringer M. B., Osborne J. R., Ozenberger B., Ramos A. H., Robinson J., Roth J. A., Rusch V., Sasaki H., Shepherd F., Sougnez C., Spitz M. R., Tsao M. S., Twomey D., Verhaak R. G., Weinstock G. M., Wheeler D. A., Winckler W., Yoshizawa A., Yu S., Zakowski M. F., Zhang Q., Beer D. G., Wistuba I. I., Watson M. A., Garraway L. A., Ladanyi M., Travis W. D., Pao W., Rubin M. A., Gabriel S. B., Gibbs R. A., Varmus H. E., Wilson R. K., Lander E. S., and Meyerson M. (2007) Characterizing the cancer genome in lung adenocarcinoma. Nature 450, 893–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fukazawa T., Guo M., Ishida N., Yamatsuji T., Takaoka M., Yokota E., Haisa M., Miyake N., Ikeda T., Okui T., Takigawa N., Maeda Y., and Naomoto Y. (2016) SOX2 suppresses CDKN1A to sustain growth of lung squamous cell carcinoma. Scientific Reports 6, 20113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Watanabe H., Ma Q., Peng S., Adelmant G., Swain D., Song W., Fox C., Francis J. M., Pedamallu C. S., Deluca D. S., Brooks A. N., Wang S., Que J., Rustgi A. K., Wong K. K., Ligon K. L., Liu X. S., Marto J. A., Meyerson M., and Bass A. J. (2014) SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J. Clin. Invest. 124, 1636–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Justilien V., Walsh M. P., Ali S. A., Thompson E. A., Murray N. R., and Fields A. P. (2014) The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 25, 139–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dalmau E., Armengol-Alonso A., Munoz M., and Segui-Palmer M. A. (2014) Current status of hormone therapy in patients with hormone receptor positive (HR+) advanced breast cancer. Breast 23, 710–720 [DOI] [PubMed] [Google Scholar]
- 40. Foley C., and Mitsiades N. (2016) Moving beyond the androgen receptor (AR): targeting AR-interacting proteins to treat prostate cancer. Hormones Cancer 7, 84–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Vansteenkiste J. F., Canon J. L., Braud F. D., Grossi F., De Pas T., Gray J. E., Su W. C., Felip E., Yoshioka H., Gridelli C., Dy G. K., Thongprasert S., Reck M., Aimone P., Vidam G. A., Roussou P., Wang Y. A., Di Tomaso E., and Soria J. C. (2015) Safety and efficacy of buparlisib (BKM120) in patients with PI3K pathway-activated non-small cell lung cancer: results from the phase II BASALT-1 study. J. Thoracic Oncol. 10, 1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Andreeff M., Kelly K. R., Yee K., Assouline S., Strair R., Popplewell L., Bowen D., Martinelli G., Drummond M. W., Vyas P., Kirschbaum M., Iyer S. P., Ruvolo V., Gonzalez G. M., Huang X., Chen G., Graves B., Blotner S., Bridge P., Jukofsky L., Middleton S., Reckner M., Rueger R., Zhi J., Nichols G., and Kojima K. (2016) Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 22, 868–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zinzani P. L., Bonthapally V., Huebner D., Lutes R., Chi A., and Pileri S. (2016) Panoptic clinical review of the current and future treatment of relapsed/refractory T-cell lymphomas: Cutaneous T-cell lymphomas. Crit. Rev. Oncol. Hematol. 99, 228–240 [DOI] [PubMed] [Google Scholar]
- 44. Cox J. L., Wilder P. J., Gilmore J. M., Wuebben E. L., Washburn M. P., and Rizzino A. (2013) The SOX2-interactome in brain cancer cells identifies the requirement of MSI2 and USP9X for the growth of brain tumor cells. PLoS ONE 8, e62857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ding J., Huang X., Shao N., Zhou H., Lee D. F., Faiola F., Fidalgo M., Guallar D., Saunders A., Shliaha P. V., Wang H., Waghray A., Papatsenko D., Sanchez-Priego C., Li D., Yuan Y., Lemischka I. R., Shen L., Kelley K., Deng H., Shen X., and Wang J. (2015) Tex10 coordinates epigenetic control of super-enhancer activity in pluripotency and reprogramming. Cell Stem Cell 16, 653–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Engelen E., Akinci U., Bryne J. C., Hou J., Gontan C., Moen M., Szumska D., Kockx C., van Ijcken W., Dekkers D. H., Demmers J., Rijkers E. J., Bhattacharya S., Philipsen S., Pevny L. H., Grosveld F. G., Rottier R. J., Lenhard B., and Poot R. A. (2011) Sox2 cooperates with Chd7 to regulate genes that are mutated in human syndromes. Nat. Genet. 43, 607–611 [DOI] [PubMed] [Google Scholar]
- 47. Fang X., Yoon J. G., Li L., Tsai Y. S., Zheng S., Hood L., Goodlett D. R., Foltz G., and Lin B. (2011) Landscape of the SOX2 protein-protein interactome. Proteomics 11, 921–934 [DOI] [PubMed] [Google Scholar]
- 48. Gao Z., Cox J. L., Gilmore J. M., Ormsbee B. D., Mallanna S. K., Washburn M. P., and Rizzino A. (2012) Determination of protein interactome of transcription factor Sox2 in embryonic stem cells engineered for inducible expression of four reprogramming factors. J. Biol. Chem. 287, 11384–11397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mallanna S. K., Ormsbee B. D., Iacovino M., Gilmore J. M., Cox J. L., Kyba M., Washburn M. P., and Rizzino A. (2010) Proteomic analysis of Sox2-associated proteins during early stages of mouse embryonic stem cell differentiation identifies Sox21 as a novel regulator of stem cell fate. Stem Cells 28, 1715–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Myers S. A., Peddada S., Chatterjee N., Friedrich T., Tomoda K., Krings G., Thomas S., Maynard J., Broeker M., Thomson M., Pollard K., Yamanaka S., Burlingame A. L., and Panning B. (2016) SOX2 O-GlcNAcylation alters its protein-protein interactions and genomic occupancy to modulate gene expression in pluripotent cells. eLife 5, e10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roux K. J., Kim D. I., Raida M., and Burke B. (2012) A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 196, 801–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Comartin D., Gupta G. D., Fussner E., Coyaud E., Hasegan M., Archinti M., Cheung S. W., Pinchev D., Lawo S., Raught B., Bazett-Jones D. P., Luders J., and Pelletier L. (2013) CEP120 and SPICE1 cooperate with CPAP in centriole elongation. Curr. Biol. 23, 1360–1366 [DOI] [PubMed] [Google Scholar]
- 53. Couzens A. L., Knight J. D., Kean M. J., Teo G., Weiss A., Dunham W. H., Lin Z. Y., Bagshaw R. D., Sicheri F., Pawson T., Wrana J. L., Choi H., and Gingras A. C. (2013) Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 6, rs15. [DOI] [PubMed] [Google Scholar]
- 54. Coyaud E., Mis M., Laurent E. M., Dunham W. H., Couzens A. L., Robitaille M., Gingras A. C., Angers S., and Raught B. (2015) BioID-based identification of Skp cullin F-box (SCF)beta-TrCP1/2 E3 ligase substrates. Mol. Cell. Proteomics 14, 1781–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dingar D., Kalkat M., Chan P. K., Srikumar T., Bailey S. D., Tu W. B., Coyaud E., Ponzielli R., Kolyar M., Jurisica I., Huang A., Lupien M., Penn L. Z., and Raught B. (2014) BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. J. Proteomics 118, 95–111 [DOI] [PubMed] [Google Scholar]
- 56. Gupta G. D., Coyaud E., Goncalves J., Mojarad B. A., Liu Y., Wu Q., Gheiratmand L., Comartin D., Tkach J. M., Cheung S. W., Bashkurov M., Hasegan M., Knight J. D., Lin Z. Y., Schueler M., Hildebrandt F., Moffat J., Gingras A. C., Raught B., and Pelletier L. (2015) A dynamic protein interaction landscape of the human centrosome-cilium interface. Cell 163, 1484–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lambert J. P., Tucholska M., Go C., Knight J. D., and Gingras A. C. (2015) Proximity biotinylation and affinity purification are complementary approaches for the interactome mapping of chromatin-associated protein complexes. J. Proteomics 118, 81–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. St-Denis N., Gupta G. D., Lin Z. Y., Gonzalez-Badillo B., Veri A. O., Knight J. D., Rajendran D., Couzens A. L., Currie K. W., Tkach J. M., Cheung S. W., Pelletier L., and Gingras A. C. (2016) Phenotypic and interaction profiling of the human phosphatases identifies diverse mitotic regulators. Cell Reports 17, 2488–2501 [DOI] [PubMed] [Google Scholar]
- 59. Kessner D., Chambers M., Burke R., Agus D., and Mallick P. (2008) ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Craig R., and Beavis R. C. (2004) TANDEM: matching proteins with tandem mass spectra. Bioinformatics 20, 1466–1467 [DOI] [PubMed] [Google Scholar]
- 61. Pedrioli P. G. (2010) Trans-proteomic pipeline: a pipeline for proteomic analysis. Methods Mol. Biol. 604, 213–238 [DOI] [PubMed] [Google Scholar]
- 62. Liu G., Zhang J., Larsen B., Stark C., Breitkreutz A., Lin Z. Y., Breitkreutz B. J., Ding Y., Colwill K., Pasculescu A., Pawson T., Wrana J. L., Nesvizhskii A. I., Raught B., Tyers M., and Gingras A. C. (2010) ProHits: integrated software for mass spectrometry-based interaction proteomics. Nat. Biotechnol. 28, 1015–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 64. Choi H., Larsen B., Lin Z. Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z. S., Tyers M., Gingras A. C., and Nesvizhskii A. I. (2011) SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Teo G., Liu G., Zhang J., Nesvizhskii A. I., Gingras A. C., and Choi H. (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J. Proteomics 100, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Blake J. A., Dolan M., Drabkin H., Hill D. P., Li N., Sitnikov D., Bridges S., Burgess S., Buza T., McCarthy F., Peddinti D., Pillai L., Carbon S., Dietze H., Ireland A., Lewis S. E., Mungall C. J., Gaudet P., Chrisholm R. L., Fey P., Kibbe W. A., Basu S., Siegele D. A., McIntosh B. K., Renfro D. P., Zweifel A. E., Hu J. C., Brown N. H., Tweedie S., Alam-Faruque Y., Apweiler R., Auchinchloss A., Axelsen K., Bely B., Blatter M., Bonilla C., Bouguerleret L., Boutet E., Breuza L., Bridge A., Chan W. M., Chavali G., Coudert E., Dimmer E., Estreicher A., Famiglietti L., Feuermann M., Gos A., Gruaz-Gumowski N., Hieta R., Hinz C., Hulo C., Huntley R., James J., Jungo F., Keller G., Laiho K., Legge D., Lemercier P., Lieberherr D., Magrane M., Martin M. J., Masson P., Mutowo-Muellenet P., O'Donovan C., Pedruzzi I., Pichler K., Poggioli D., Porras Millan P., Poux S., Rivoire C., Roechert B., Sawford T., Schneider M., Stutz A., Sundaram S., Tognolli M., Xenarios I., Foulgar R., Lomax J., Roncaglia P., Khodiyar V. K., Lovering R. C., Talmud P. J., Chibucos M., Giglio M. G., Chang H., Hunter S., McAnulla C., Mitchell A., Sangrador A., Stephan R., Harris M. A., Oliver S. G., Rutherford K., Wood V., Bahler J., Lock A., Kersey P. J., McDowall D. M., Staines D. M., Dwinell M., Shimoyama M., Laulederkind S., Hayman T., Wang S., Petri V., Lowry T., D'Eustachio P., Matthews L., Balakrishnan R., Binkley G., Cherry J. M., Costanzo M. C., Dwight S. S., Engel S. R., Fisk D. G., Hitz B. C., Hong E. L., Karra K., Miyasato S. R., Nash R. S., Park J., Skrzypek M. S., Weng S., Wong E. D., Berardini T. Z., Huala E., Mi H., Thomas P. D., Chan J., Kishore R., Sternberg P., Van Auken K., Howe D., and Westerfield M. (2013) Gene Ontology annotations and resources. Nucleic Acids Res. 41, D530–D535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mi H., Muruganujan A., Casagrande J. T., and Thomas P. D. (2013) Large-scale gene function analysis with the PANTHER classification system. Nat. Protocols 8, 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Mootha V. K., Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., and Groop L. C. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 [DOI] [PubMed] [Google Scholar]
- 69. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matsui H., Randell S. H., Peretti S. W., Davis C. W., and Boucher R. C. (1998) Coordinated clearance of periciliary liquid and mucus from airway surfaces. J. Clin. Invest. 102, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Liu C., and Tsao M. S. (1993) Proto-oncogene and growth factor/receptor expression in the establishment of primary human non-small cell lung carcinoma cell lines. Am. J. Pathol. 142, 413–423 [PMC free article] [PubMed] [Google Scholar]
- 73. John T., Kohler D., Pintilie M., Yanagawa N., Pham N. A., Li M., Panchal D., Hui F., Meng F., Shepherd F. A., and Tsao M. S. (2011) The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin. Cancer Res. 17, 134–141 [DOI] [PubMed] [Google Scholar]
- 74. Wang D., Pham N. A., Tong J., Sakashita S., Allo G., Kim L., Yanagawa N., Raghavan V., Wei Y., To C., Trinh Q. M., Starmans M. H., Chan-Seng-Yue M. A., Chadwick D., Li L., Zhu C. Q., Liu N., Li M., Lee S., Ignatchenko V., Strumpf D., Taylor P., Moghal N., Liu G., Boutros P. C., Kislinger T., Pintilie M., Jurisica I., Shepherd F. A., McPherson J. D., Muthuswamy L., Moran M. F., and Tsao M. S. (2017) Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int. J. Cancer 140, 662–673 [DOI] [PubMed] [Google Scholar]
- 75. Uhlen M., Bjorling E., Agaton C., Szigyarto C. A., Amini B., Andersen E., Andersson A. C., Angelidou P., Asplund A., Asplund C., Berglund L., Bergstrom K., Brumer H., Cerjan D., Ekstrom M., Elobeid A., Eriksson C., Fagerberg L., Falk R., Fall J., Forsberg M., Bjorklund M. G., Gumbel K., Halimi A., Hallin I., Hamsten C., Hansson M., Hedhammar M., Hercules G., Kampf C., Larsson K., Lindskog M., Lodewyckx W., Lund J., Lundeberg J., Magnusson K., Malm E., Nilsson P., Odling J., Oksvold P., Olsson I., Oster E., Ottosson J., Paavilainen L., Persson A., Rimini R., Rockberg J., Runeson M., Sivertsson A., Skollermo A., Steen J., Stenvall M., Sterky F., Stromberg S., Sundberg M., Tegel H., Tourle S., Wahlund E., Walden A., Wan J., Wernerus H., Westberg J., Wester K., Wrethagen U., Xu L. L., Hober S., and Ponten F. (2005) A human protein atlas for normal and cancer tissues based on antibody proteomics. Mol. Cell. Proteomics 4, 1920–1932 [DOI] [PubMed] [Google Scholar]
- 76. Uhlen M., Fagerberg L., Hallstrom B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., Odeberg J., Djureinovic D., Takanen J. O., Hober S., Alm T., Edqvist P. H., Berling H., Tegel H., Mulder J., Rockberg J., Nilsson P., Schwenk J. M., Hamsten M., von Feilitzen K., Forsberg M., Persson L., Johansson F., Zwahlen M., von Heijne G., Nielsen J., and Ponten F. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 77. Uhlen M., Oksvold P., Fagerberg L., Lundberg E., Jonasson K., Forsberg M., Zwahlen M., Kampf C., Wester K., Hober S., Wernerus H., Bjorling L., and Ponten F. (2010) Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol. 28, 1248–1250 [DOI] [PubMed] [Google Scholar]
- 78. Cole A., Wang Z., Coyaud E., Voisin V., Gronda M., Jitkova Y., Mattson R., Hurren R., Babovic S., Maclean N., Restall I., Wang X., Jeyaraju D. V., Sukhai M. A., Prabha S., Bashir S., Ramakrishnan A., Leung E., Qia Y. H., Zhang N., Combes K. R., Ketela T., Lin F., Houry W. A., Aman A., Al-Awar R., Zheng W., Wienholds E., Xu C. J., Dick J., Wang J. C., Moffat J., Minden M. D., Eaves C. J., Bader G. D., Hao Z., Kornblau S. M., Raught B., and Schimmer A. D. (2015) Inhibition of the mitochondrial protease ClpP as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 27, 864–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Kondoh H., and Kamachi Y. (2010) SOX-partner code for cell specification: Regulatory target selection and underlying molecular mechanisms. Int. J. Biochem. Cell Biol. 42, 391–399 [DOI] [PubMed] [Google Scholar]
- 80. Lin Y. C., Boone M., Meuris L., Lemmens I., Van Roy N., Soete A., Reumers J., Moisse M., Plaisance S., Drmanac R., Chen J., Speleman F., Lambrechts D., Van de Peer Y., Tavernier J., and Callewaert N. (2014) Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nat. Commun. 5, 4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shaw G., Morse S., Ararat M., and Graham F. L. (2002) Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16, 869–871 [DOI] [PubMed] [Google Scholar]
- 82. Kagey M. H., Newman J. J., Bilodeau S., Zhan Y., Orlando D. A., van Berkum N. L., Ebmeier C. C., Goossens J., Rahl P. B., Levine S. S., Taatjes D. J., Dekker J., and Young R. A. (2010) Mediator and cohesin connect gene expression and chromatin architecture. Nature 467, 430–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nitzsche A., Paszkowski-Rogacz M., Matarese F., Janssen-Megens E. M., Hubner N. C., Schulz H., de Vries I., Ding L., Huebner N., Mann M., Stunnenberg H. G., and Buchholz F. (2011) RAD21 cooperates with pluripotency transcription factors in the maintenance of embryonic stem cell identity. PLoS ONE 6, e19470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chen X., Xu H., Yuan P., Fang F., Huss M., Vega V. B., Wong E., Orlov Y. L., Zhang W., Jiang J., Loh Y. H., Yeo H. C., Yeo Z. X., Narang V., Govindarajan K. R., Leong B., Shahab A., Ruan Y., Bourque G., Sung W. K., Clarke N. D., Wei C. L., and Ng H. H. (2008) Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 133, 1106–1117 [DOI] [PubMed] [Google Scholar]
- 85. Fang F., Xu Y., Chew K. K., Chen X., Ng H. H., and Matsudaira P. (2014) Coactivators p300 and CBP maintain the identity of mouse embryonic stem cells by mediating long-range chromatin structure. Stem Cells 32, 1805–1816 [DOI] [PubMed] [Google Scholar]
- 86. Singhal N., Graumann J., Wu G., Arauzo-Bravo M. J., Han D. W., Greber B., Gentile L., Mann M., and Scholer H. R. (2010) Chromatin-Remodeling Components of the BAF Complex Facilitate Reprogramming. Cell 141, 943–955 [DOI] [PubMed] [Google Scholar]
- 87. Card D. A., Hebbar P. B., Li L., Trotter K. W., Komatsu Y., Mishina Y., and Archer T. K. (2008) Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol. Cell. Biol. 28, 6426–6438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wang J., Park J. W., Drissi H., Wang X., and Xu R. H. (2014) Epigenetic regulation of miR-302 by JMJD1C inhibits neural differentiation of human embryonic stem cells. J. Biol. Chem. 289, 2384–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Gearhart M. D., Corcoran C. M., Wamstad J. A., and Bardwell V. J. (2006) Polycomb group and SCF ubiquitin ligases are found in a novel BCOR complex that is recruited to BCL6 targets. Mol. Cell. Biol. 26, 6880–6889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Fantes J., Ragge N. K., Lynch S. A., McGill N. I., Collin J. R., Howard-Peebles P. N., Hayward C., Vivian A. J., Williamson K., van Heyningen V., and FitzPatrick D. R. (2003) Mutations in SOX2 cause anophthalmia. Nat. Genet. 33, 461–463 [DOI] [PubMed] [Google Scholar]
- 91. Ng D., Thakker N., Corcoran C. M., Donnai D., Perveen R., Schneider A., Hadley D. W., Tifft C., Zhang L., Wilkie A. O., van der Smagt J. J., Gorlin R. J., Burgess S. M., Bardwell V. J., Black G. C., and Biesecker L. G. (2004) Oculofaciocardiodental and Lenz microphthalmia syndromes result from distinct classes of mutations in BCOR. Nat. Genet. 36, 411–416 [DOI] [PubMed] [Google Scholar]
- 92. Ben-Porath I., Thomson M. W., Carey V. J., Ge R., Bell G. W., Regev A., and Weinberg R. A. (2008) An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Soderberg O., Gullberg M., Jarvius M., Ridderstrale K., Leuchowius K. J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L. G., and Landegren U. (2006) Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 3, 995–1000 [DOI] [PubMed] [Google Scholar]
- 94. Luk C., Tsao M. S., Bayani J., Shepherd F., and Squire J. A. (2001) Molecular cytogenetic analysis of non-small cell lung carcinoma by spectral karyotyping and comparative genomic hybridization. Cancer Genet. Cytogenet. 125, 87–99 [DOI] [PubMed] [Google Scholar]
- 95. Wang F., Marshall C. B., and Ikura M. (2013) Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: structural and functional versatility in target recognition. Cell. Mol. Life Sci. 70, 3989–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Delvecchio M., Gaucher J., Aguilar-Gurrieri C., Ortega E., and Panne D. (2013) Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 20, 1040–1046 [DOI] [PubMed] [Google Scholar]
- 97. Balasubramanyam K., Swaminathan V., Ranganathan A., and Kundu T. K. (2003) Small molecule modulators of histone acetyltransferase p300. J. Biol. Chem. 278, 19134–19140 [DOI] [PubMed] [Google Scholar]
- 98. Balasubramanyam K., Varier R. A., Altaf M., Swaminathan V., Siddappa N. B., Ranga U., and Kundu T. K. (2004) Curcumin, a novel p300/CREB-binding protein-specific inhibitor of acetyltransferase, represses the acetylation of histone/nonhistone proteins and histone acetyltransferase-dependent chromatin transcription. J. Biol. Chem. 279, 51163–51171 [DOI] [PubMed] [Google Scholar]
- 99. Bowers E. M., Yan G., Mukherjee C., Orry A., Wang L., Holbert M. A., Crump N. T., Hazzalin C. A., Liszczak G., Yuan H., Larocca C., Saldanha S. A., Abagyan R., Sun Y., Meyers D. J., Marmorstein R., Mahadevan L. C., Alani R. M., and Cole P. A. (2010) Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 17, 471–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Conery A. R., Centore R. C., Neiss A., Keller P. J., Joshi S., Spillane K. L., Sandy P., Hatton C., Pardo E., Zawadzke L., Bommi-Reddy A., Gascoigne K. E., Bryant B. M., Mertz J. A., and Sims R. J. (2016) Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hammitzsch A., Tallant C., Fedorov O., O'Mahony A., Brennan P. E., Hay D. A., Martinez F. O., Al-Mossawi M. H., de Wit J., Vecellio M., Wells C., Wordsworth P., Muller S., Knapp S., and Bowness P. (2015) CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc. Natl. Acad. Sci. U.S.A. 112, 10768–10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hay D. A., Fedorov O., Martin S., Singleton D. C., Tallant C., Wells C., Picaud S., Philpott M., Monteiro O. P., Rogers C. M., Conway S. J., Rooney T. P., Tumber A., Yapp C., Filippakopoulos P., Bunnage M. E., Muller S., Knapp S., Schofield C. J., and Brennan P. E. (2014) Discovery and optimization of small-molecule ligands for the CBP/p300 bromodomains. J. Am. Chem. Soc. 136, 9308–9319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Picaud S., Fedorov O., Thanasopoulou A., Leonards K., Jones K., Meier J., Olzscha H., Monteiro O., Martin S., Philpott M., Tumber A., Filippakopoulos P., Yapp C., Wells C., Che K. H., Bannister A., Robson S., Kumar U., Parr N., Lee K., Lugo D., Jeffrey P., Taylor S., Vecellio M. L., Bountra C., Brennan P. E., O'Mahony A., Velichko S., Muller S., Hay D., Daniels D. L., Urh M., La Thangue N. B., Kouzarides T., Prinjha R., Schwaller J., and Knapp S. (2015) Generation of a Selective Small Molecule Inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 75, 5106–5119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Selvi R. B., Swaminathan A., Chatterjee S., Shanmugam M. K., Li F., Ramakrishnan G. B., Siveen K. S., Chinnathambi A., Zayed M. E., Alharbi S. A., Basha J., Bhat A., Vasudevan M., Dharmarajan A., Sethi G., and Kundu T. K. (2015) Inhibition of p300 lysine acetyltransferase activity by luteolin reduces tumor growth in head and neck squamous cell carcinoma (HNSCC) xenograft mouse model. Oncotarget 6, 43806–43818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Shirakawa K., Wang L., Man N., Maksimoska J., Sorum A. W., Lim H. W., Lee I. S., Shimazu T., Newman J. C., Schroder S., Ott M., Marmorstein R., Meier J., Nimer S., and Verdin E. (2016) Salicylate, diflunisal and their metabolites inhibit CBP/p300 and exhibit anticancer activity. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Baltus G. A., Kowalski M. P., Zhai H., Tutter A. V., Quinn D., Wall D., and Kadam S. (2009) Acetylation of sox2 induces its nuclear export in embryonic stem cells. Stem Cells 27, 2175–2184 [DOI] [PubMed] [Google Scholar]
- 107. Nowling T., Bernadt C., Johnson L., Desler M., and Rizzino A. (2003) The co-activator p300 associates physically with and can mediate the action of the distal enhancer of the FGF-4 gene. J. Biol. Chem. 278, 13696–13705 [DOI] [PubMed] [Google Scholar]
- 108. Nowling T. K., Johnson L. R., Wiebe M. S., and Rizzino A. (2000) Identification of the transactivation domain of the transcription factor Sox-2 and an associated co-activator. J. Biol. Chem. 275, 3810–3818 [DOI] [PubMed] [Google Scholar]
- 109. Debeb B. G., Zhang X., Krishnamurthy S., Gao H., Cohen E., Li L., Rodriguez A. A., Landis M. D., Lucci A., Ueno N. T., Robertson F., Xu W., Lacerda L., Buchholz T. A., Cristofanilli M., Reuben J. M., Lewis M. T., and Woodward W. A. (2010) Characterizing cancer cells with cancer stem cell-like features in 293T human embryonic kidney cells. Mol. Cancer 9, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ojesina A. I., Lichtenstein L., Freeman S. S., Pedamallu C. S., Imaz-Rosshandler I., Pugh T. J., Cherniack A. D., Ambrogio L., Cibulskis K., Bertelsen B., Romero-Cordoba S., Trevino V., Vazquez-Santillan K., Guadarrama A. S., Wright A. A., Rosenberg M. W., Duke F., Kaplan B., Wang R., Nickerson E., Walline H. M., Lawrence M. S., Stewart C., Carter S. L., McKenna A., Rodriguez-Sanchez I. P., Espinosa-Castilla M., Woie K., Bjorge L., Wik E., Halle M. K., Hoivik E. A., Krakstad C., Gabino N. B., Gomez-Macias G. S., Valdez-Chapa L. D., Garza-Rodriguez M. L., Maytorena G., Vazquez J., Rodea C., Cravioto A., Cortes M. L., Greulich H., Crum C. P., Neuberg D. S., Hidalgo-Miranda A., Escareno C. R., Akslen L. A., Carey T. E., Vintermyr O. K., Gabriel S. B., Barrera-Saldana H. A., Melendez-Zajgla J., Getz G., Salvesen H. B., and Meyerson M. (2014) Landscape of genomic alterations in cervical carcinomas. Nature 506, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Chen M. K., Cai M. Y., Luo R. Z., Tian X., Liao Q. M., Zhang X. Y., and Han J. D. (2015) Overexpression of p300 correlates with poor prognosis in patients with cutaneous squamous cell carcinoma. Br. J. Dermatol. 172, 111–119 [DOI] [PubMed] [Google Scholar]
- 112. Chen Y. F., Luo R. Z., Li Y., Cui B. K., Song M., Yang A. K., and Chen W. K. (2013) High expression levels of COX-2 and P300 are associated with unfavorable survival in laryngeal squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 270, 1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Li Y., Yang H. X., Luo R. Z., Zhang Y., Li M., Wang X., and Jia W. H. (2011) High expression of p300 has an unfavorable impact on survival in resectable esophageal squamous cell carcinoma. Ann. Thorac. Surg. 91, 1531–1538 [DOI] [PubMed] [Google Scholar]
- 114. Ashton K. J., Weinstein S. R., Maguire D. J., and Griffiths L. R. (2003) Chromosomal aberrations in squamous cell carcinoma and solar keratoses revealed by comparative genomic hybridization. Arch. Dermatol. 139, 876–882 [DOI] [PubMed] [Google Scholar]
- 115. Patmore H. S., Ashman J. N., Stafford N. D., Berrieman H. K., MacDonald A., Greenman J., and Cawkwell L. (2007) Genetic analysis of head and neck squamous cell carcinoma using comparative genomic hybridisation identifies specific aberrations associated with laryngeal origin. Cancer Lett. 258, 55–62 [DOI] [PubMed] [Google Scholar]
- 116. Van de Laar E., Clifford M., Hasenoeder S., Kim B., Wang D., Lee S., Paterson J., Vu N. M., Waddell T. K., Keshavjee S., Tsao M. S., Ailles L., and Moghal N. (2014) Cell surface marker profiling of human tracheal basal cells reveals distinct subpopulations, identifies MST1/MSP as a mitogenic signal, and identifies new biomarkers for lung squamous cell carcinomas. Respiratory Res. 15, 1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rabson A. S., Rosen S. W., Tashijan A. H. Jr, and Weintraub B. D. (1973) Production of human chorionic gonadotropin in vitro by a cell line derived from a carcinoma of the lung. J. Natl. Cancer Inst. 50, 669–674 [DOI] [PubMed] [Google Scholar]
- 118. Wilbertz T., Wagner P., Petersen K., Stiedl A. C., Scheble V. J., Maier S., Reischl M., Mikut R., Altorki N. K., Moch H., Fend F., Staebler A., Bass A. J., Meyerson M., Rubin M. A., Soltermann A., Lengerke C., and Perner S. (2011) SOX2 gene amplification and protein overexpression are associated with better outcome in squamous cell lung cancer. Mod. Pathol. 24, 944–953 [DOI] [PubMed] [Google Scholar]
- 119. Eckner R., Ewen M. E., Newsome D., Gerdes M., DeCaprio J. A., Lawrence J. B., and Livingston D. M. (1994) Molecular cloning and functional analysis of the adenovirus E1A-associated 300-kD protein (p300) reveals a protein with properties of a transcriptional adaptor. Genes Dev. 8, 869–884 [DOI] [PubMed] [Google Scholar]
- 120. Whyte P., Williamson N. M., and Harlow E. (1989) Cellular targets for transformation by the adenovirus E1A proteins. Cell 56, 67–75 [DOI] [PubMed] [Google Scholar]
- 121. Turnell A. S., and Mymryk J. S. (2006) Roles for the coactivators CBP and p300 and the APC/C E3 ubiquitin ligase in E1A-dependent cell transformation. Br. J. Cancer 95, 555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. TCGA. (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Boers J. E., Ambergen A. W., and Thunnissen F. B. (1998) Number and proliferation of basal and parabasal cells in normal human airway epithelium. Am. J. Respir. Crit. Care Med. 157, 2000–2006 [DOI] [PubMed] [Google Scholar]
- 124. Dai X., Liu P., Lau A. W., Liu Y., and Inuzuka H. (2014) Acetylation-dependent regulation of essential iPS-inducing factors: a regulatory crossroad for pluripotency and tumorigenesis. Cancer Med. 3, 1211–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Looijenga L. H., Stoop H., de Leeuw H. P., de Gouveia Brazao C. A., Gillis A. J., van Roozendaal K. E., van Zoelen E. J., Weber R. F., Wolffenbuttel K. P., van Dekken H., Honecker F., Bokemeyer C., Perlman E. J., Schneider D. T., Kononen J., Sauter G., and Oosterhuis J. W. (2003) POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 63, 2244–2250 [PubMed] [Google Scholar]
- 126. Sterlacci W., Savic S., Fiegl M., Obermann E., and Tzankov A. (2014) Putative stem cell markers in non-small-cell lung cancer: a clinicopathologic characterization. J. Thoracic Oncol. 9, 41–49 [DOI] [PubMed] [Google Scholar]
- 127. Pasqualucci L., Dominguez-Sola D., Chiarenza A., Fabbri G., Grunn A., Trifonov V., Kasper L. H., Lerach S., Tang H., Ma J., Rossi D., Chadburn A., Murty V. V., Mullighan C. G., Gaidano G., Rabadan R., Brindle P. K., and Dalla-Favera R. (2011) Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 471, 189–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Kimbrel E. A., Lemieux M. E., Xia X., Davis T. N., Rebel V. I., and Kung A. L. (2009) Systematic in vivo structure-function analysis of p300 in hematopoiesis. Blood 114, 4804–4812 [DOI] [PubMed] [Google Scholar]
- 129. Bryan E. J., Jokubaitis V. J., Chamberlain N. L., Baxter S. W., Dawson E., Choong D. Y., and Campbell I. G. (2002) Mutation analysis of EP300 in colon, breast and ovarian carcinomas. Int. J. Cancer 102, 137–141 [DOI] [PubMed] [Google Scholar]
- 130. Rebel V. I., Kung A. L., Tanner E. A., Yang H., Bronson R. T., and Livingston D. M. (2002) Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc. Natl. Acad. Sci. U.S.A. 99, 14789–14794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Gao X. N., Lin J., Ning Q. Y., Gao L., Yao Y. S., Zhou J. H., Li Y. H., Wang L. L., and Yu L. (2013) A histone acetyltransferase p300 inhibitor C646 induces cell cycle arrest and apoptosis selectively in AML1-ETO-positive AML cells. PLoS ONE 8, e55481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Wang L., Gural A., Sun X. J., Zhao X., Perna F., Huang G., Hatlen M. A., Vu L., Liu F., Xu H., Asai T., Deblasio T., Menendez S., Voza F., Jiang Y., Cole P. A., Zhang J., Melnick A., Roeder R. G., and Nimer S. D. (2011) The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333, 765–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Nuernberg B., Koehler G., and Brune K. (1991) Pharmacokinetics of diflunisal in patients. Clin. Pharmacokinet. 20, 81–89 [DOI] [PubMed] [Google Scholar]
- 134. Gustafson A. M., Soldi R., Anderlind C., Scholand M. B., Qian J., Zhang X., Cooper K., Walker D., McWilliams A., Liu G., Szabo E., Brody J., Massion P. P., Lenburg M. E., Lam S., Bild A. H., and Spira A. (2010) Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci. Transl. Med. 2, 26ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Han W., Gills J. J., Memmott R. M., Lam S., and Dennis P. A. (2009) The chemopreventive agent myoinositol inhibits Akt and extracellular signal-regulated kinase in bronchial lesions from heavy smokers. Cancer Prevention Res. 2, 370–376, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Lam S., McWilliams A., LeRiche J., MacAulay C., Wattenberg L., and Szabo E. (2006) A phase I study of myo-inositol for lung cancer chemoprevention. Cancer Epidemiol. Biomarkers Prev. 15, 1526–1531 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry data have been uploaded to the public MassIVE archive (www.massive.ucsd.edu) with ID: MSV000080175 (SOX2_BioID_Interactome).