

Summary
Peptide recognition through the MHC class I molecule by cytotoxic T lymphocytes (CTLs) leads to the killing of cancer cells. A potential challenge for T‐cell immunotherapy is that dendritic cells (DCs) are exposed to the MHC class I–peptide complex for an insufficient amount of time. To improve tumour antigen presentation to T cells and thereby initiate a more effective T‐cell response, we generated artificial antigen‐presenting cells (aAPCs) by incubating human immature DCs (imDCs) with poly(lactic‐co‐glycolic) acid nanoparticles (PLGA‐NPs) encapsulating tumour antigenic peptides, followed by maturation with lipopolysaccharide. Tumour antigen‐specific CTLs were then induced using either peptide‐loaded mature DCs (mDCs) or aAPCs, and their activities were analysed using both ELISpot and cytotoxicity assays. We found that the aAPCs induced significantly stronger tumour antigen‐specific CTL responses than the controls, which included both mDCs and aAPCs loaded with empty nanoparticles. Moreover, frozen CTLs that were generated by exposure to aAPCs retained the capability to eradicate HLA‐A2‐positive tumour antigen‐bearing cancer cells. These results indicated that aAPCs are superior to DCs when inducing the CTL response because the former are capable of continuously presenting tumour antigens to T cells in a sustained manner. The development of aAPCs with PLGA‐NPs encapsulating tumour antigenic peptides is a promising approach for the generation of effective CTL responses in vitro and warrants further assessments in clinical trials.
Keywords: artificial antigen‐presenting cells, cytotoxic T lymphocytes, dendritic cells, peptide, poly(lactide‐co‐glycolide) acid
Abbreviations
- aAPC
artificial antigen‐presenting cell
- CNPs
control nanoparticles
- CTL
cytotoxic T lymphocyte
- DC
dendritic cell
- gp100
glycoprotein 100
- gp209
glycoprotein 209
- HLA
human histocompatibility leucocyte antigen
- IL
interleukin
- imDC
immature dendritic cell
- Mart‐1
melanoma antigen recognized by T cells 1
- mDC
mature dendritic cell
- MHC
major histocompatibility complex
- NP
nanoparticle
- PBMC
peripheral blood mononuclear cell
- PLGA
poly(lactic‐co‐glycolic) acid
- SV95
survivin95–104
- TIL
tumour‐infiltrating lymphocyte
Introduction
The host immune system, if better harnessed, could be a powerful tool that could significantly enhance the efficacy of cytotoxic cell therapy and prolong the lives of cancer patients.1 Various cancer immunotherapy strategies, including therapeutic non‐cellular cancer vaccines (vector‐based or subunits), dendritic cell (DC) vaccines, engineered T cells and immune checkpoint blockade agents, are emerging in both laboratory studies and clinical trials.2
Despite the remarkable progress made in cancer immunotherapy in recent decades, cancer peptide vaccines have not been successful because of the poor immunogenicity of the antigens and limitations in traditional systemic delivery methods.3 In 2010, the US Food and Drug Administration (FDA) approval of Sipuleucel‐T, a cell‐based cancer immunotherapy for prostate cancer, became a landmark in cancer immunotherapy, making it the first cancer “vaccine” for use in clinic. This event revived the interest in DC‐based cancer immunotherapy.
Dendritic cells are the most potent type of antigen‐presenting cell (APC). They form a cellular network that is involved in immune surveillance, antigen capture, processing and antigen presentation to T cells, making DCs an essential component of cancer immunotherapy.4, 5, 6 In early clinical trials, human DCs were loaded with tumour lysates7 or tumour antigens.8 Because MHC class I–peptide complexes only last for a few hours on DCs,9 the antigen presentation in vivo was probably suboptimal, which led to low response rates. This disadvantage, however, can be overcome when the DCs are produced using continuously present endogenously processed antigens.
With the exception of Sipuleucel‐T, DC‐based vaccines have shown poor response rates in treating advanced cancer. A pooled analysis of 32 clinical studies of advanced colorectal cancer showed an overall response rate of 0·9% for those who underwent active specific immunization with a broad variety of substances (e.g. autologous tumour cells, peptide vaccines, DCs, idiotypic antibodies and virus‐based vaccines).10 Hodi et al.11 reported that in 136 HLA‐A*0201‐positive patients with unresectable stage III/IV melanoma who received a single gp100 peptide in incomplete Freund's adjuvant (Montanide ISA‐51) an objective response of 1·5% was observed. Draube et al.12 summarized 29 clinical trials that involved a total of 906 patients with either prostate cancer (17 trials) or renal cell carcinoma (12 trials) who received DC treatment as a cellular adjuvant, and reported that the objective response rates were 7·7% in prostate cancer and 12·7% in renal cell carcinoma. Recently, Noguchi et al.13 demonstrated that the vaccine arm of a personalized peptide vaccine led to a successful cytotoxic T‐lymphocyte (CTL) response, with an increased overall survival compared with the control arm. Moreover, Kibe et al.14 conducted a phase II study of a personalized peptide vaccine with a maximum of four HLA‐matched peptides in patients with advanced colorectal cancer who had failed at least one regimen of standard chemotherapy and/or targeted therapy. The subjects who were treated with the personalized peptide vaccine demonstrated specific IgG responses of 49%, CTL responses of 63%, and 1‐ and 2‐year survival rates of 53% and 22%, respectively.
Since the approval of the first DC‐based cancer vaccine (Sipuleucel‐T) by the FDA in 2010, there has been an increased interest in harnessing DCs for cancer treatment. However, DC‐based vaccination strategies still need to be optimized to improve the response and survival rates of people with cancer. Multifunctional nanoparticles possess considerable advantages, including targeted delivery to immune cells, co‐delivery of therapeutic agents, reduction of adverse effects, blockade of immune checkpoint molecules, and amplification of immune activation through the use of stimulus‐responsive or immunostimulatory materials.3, 15 Hence, nanotechnology‐based delivery systems provide powerful tools for the development of cancer vaccines.
In this study, we aimed to enhance the efficacy of antigen presentation by generating artificial antigen‐presenting cells (aAPCs) that were capable of continuously presenting tumour antigens in a sustained manner. We used human DCs that were loaded with poly(lactic‐co‐glycolic) acid nanoparticles (PLGA‐NPs) encapsulating peptides of Mart‐1 or survivin as the aAPCs to induce tumour antigen‐specific CTLs. We demonstrated that the aAPCs induced a more robust and effective CTL response than that of the peptide‐pulsed human DCs.
Materials and methods
Peptides and PLGA polymer
HLA‐A*0201 restricted peptides of Mart‐127–35 (Mart‐1, AAGIGILTV),16 Survivin95–104 (SV95, ELTLGEFLKL)17 were synthesized by GenScript Corporation (Piscataway, NJ). The purity and identity of the peptides were determined using HPLC analysis and mass spectrometry. PLGA (MW 23 000, co‐polymer ratio 50 : 50) was purchased from Birmingham Polymers (Birmingham, AL). Polyvinyl alcohol (average MW 30 000–70 000) and lipopolysaccharide were purchased from Sigma‐Aldrich (St Louis, MO). Coumarin 6 was from Polysciences Inc. (Warrington, PA).
Human cell lines and culture media
The cancer cell lines T2, LN‐18, MCF7 and LNCap‐A2 are HLA‐A2 positive whereas LN‐229, SK‐BR‐3 and DU145 are HLA‐A2 negative. All of these cell lines were purchased from ATCC (Manassas, VA). Melanoma cell lines 624 and 1351 and a tumour‐infiltrating lymphocyte (TIL) cell line TIL1235 were kindly provided by Dr John Wunderlich (NIH/NCI, Bethesda, MD). RPMI‐1640 medium was supplemented with either 10% fetal bovine serum for the cancer cell lines or 10% human AB serum (Omega Scientific, Tarzana, CA), interleukin‐2 (IL‐2) (20 U/ml) and IL‐7 (30 U/ml) (R&D, Minneapolis, MN) for the TIL cell line.
Isolation of CD8+ cells from PBMCs
HLA‐A2‐positive buffy‐coat (purchased from San Diego Blood Bank, San Diego, CA) was mixed with Ca2+‐free and Mg2+‐free 1 × PBS to harvest the peripheral blood mononuclear cells (PBMCs) by centrifugation at 930 g for 30 min at 20° with maximum acceleration and free deceleration. CD8+ T cells were positively selected by labelling with CD8 MicroBeads in a MACS column (Miltenyi Biotec, Auburn, CA) according to the manufacturer's instructions.
Preparation and characterization of peptide‐loaded PLGA‐NPs
Peptide‐loaded PLGA‐NPs were formulated using a double emulsion‐solvent evaporation method as previously described18 with minor optimization modifications. Briefly, 60 mg PLGA, 1·2 mg tumour antigenic peptides, and 200 μg of coumarin 6 in 2 ml of chloroform were sonicated on ice using a microtip probe sonicator at a 55‐Watt output (SSE‐1, Branson Digital Sonifier, Cleveland, OH), followed by the further emulsion in 12 ml of 2% polyvinyl alcohol. After the removal of the chloroform, polyvinyl alcohol and un‐encapsulated peptides, the peptide‐loaded PLGA‐NPs were then collected by centrifugation at 108 568 g for 30 min at 4° and then lyophilized at −80°. To characterize the formulated PLGA‐NPs, the samples were sputter‐coated with gold/palladium and imaged using a scanning electron microscope (Phillips XL30, FEI, OR). In addition, the size distribution of the PLGA‐NPs was analysed using a Zetasizer® Nano ZS90 (Malvern Instruments, Worcestershire, UK), and the ζ potential of the PLGA‐NPs was measured in 0·001 m HEPES buffer (pH 7·4) using a ZetaPlus™ instrument (Brookhaven Instruments Corporation, Holtsville, NY).
Study of in vitro sustained peptide release
The in vitro controlled release of peptide was completed as we previously described with minor modification.18 Mart‐127–35 or Survivin95–104 peptide‐loaded PLGA‐NPs aliquots were suspended in PBS (pH 7·4) containing 0·01% sodium azide, and the suspension was incubated at 37° in a shaking incubator (60 r.p.m.). Triplicates were set up for each time‐point. At predetermined time‐points (6 hr, 12 hr, and 1, 3, 7, 14, 21, 28, 35, 42, 49 and 56 days), the tubes were centrifuged for the collection of the supernatants followed by re‐suspending the PLGA‐NP pellet in fresh PBS. The peptide concentration in the supernatant was determined by the absorbance at the wavelength of 280 nm by comparing the concentration with a previously constructed standard calibration curve, as described elsewhere.15
Generation of human DCs and aAPCs
CD14 MicroBeads (Miltenyi Biotech, Auburn, CA) were used to isolate the monocytes from the HLA‐A2‐positive blood samples, which were purchased from the San Diego Blood Bank. Human DCs were generated by culturing the enriched monocytes in complete RPMI‐1640 medium supplemented with granulocyte–macrophage colony‐stimulating factor (1000 U/ml) and recombinant IL‐4 (400 U/ml) (R&D) at 37° in 5% CO2. The medium was replaced every other day. On day 7, the immature DCs (imDCs) were harvested and either stimulated with lipopolysaccharide (Sigma‐Aldrich) at 100 ng/ml for two additional days to generate mature DCs (mDCs) or incubated with the PLGA‐NPs encapsulating tumour antigenic peptides at 37° for 45 min, followed by two additional days of lipopolysaccharide stimulation to generate aAPCs. The mDCs and the aAPCs were harvested and kept frozen in a liquid nitrogen tank for subsequent use.
Intracellular uptake of PLGA‐NPs by human imDCs
Human imDCs at 50 000 cells/ml were incubated with coumarin 6 (a lipophilic fluorescent dye) ‐labelled PLGA‐NPs at 100 μg/ml in a 12‐well plate for 45 min. After removing the uninternalized PLGA‐NPs by washing with 1 × PBS, the imDCs were stained with Hoechst 33342 (Invitrogen, Carlsbad, CA), analysed by flow cytometry (FACSCalibur, BD, San Jose, CA), and imaged using a confocal microscope (Leica TCS SP2, Leica Microsystems Inc., Buffalo Grove, IL).
Generation of tumour antigen‐specific CTLs
Mart‐1 or SV95 peptide‐pulsed human mDCs (mDC‐Mart‐1 or mDC‐SV95) were generated by pulsing human mDCs (2 × 106) with 100 ng/ml of Mart‐1 or SV95 peptide and β 2‐microglobulin at 5 μg/ml (EMD, San Diego, CA) at 37° for 3 hr. Both the Mart‐1 and SV95 peptide‐pulsed human mDCs and the corresponding aAPCs (2 × 106) that were generated from either Mart‐1 or SV95 peptide‐loaded PLGA‐NPs were used as APCs to induce the tumour antigen‐specific CTLs. Each type of APC was added to the isolated HLA‐A2‐positive CD8+ T cells at a ratio of 1 to 10 (APCs: CD8+ T cells) in CTL medium (RPMI‐1640 with 10% human AB serum). Both IL‐2 (20 U/ml) and IL‐7 (30 U/ml) cytokines were added to the culture medium on day 3, and the first re‐stimulation was carried out on day 7 with stimulator cells. To generate the stimulator cells, autologous CD8− cells were irradiated with gamma radiation at 50 Gray and incubated with the addition of β 2‐microglobulin and the peptide at 37° for 2 hr. The culture medium was replaced with 4 ml of fresh Leibovitz medium (Sigma‐Aldrich) containing 1% human albumin (Human Albumin Grifols®, Barcelona, Spain), β 2‐microglobulin and peptide, and the culture was further incubated for 1·5 hr at 25°. The second re‐stimulation was carried out on day 14. The tumour antigen‐specific CTLs were harvested and analysed on day 21 using both ELISpot and cytotoxicity assays.
ELISpot assay
Interferon‐γ (IFN‐γ) ELISpot assays were performed according to the protocol as previously described.15 Anti‐human IFN‐γ monoclonal antibody (10 μg/ml; Mabtech, Cincinnati, OH) ‐coated ELISpot plates were blocked with 200 μl of human AB serum (Omega Scientific, Tarzana, CA) for 2 hr at 37°. Either Mart‐1 peptide (100 ng/ml) ‐pulsed human mDCs or the aAPCs (NP‐Mart‐1) at 2 × 104 cells in 100 μl CTL media were analysed in triplicate by mixing with 2 × 104 TIL1235 cells in 100 μl CTL media for 24 hr at 37°. After the plates were washed with 0·05% Tween 20‐PBS (PBST), 100 μl of biotinylated mouse anti‐human IFN‐γ monoclonal antibody at 2 μg/ml was added to each well. The mixtures were incubated for 2 hr at room temperature followed by the addition of 100 μl for the avidin–biotinylated horseradish peroxidase complex. After a 1‐hr incubation at room temperature, the substrate solution (100 μl) was added for colour development, and the reaction was stopped with running tap water. Finally, the IFN‐γ‐secreting T cells (spot‐forming cells) on the membrane were counted using an ELISpot reader system (CTL Analyzers LLC, Cleveland, OH). Activated DCs have a lifespan of only a few days. To investigate whether the aAPCs present antigens to T cells in a controlled release manner, we performed ELISpot assays to compare the abilities of aAPCs and peptide‐pulsed mDCs to stimulate T cells with different incubation times (24, 36 and 48 hr). Each assay was independently repeated three times, and each condition was analysed in triplicate.
Cytotoxicity assay
The target cells, including T2 cells, peptide‐pulsed T2 cells, prostate cancer cell lines (LNCap‐A2 and DU145), breast cancer cell lines (MCF‐7 and SK‐BR‐3) and melanoma cell lines (624 and 1351), were used to test the potency and selectivity of both the Mart‐1‐specific and SV95‐specific CTLs. The assays were performed following the manufacturer's instructions (CytoTox 96® Non‐Radioactive Cytotoxicity Assay, G1780). In each corresponding well, the CTL cells (1 × 106) in 100 μl of medium were briefly mixed with 50 μl of RPMI‐1640 medium containing 5% human AB serum (5% complete medium) and one of the types of target cells at 10 000 cells in 50 μl of medium to achieve ratios of the effector to target cells (E : T) of 50 : 1, 25 : 1 and 12·5 : 1, in a final volume of 100 μl. The plate was then incubated for 3·25 hr at 37° and 5% CO2. Ten microlitres of lysis buffer was then added to the wells of the volume correction control and the wells to assess the target maximum release. The plate was incubated for another 45 min at 37°. After centrifugation, 50 μl of the supernatant was mixed with 50 μl of the substrate mix solution in an ELISA plate and incubated for 30 min in the dark (to avoid light) at room temperature. The reaction was stopped with 50 μl stop buffer (4°). The absorbance was measured at a wavelength of 490 nm. Each cytotoxicity assay was independently repeated three times, and each condition was analysed in triplicate. The percentage cytotoxicity was calculated based on the following equation: Lysis specific (%) = 100 × (Experimental release – Effector spontaneous release – Target spontaneous release)/(Target maximum release – Target spontaneous release).15
MHC restriction assays
SV95‐specific CTLs induced with the aAPCs (NP‐SV95), blocking antibodies against MHC class I molecule (anti‐HLA‐A2, BB7.2, BD Biosciences, San Diego, CA), and the MHC class II molecule (anti‐MHC class II, IVA 12, BD Biosciences) were used in this assay. MHC restriction assays were carried out using the same protocol of the cytotoxicity assay as described above except for the addition of the blocking antibodies. Each assay was independently repeated three times, and each condition was run in replicate.
Statistical analyses
One‐way analysis of variance was used to compare (i) the means of the values for antigen presentation to T cells among the groups of mDC‐Mart‐1 and aAPCs (NP‐Mart‐1), (ii) the difference in the average cytotoxic activities of the CTLs generated using both mDC‐Mart‐1 and aAPCs (NP‐Mart‐1) and the freshly made and frozen CTLs generated with the aAPCs (NP‐SV95). Values of P < 0·05 were considered statistically significant, and ‘high statistical significance’ was defined as P < 0·01.
Results
Characterization of the PLGA‐NPs encapsulating tumour antigen peptide
Biodegradable polymer‐formulated nanoparticles that range from 10 to 1000 nm in diameter are a type of colloidal drug delivery system.19 Figure 1(a) presents a representative image of the PLGA‐NPs produced using the emulsion‐solvent evaporation method. The size distribution of the unfractionated PLGA‐NPs ranged from 70 to 795 nm, with 80% displaying diameters between 150 and 500 nm, as we previously reported.15 The charge of the PLGA‐NPs at pH 7·4 averaged −15·53 mV with a standard deviation (SD) of 0·71 mV (Fig. 1b). The Zetasizer® Nano ZS90 analyser showed a polydispersity index of 0·308 ± 0·034. Based on the definitions and the results of the HPLC analysis, the PLGA‐NP peptide loading was 3·176 ± 0·144 μg and the peptide encapsulation efficiency was 82·34 ± 8·4%.
Figure 1.
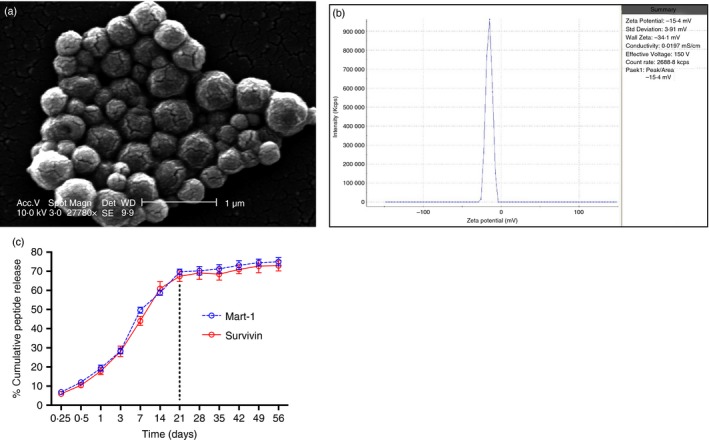
Characterization of peptide loaded poly(lactic‐co‐glycolic) acid nanoparticles (PLGA‐NPs). PLGA‐NPs were sputter‐coated for 100 seconds with gold/palladium using a Cressington 108 auto sputter‐coater (Cressington, Watford, UK), and images were taken using an SEM (Phillips XL 30, FEI, Hillsboro, OR) at 10 kV. (a) SEM image of the PLGA‐NPs (magnification 27 780 ×, scale bar = 1 μm). The particle size distribution of sonicated suspension of PLGA‐NPs (100 μg/ml) in double distilled water was analysed using a Zetasizer® Nano ZS90 (Malvern Instruments, Worcestershire, UK). (b) The zeta potential of PLGA‐NPs in 0·001 m HEPES buffer (pH 7·4) was measured using ZetaPlus™. (c) Sustained peptide release from PLGA‐NPs. A burst peptide release was observed and approximately 68% of peptide was released within 21 days, then reached a plateau in the next 35 days with a cumulative release of 74%. [Colour figure can be viewed at wileyonlinelibrary.com]
Sustained peptide release from the PLGA‐NPs
The peptide release kinetics study was evaluated in PBS at pH 7·4. A biphasic release pattern was observed in this study. In the first 21 days, an initial rapid release showed with approximately 68% of peptide being released. For the second part of kinetics, peptide release reached a plateau in the next 35 days, with a cumulative release of 74% (Fig. 1c). The burst release of peptide might be due to the rapid release of the peptide deposited at the NP interface via diffusion through the water channels. The peptide release during phase 2 might be due to the degradation of the PLGA‐NP matrix.
Cellular uptake and co‐localization of PLGA‐NPs in human DCs
To evaluate the kinetics of the uptake and localization of PLGA‐NPs in DCs, we incubated PLGA‐NPs labelled with coumarin 6 with human imDCs and performed a flow cytometric analysis. Figure 2(a) shows that PLGA‐NPs can be taken up in as little as 15 min of incubation and reached saturation at 45 min.
Figure 2.
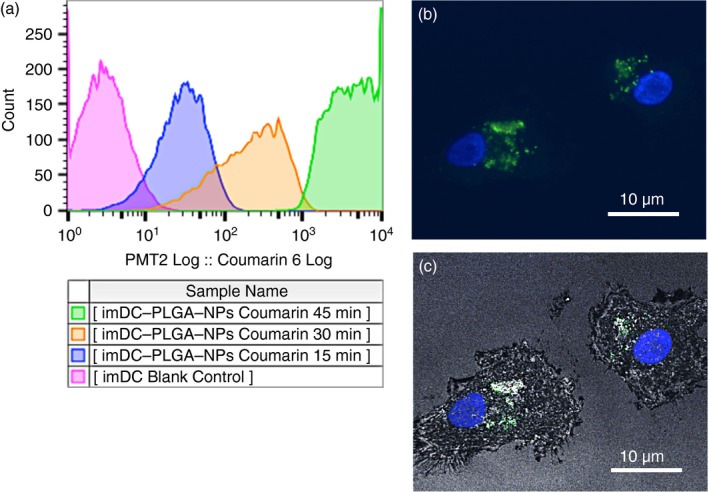
Cellular uptake and colocalization of poly(lactic‐co‐glycolic) acid nanoparticles (PLGA‐NPs) in human dendritic cells (DCs). Human immature DCs (imDCs) at 50 000/ml were cultured in 12‐well plates with 100 μg/ml of the PLGA‐NP suspension that was loaded with the fluorescent probe coumarin 6 for 15, 30 and 45 min. The cells were collected and washed three times with Hanks’ balanced salt solution to remove the uninternalized PLGA‐NPs. (a) Flow cytometry was used to analyse the kinetics of the internalization of the PLGA‐NPs by the imDCs. (b) Human imDCs incubated with PLGA‐NPs for 45 min were analysed using a confocal microscope (Leica TCS SP2). Confocal images of imDCs loaded with PLGA‐NPs; the DAPI and FITC channels were overlaid. (c) Confocal images of human imDCs loaded with PLGA‐NPs; the DAPI, FITC and reflection channels were overlaid. Scale bar = 10 μm. The flow cytometry analysis and confocal image stack scanning were independently repeated three times, respectively; each condition was performed in triplicate. [Colour figure can be viewed at wileyonlinelibrary.com]
To investigate the localization of PLGA‐NPs in human DCs, we incubated the PLGA‐NPs with imDCs for 45 min and then stained the imDCs with Hoechst 33342. The confocal microscopy (Leica TCS SP2, Buffalo Grove, IL) images show that the PLGA‐NPs were localized mainly in the cytoplasm of the generated human DCs (Fig. 2b, DAPI and FITC channels overlaid); Fig. 2(c) (DAPI, FITC and reflection channels overlaid) shows that the PLGA‐NPs appear to be localized in vesicles.
aAPCs present antigens in a sustained manner and more efficiently than mDCs
To compare the antigen presentation efficiency between mDC‐Mart‐1 and aAPCs (NP‐Mart‐1), TIL1235 cells were used as the effector cells in the ELISpot assays. Figure 3(a) shows a representative ELISpot image. The results show that the aAPCs (NP‐Mart‐1) resulted in a substantial increase in the IFN‐γ spots compared with the mDC‐Mart‐1 (Fig. 3b, P = 0·0004). This result demonstrated that the aAPCs (NP‐Mart‐1) presented significantly more Mart‐1 antigen to the TIL1235 cells than the mDC‐Mart‐1.
Figure 3.
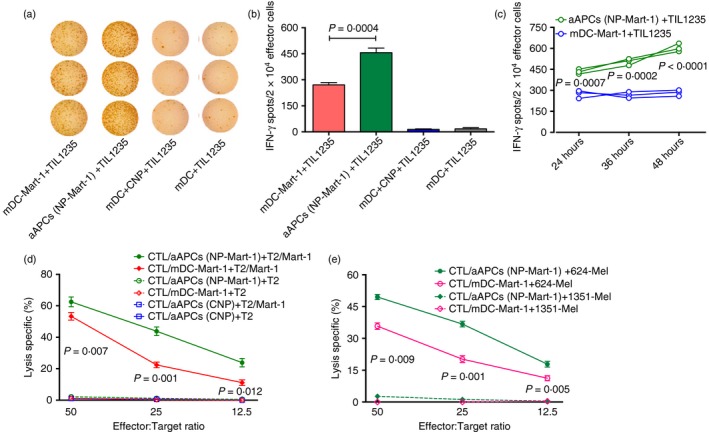
Artificial antigen‐presenting cells (aAPCs) (NP‐Mart‐1) are superior to mature dendritic cells (mDCs) ‐Mart‐1 when presenting antigen and inducing cytotoxic T‐lymphocyte (CTL) response. ELISpot and cytotoxicity assays were performed to assess the Mart‐1 antigen presentation and CTL cytotoxic activities. Both the ELISpot and cytotoxicity assays were independently repeated three times, and each condition was performed in triplicate. (a) Representative ELISpot images. (b) Comparison of the Mart‐1 antigen presentation between mDC‐Mart‐1 and aAPCs (NP‐Mart‐1) using the ELISpot assay. Empty aAPCs (mDCs + CNPs) and mDCs were also included as the controls. Significantly more Mart‐1 antigen was presented to the TIL 1235 cells by the aAPCs (NP‐Mart‐1) than by the mDC‐Mart‐1 (P = 0·0004). (c) aAPCs presented antigen to T cells in a sustained manner. ELISpot assays were performed at different incubation times of 24, 36 and 48 hr. Interferon‐γ (IFN‐γ) spots in the aAPC group were more significant than those in the peptide‐pulsed mDCs group at the above incubation time‐points (P < 0·001). (d) The Mart‐1‐specific CTLs generated by both aAPCs (NP‐Mart‐1) and mDC‐Mart‐1 were capable of lysing Mart‐1 peptide‐pulsed T2 cells [solid green line and solid red line in (c)]. The cytotoxic activity of the CTLs generated with the aAPCs (NP‐Mart‐1) was significantly higher than that of the CTLs generated with mDC‐Mart‐1 (P values of 0·007, 0·001 and 0·012 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively). (e) The Mart‐1‐specific CTLs generated by both aAPCs (NP‐Mart‐1) and mDC‐Mart‐1 were also able to lyse melanoma 624 cells [solid green line and solid pink line in (e)]. The cytotoxic activity of the CTLs generated with aAPCs (NP‐Mart‐1) was significantly higher than that of the CTLs generated by mDC‐Mart‐1 (P values of 0·009, 0·001 and 0·005 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively). In addition, the CTLs induced by aAPCs (CNPs) were also included as a control, and the results showed that they failed to recognize and kill the target cells of both T2 cells and Mart‐1 peptide‐pulsed T2 cells [solid and broken blue lines in (d)]. Each assay was independently repeated three times, and each condition was performed in triplicate. [Colour figure can be viewed at wileyonlinelibrary.com]
We have demonstrated a sustained peptide release from the PLGA‐NPs in the study of peptide release kinetics (Fig. 1c). To investigate whether aAPCs present antigen to T cells in a sustained manner, we also performed ELISpot assays to compare the abilities of aAPCs and peptide‐pulsed mDCs to stimulate T cells over time. The ELISpot assay results indicated that the IFN‐γ spots in the aAPC group were more significant than those in the peptide‐pulsed mDC group at the incubation times of 24, 36 and 48 hr (Fig. 3c, P < 0·001).
To evaluate the efficiency of the CTLs generated by using both aAPCs and peptide‐pulsed mDCs, cytotoxicity assays were performed. The results showed that the Mart‐1‐specific CTLs are capable of lysing both Mart‐1 peptide‐pulsed T2 cells (Fig. 3d) and melanoma cell line 624 cells (Fig. 3e). The cytotoxic activities of the Mart‐1‐specific CTLs generated by the aAPCs (NP‐Mart‐1) were significantly higher than those produced by the mDC‐Mart‐1 (solid green line versus solid red line in Fig. 3d, e). Furthermore, these CTLs failed to recognize and kill the controls, which were T2 and the melanoma 1351 cells that lack the Mart‐1 antigen on the cell surfaces (broken green line and broken red line in Fig. 3d, e). These results demonstrated the antigen‐specificity of the CTL response.
As additional controls, the cytotoxic activities of the CTLs generated from control aAPCs (CNPs) and aAPCs (NP‐Mart‐1) were investigated. The CTLs induced by control aAPCs (CNPs) failed to recognize and kill the target cells of both T2 cells (solid blue line in Fig. 3d) and Mart‐1 peptide‐pulsed T2 cells (broken blue line in Fig. 3d).
To investigate the superiority between the aAPCs and peptide‐pulsed mDCs, in addition to the above comparison between aAPCs (NP‐Mart‐1) and mDCs‐Mart‐1 (Fig. 3), we also compared aAPCs (NP‐SV95) with mDCs‐SV95. The results showed that the aAPCs (NP‐SV95) induced a more effective and enhanced T‐cell response in comparison to the mDCs‐SV95 (solid green line versus solid blue line, Fig. 4a).
Figure 4.
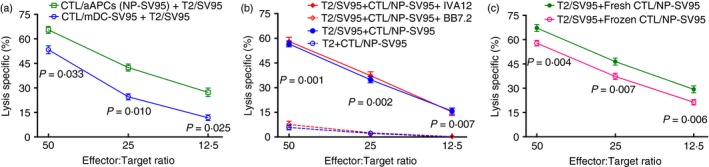
Artificial antigen‐presenting cells (aAPCs) (NP‐SV95) are superior to mature dendritic cells (mDCs) ‐SV95, freshly prepared cytotoxic T lymphocytes (CTLs) are more potent than frozen CTLs, and the CTLs eradicate the target cells in an MHC I‐restricted manner. The cytolytic activities were compared between the CTLs that were induced with both aAPCs and mDCs. The aAPCs induced more effective and enhanced T‐cell responses in comparison to the mDCs [solid green line versus solid blue line, (a)]. The cytotoxicity of the SV95‐specific CTLs was significantly blocked by the addition of BB7.2 [broken red line versus solid red line; P values of 0·001, 0·002 and 0·007 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively, (b)]. In addition, the fresh CTLs had significantly greater activities than the frozen CTLs [solid green line versus solid red line; P values of 0·004, 0·007 and 0·006 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively, (c)]. Each cytotoxicity assay was independently repeated three times, and each condition was performed in triplicate. [Colour figure can be viewed at wileyonlinelibrary.com]
Frozen aAPCs induce tumour antigen‐specific CTLs
Because it would save time and be more cost‐effective to be able to generate a large batch of aAPCs for immunotherapy, we compared the capacities of frozen aAPCs with freshly produced aAPCs to induce tumour antigen‐specific CTLs. The cytotoxicity assay results demonstrated that the cryopreservation of aAPCs did not impair their abilities to induce antigen‐specific immune responses. Most of the CTLs used in this study were generated using frozen aAPCs, including some CTLs in Fig. 3(d) (both solid and broken green lines), some CTLs in Fig. 3(e) (both solid and broken green lines), some CTLS in Fig. 4(a) (solid green line), and all of the CTLs used in Figs 4(b, c) and 5.
Figure 5.
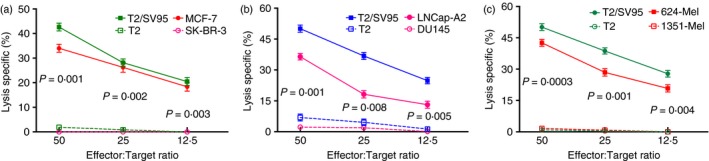
Frozen SV95‐specific cytotoxic T lymphocytes (CTLs) were capable of recognizing and killing cancer cells. Cytotoxicity assays were performed to assess the cytotoxic activities of the frozen SV95‐specific CTLs. T2 cells were used as an internal control, and SV95 peptide‐pulsed T2 cells were used as a positive control. HLA‐A2‐negative cell lines including SK‐BR‐3, DU145 and Melanoma 1351 were used as negative controls. (a) Breast cancer MCF7 cells were significantly recognized and killed (P values of 0·001, 0·002 and 0·003 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively). (b) Prostate cancer LNCap‐A2 cells were significantly recognized and killed (P values of 0·001, 0·008 and 0·005 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively). (c) Human melanoma 624 cells were also significantly killed by the frozen SV95‐specific CTLs (P values of 0·0003, 0·001 and 0·004 at the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively). Each assay was independently repeated three times, and each condition was performed in triplicate. [Colour figure can be viewed at wileyonlinelibrary.com]
Activity of MHC class I‐dependent CTLs against target cells
We further examined the capacities of anti‐MHC class I and anti‐MHC class II monoclonal antibodies to inhibit the cytolytic activities of the SV95‐specific CTLs. Lysis of SV95 peptide‐pulsed T2 cells by SV95‐specific CTLs was inhibited by the anti‐MHC class I monoclonal antibody BB7.2 (broken red line in Fig. 4b), but not by the anti‐MHC class II antibody IVA12 (solid red line in Fig. 4b). This inhibition of SV95‐CTLs caused by the addition of BB7.2 was extremely significant (P values of 0·001, 0·002 and 0·007 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively, Fig. 4b).
Frozen SV95‐specific CTLs are capable of killing HLA‐A2‐positive cancer cells
We further compared the cytotoxic activities between freshly made and frozen CTLs. To test whether the CTLs were still functional after cryopreservation, the SV95‐specific CTLs generated using the aAPCs (NP‐SV95) were stored frozen for 60 days in a liquid nitrogen tank. The results showed that fresh CTLs had more potent cytotoxic activity than that of frozen CTLs (P values of 0·004, 0·007 and 0·006 for the E : T ratios of 50 : 1, 25 : 1 and 12·5 : 1, respectively, Fig. 4c).
Nevertheless, the cryopreserved CTLs remained significantly effective in killing the HLA‐A2‐positive cancer cells of the MCF7 (solid red line in Fig. 5a), LNCap‐A2 (solid red line in Fig. 5b) and melanoma 624 cell lines (solid red line in Fig. 5c), but not the HLA‐A2‐negative cell lines of the breast cancer SK‐BR‐3 (broken pink line in Fig. 5a), prostate cancer DU‐145 (broken pink line in Fig. 5b), and melanoma 1351 cell lines (broken red line in Fig. 5c). T2 cells were used as an internal control, and SV95 peptide‐pulsed T2 cells were used as a positive control.
Discussion
Dendritic cells play an important role in inhibiting tumour growth by priming the T‐cell immune response through the expression of molecules that are used for the interaction between DCs and T cells.20 In DC‐based vaccines, DCs generated in vitro and loaded with tumour antigens have been used as a therapeutic strategy for multiple cancers, including melanoma, renal cell carcinoma, lymphoma, colorectal cancer, prostate cancer and paediatric and adult brain cancers.21 Nevertheless, insufficient induction of immune responses with cancer vaccines hinders the successful treatment and eradication of tumours.22 Several clinical trials have shown that DC‐based immunotherapy increases the antitumour T‐cell reactivity in patients experiencing partial responses of blood and solid cancers.23, 24, 25 Tumour lysate‐pulsed DC immunotherapy has also been shown to be clinically effective.26, 27, 28 However, the response rate was still less than 10%,22 and some cases failed to show any response.29
One major reason for why vaccination using these traditional peptide‐pulsed DCs fails to effectively elicit an immune response is that the presentation of MHC class I–peptide complexes on the cell surface only lasts for a few hours. The controlled release of peptide in PLGA‐NPs is an interesting attribute property for enhancing the presentation of peptide to APCs as well as a vaccine candidate as this may reduce the immunization number. In this study, we generated aAPCs by incubating human DCs with PLGA‐NPs encapsulating tumour antigenic peptides. Our results showed that the aAPCs induced a more effective tumour antigen‐specific CTL response than that of the peptide‐pulsed human mDCs. These findings suggested that the tumour peptides can be continuously released from the PLGA‐NPs into the cytosol of the DCs and are then transported to the rough endoplasmic reticulum where they bind to the cleft of the MHC class I molecules. Via the Golgi apparatus, the MHC class I–peptide complexes are subsequently transported to the surface of the aAPCs. These tumour antigenic peptides are therefore continuously presented to T cells and elicit an effective immune response.
To achieve better immunization efficacy, various strategies have been proposed, including the injection of the vaccine into lymph nodes30 and the vaccination of patients with tumour RNA‐transfected DCs by electroporation and lipofection.31, 32 In this study, we attempted to modify the antigen‐presenting abilities of the DCs. Although the amount of peptide loaded in PLGA‐NPs was 63 times less than that provided by emulsification in incomplete Freund's adjuvant, a more robust CTL response was induced by the peptide‐loaded PLGA‐NPs than that induced by the peptide emulsified in incomplete Freund's adjuvant.15 These results suggest that it is crucial to retain the peptide–MHC class I complex on the surface of APCs to elicit an effective T‐cell response.
The surfaces of nanomaterials can be engineered to display antigens and co‐stimulatory ligands to serve as aAPCs and activate T cells. Our functional studies showed that significantly more Mart‐1 antigens were presented to TIL1235 cells by the aAPCs (NP‐Mart‐1) than by the mDC‐Mart‐1 (Fig. 3b), and they were presented in a sustained manner (Fig. 3c). In addition, the aAPCs (NP‐Mart‐1)‐induced Mart‐1‐specific CTLs demonstrated significantly higher cytotoxic activities than the mDC‐Mart‐1 (Fig. 3d, e). These results suggested that the PLGA‐NPs continuously provide the antigenic tumour peptides to DCs, which led to increased antigen presentation to the T cells and therefore resulted in more effective tumour antigen‐specific CTLs.
Survivin is aberrantly expressed in most solid cancers33 and haematopoietic malignancies34 but is undetectable in terminally differentiated normal adult tissues35 with the exceptions of testis, thymus and placenta.36 Our results demonstrate that the activation of SV95‐specific CTLs by aAPCs with the SV95 peptide as the presented antigen led to increased cytotoxicity against multiple HLA‐A2‐positive cancer types including breast cancer (Fig. 5a), prostate cancer (Fig. 5b), melanoma (Fig. 5c) and glioma cells.37
The remarkable ability of DCs to elicit an immune response and the feasibility of culturing DCs in vitro have led to improvements in cancer immunotherapy. However, this approach has the following limitations: (i) only patients with HLA‐A2‐positive tumours can be treated, (ii) the current approach is far from optimal, because many patients treated with peptide‐ or tumour lysate‐pulsed DCs show little or no response, (iii) the culturing of DCs is time‐consuming and costly and may also increase the risk of infection for patients. Nevertheless, some advantages of the approach described herein exist, e.g. the avoidance of DC culture in vitro, the enhancement of DC vaccination potency, and the activation in vivo of DCs by the nano‐vaccines.
Our previous preclinical data showed delayed tumour development in mice that were immunized with the PLGA‐NPs encapsulating mouse six‐transmembrane epithelial antigen of the prostate (STEAP) peptide. Furthermore, the tumour sizes in the treated mice were significantly smaller than those in the control group immunized with empty PLGA‐NPs.15
The results of this study demonstrated that the generated aAPCs were capable of inducing a more robust and effective CTL response than that of the controls, including both mDCs and aAPCs loaded with empty PLGA‐NPs. More recently, Cruz et al.38 demonstrated that PLGA‐encapsulated antigen showed an enhanced T‐cell response when compared with soluble antigen in a DC mix. Although the DC‐based Sipuleucel‐T vaccine for metastatic castration‐resistant prostate cancer was recently approved by the FDA, no clinically effective DC‐based immunotherapies have been approved for other human cancers. More strategies are required for the development of a potent “next‐generation” DC vaccine for cancer immunotherapy.
Funding
This study was supported by the Natural Science Foundation of Zhejiang Province, China (LY14H160015), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, Ministry of Education of China, the Huzhou Municipal Bureau of Science and Technology (2014GY08) and the Public Technology Program of Zhejiang Province, China (2016C37126).
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgements
We would like to acknowledge Dr Davorka Messmer, Dr Christina Jamieson and Ms Amy Yiming Ma at Moores Cancer Center, University of California San Diego for their help with critical reading and editing.
Contributor Information
Boris Minev, Email: bminev@ucsd.edu.
Wenxue Ma, Email: wma@ucsd.edu.
References
- 1. Palucka AK, Coussens LM. The basis of oncoimmunology. Cell 2016; 164:1233–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jeanbart L, Swartz MA. Engineering opportunities in cancer immunotherapy. Proc Natl Acad Sci U S A 2015; 112:14467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saleh T, Shojaosadati SA. Multifunctional nanoparticles for cancer immunotherapy. Hum Vaccin Immunother 2016; 12:1863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Joffre OP, Segura E, Savina A, Amigorena S. Cross‐presentation by dendritic cells. Nat Rev Immunol 2012; 12:557–69. [DOI] [PubMed] [Google Scholar]
- 5. Palucka K, Banchereau J. SnapShot: cancer vaccines. Cell 2014; 157:516–e1. [DOI] [PubMed] [Google Scholar]
- 6. Cintolo JA, Datta J, Mathew SJ, Czerniecki BJ. Dendritic cell‐based vaccines: barriers and opportunities. Future Oncol 2012; 8:1273–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matsushita H, Enomoto Y, Kume H, Nakagawa T, Fukuhara H, Suzuki M et al A pilot study of autologous tumor lysate‐loaded dendritic cell vaccination combined with sunitinib for metastatic renal cell carcinoma. J Immunother Cancer 2014; 2:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sabado RL, Miller E, Spadaccia M, Vengco I, Hasan F, Bhardwaj N. Preparation of tumor antigen‐loaded mature dendritic cells for immunotherapy. J Vis Exp 2013; 78:e50085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Waeckerle‐Men Y, Groettrup M. PLGA microspheres for improved antigen delivery to dendritic cells as cellular vaccines. Adv Drug Deliv Rev 2005; 57:475–82. [DOI] [PubMed] [Google Scholar]
- 10. Nagorsen D, Thiel E. Clinical and immunologic responses to active specific cancer vaccines in human colorectal cancer. Clin Cancer Res 2006; 12:3064–9. [DOI] [PubMed] [Google Scholar]
- 11. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB et al Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 2010; 363:711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Draube A, Klein‐Gonzalez N, Mattheus S, Brillant C, Hellmich M, Engert A et al Dendritic cell based tumor vaccination in prostate and renal cell cancer: a systematic review and meta‐analysis. PLoS ONE 2011; 6:e18801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Noguchi M, Matsumoto K, Uemura H, Arai G, Eto M, Naito S, et al An open‐label, randomized phase ii trial of personalized peptide vaccination in patients with bladder cancer that progressed after platinum‐based chemotherapy. Clin Cancer Res 2016; 22:54–60. [DOI] [PubMed] [Google Scholar]
- 14. Kibe S, Yutani S, Motoyama S, Nomura T, Tanaka N, Kawahara A et al Phase II study of personalized peptide vaccination for previously treated advanced colorectal cancer. Cancer Immunol Res 2014; 2:1154–62. [DOI] [PubMed] [Google Scholar]
- 15. Ma W, Chen M, Kaushal S, McElroy M, Zhang Y, Ozkan C et al PLGA nanoparticle‐mediated delivery of tumor antigenic peptides elicits effective immune responses. Int J Nanomedicine 2012; 7:1475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Minev BR, Chavez FL, Dudouet BM, Mitchell MS. Synthetic insertion signal sequences enhance MHC class I presentation of a peptide from the melanoma antigen MART‐1. Eur J Immunol 2000; 30:2115–24. [DOI] [PubMed] [Google Scholar]
- 17. Andersen MH, Pedersen LO, Becker JC, Straten PT. Identification of a cytotoxic T lymphocyte response to the apoptosis inhibitor protein survivin in cancer patients. Cancer Res 2001; 61:869–72. [PubMed] [Google Scholar]
- 18. Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin‐conjugated paclitaxel‐loaded nanoparticles in a murine model of prostate cancer. Int J Cancer 2004; 112:335–40. [DOI] [PubMed] [Google Scholar]
- 19. Singh R, Lillard JW Jr. Nanoparticle‐based targeted drug delivery. Exp Mol Pathol 2009; 86:215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tel J, Anguille S, Waterborg CE, Smits EL, Figdor CG, de Vries IJ. Tumoricidal activity of human dendritic cells. Trends Immunol 2014; 35:38–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012; 12:265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med 2004; 10:909–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Nicola M, Zappasodi R, Carlo‐Stella C, Mortarini R, Pupa SM, Magni M et al Vaccination with autologous tumor‐loaded dendritic cells induces clinical and immunologic responses in indolent B‐cell lymphoma patients with relapsed and measurable disease: a pilot study. Blood 2009; 113:18–27. [DOI] [PubMed] [Google Scholar]
- 24. Comber JD, Philip R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther Adv Vaccines 2014; 2:77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chandramohan V, Mitchell DA, Johnson LA, Sampson JH, Bigner DD. Antibody, T‐cell and dendritic cell immunotherapy for malignant brain tumors. Future Oncol 2013; 9:977–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lasky JL 3rd, Panosyan EH, Plant A, Davidson T, Yong WH, Prins RM et al Autologous tumor lysate‐pulsed dendritic cell immunotherapy for pediatric patients with newly diagnosed or recurrent high‐grade gliomas. Anticancer Res 2013; 33:2047–56. [PMC free article] [PubMed] [Google Scholar]
- 27. Gao D, Li C, Xie X, Zhao P, Wei X, Sun W et al Autologous tumor lysate‐pulsed dendritic cell immunotherapy with cytokine‐induced killer cells improves survival in gastric and colorectal cancer patients. PLoS ONE 2014; 9:e93886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiang CL, Hagemann AR, Leskowitz R, Mick R, Garrabrant T, Czerniecki BJ et al Day‐4 myeloid dendritic cells pulsed with whole tumor lysate are highly immunogenic and elicit potent anti‐tumor responses. PLoS ONE 2011; 6:e28732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Redman BG, Chang AE, Whitfield J, Esper P, Jiang G, Braun T et al Phase Ib trial assessing autologous, tumor‐pulsed dendritic cells as a vaccine administered with or without IL‐2 in patients with metastatic melanoma. J Immunother 2008; 31:591–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamanaka R. Dendritic‐cell‐ and peptide‐based vaccination strategies for glioma. Neurosurg Rev 2009; 32:265–73. [DOI] [PubMed] [Google Scholar]
- 31. Weise JB, Csiszar K, Gottschlich S, Hoffmann M, Schmidt A, Weingartz U et al Vaccination strategy to target lysyl oxidase‐like 4 in dendritic cell based immunotherapy for head and neck cancer. Int J Oncol 2008; 32:317–22. [PubMed] [Google Scholar]
- 32. Bontkes HJ, Kramer D, Ruizendaal JJ, Meijer CJ, Hooijberg E. Tumor associated antigen and interleukin‐12 mRNA transfected dendritic cells enhance effector function of natural killer cells and antigen specific T‐cells. Clin Immunol 2008; 127:375–84. [DOI] [PubMed] [Google Scholar]
- 33. Altieri DC. Survivin, versatile modulation of cell division and apoptosis in cancer. Oncogene 2003; 22:8581–9. [DOI] [PubMed] [Google Scholar]
- 34. Cong XL, Han ZC. Survivin and leukemia. Int J Hematol 2004; 80:232–8. [DOI] [PubMed] [Google Scholar]
- 35. Hirohashi Y, Torigoe T, Maeda A, Nabeta Y, Kamiguchi K, Sato T et al An HLA‐A24‐restricted cytotoxic T lymphocyte epitope of a tumor‐associated protein, survivin. Clin Cancer Res 2002; 8:1731–9. [PubMed] [Google Scholar]
- 36. Idenoue S, Hirohashi Y, Torigoe T, Sato Y, Tamura Y, Hariu H et al A potent immunogenic general cancer vaccine that targets survivin, an inhibitor of apoptosis proteins. Clin Cancer Res 2005; 11:1474–82. [DOI] [PubMed] [Google Scholar]
- 37. Shao S, Risch E, Burner D, Lu L, Minev B, Ma W. IFN‐γ enhances cytotoxic efficiency of the cytotoxic T lymphocytes against human glioma cells. Int Immunopharmacol 2017; 47:159–65. [DOI] [PubMed] [Google Scholar]
- 38. Cruz LJ, Tacken PJ, Eich C, Rueda F, Torensma R, Figdor CG. Controlled release of antigen and Toll‐like receptor ligands from PLGA nanoparticles enhances immunogenicity. Nanomedicine (Lond) 2017; 12:491–510. [DOI] [PubMed] [Google Scholar]