

Summary
CD11c+ dendritic cells (DCs) exert a critical role as antigen‐presenting cells in regulating pathogenic T cells in multiple sclerosis (MS). To determine whether the therapeutic benefit of interferon‐β (IFN‐β) treatment for MS is in part influenced by IFN regulation of DC function, we examined the immunophenotype of DCs derived from IFN‐β +/+ and IFN‐β −/− mice using a myelin oligodendrocyte glycoprotein (MOG) peptide‐induced mouse model of MS, experimental autoimmune encephalomyelitis (EAE). Our earlier work identified that IFN‐β −/− mice exhibit earlier onset and more rapid progression of neurological impairment compared with IFN‐β +/+ mice. In this study we show that lipopolysaccharide‐/MOG peptide‐stimulated IFN‐β −/− DCs secrete cytokines associated with pathological T helper type 17 rather than regulatory T‐cell polarization and exhibit increased CD80 and MHCII expression when compared with stimulated IFN‐β +/+ DCs. IFN‐β −/− DCs from mice immunized to develop EAE induce greater proliferation of MOG‐transgenic CD4+ T cells and promote interleukin‐17 production by these T cells. Adoptive transfer of MOG peptide‐primed IFN‐β −/− DCs into IFN‐β +/+ and IFN‐β −/− mice immunized to develop EAE resulted in their rapid migration into the central nervous system of recipient mice, before onset of disease, which we attribute to failed signal transducer and activator of transcription 1‐mediated inhibition of CCR7. Taken together, our data support immunoregulatory roles for IFN‐β in the activation and migration of DCs during EAE.
Keywords: dendritic cells, interferon‐β, multiple sclerosis, T helper type 17 cells
Introduction
Multiple sclerosis (MS) is an autoimmune disorder characterized by central nervous system (CNS) damage. Approximately 85% of patients with MS suffer from a relapsing–remitting form of MS.1 Relapsing–remitting MS is associated with unpredictable, self‐limiting bouts of CNS dysfunction, which vary in their frequency and duration. MS is a complex disease with an equally complex pathophysiology, as indicated by the heterogeneity in the level of clinical presentations as well as morphological differences in brain lesions.2
Multiple sclerosis is characterized by a profound inflammatory cell infiltrate into the CNS. The migration of inflammatory T cells to the CNS is critical to the development of MS and mice deficient in CD4 or CD8 T cells exhibit a reduced incidence of experimental autoimmune encephalomyelitis (EAE), an animal model of MS.3 Recruitment of inflammatory cells into the CNS requires cells to cross the blood–brain barrier, with its breakdown being one of the initial steps in the development of MS. Chemokine and chemokine receptor expression is critical for inflammatory cell migration, recruitment to the CNS and subsequent disease pathology.4 The chemokine receptors CCR2 and CCR7 are of particular importance in MS and EAE. CCR2−/− mice are resistant to EAE,5 CCR2 expression is observed on CNS‐infiltrating dendritic cells (DCs)6 and elevated levels of CCR2 have been found in CNS tissue of patients with MS.7 It is well established that CCR7 is required for lymphocyte retention in lymphoid tissues such as lymph nodes (LNs) and several reports suggest the importance of CCR7 expression on lymphocytes in EAE8 and in MS9 for CNS recruitment.
Both T helper type 1 (Th1) and Th17 cells are posited to be pathogenic/pro‐inflammatory CD4+ T‐cell subsets in MS.10 Dendritic cells facilitate recruitment of T cells into the CNS,11 and are critical antigen‐presenting cells in EAE, driving the Th17 lineage.12 The DCs secrete interleukin‐12 (IL‐12), associated with Th1 lineage commitment, and transforming growth factor‐β and IL‐6, associated with Th17 polarization.10 In MS, DCs are abundant in brain lesions and, together with microglia, function to reactivate CD4+ T cells as they enter the CNS, driving myelin‐specific Th17/Th1 polarization associated with neuroinflammation, demyelination and neuronal damage. A predominant role for DCs in the pathogenesis of MS is underscored by the association of specific HLA class II molecules accounting for 10–60% of the genetic risk linked to MS.13 Dendritic cells are sufficient to induce autoimmunity in the CNS;14 increasing their numbers exacerbates EAE,15 and their depletion ablates the induction of EAE.6 By contrast, some reports have suggested a tolerogenic role for DCs in the context of T‐cell priming and activation in EAE.16, 17 Viewed together, these findings highlight the importance of DCs in CNS autoimmunity.
Interferon‐β (IFN‐β) remains one of the first‐line treatments for patients with relapsing–remitting MS. Treatment with IFN has shown benefits in outcome measures related to relapses, progression of disability and magnetic resonance imaging. It exhibits pleiotropic effects in MS, including modulation of adhesion molecule expression, inhibition of matrix metalloproteinase activity and regulation of leucocyte trafficking.18 In EAE, IFN‐β reduces relapse rates and leads to improvements in clinical scores.19 Mice that are IFN‐β −/− have been used in EAE studies to validate the importance of IFN‐β treatment in limiting the pathogenesis of the disease20, 21 and in relapsing–remitting EAE, where there was evidence of increased frequency of relapses.20 We and others have shown regulatory roles for IFN‐β in Th17 cell polarization.22, 23 Treatment with IFN‐β decreases IL‐17 gene and protein expression in proliferating murine CD4+ cells and prevents the elevation of IL‐17 mRNA in cells from the CNS draining LNs.24 In this report we describe a role for IFN‐β in regulating the DC immunophenotype, affecting DC effects on T‐cell activation, Th17 lineage polarization and DC migration in EAE.
Materials and methods
Mice
The IFN‐β +/+ and IFN‐β −/− mice25 were bred and maintained in a specific pathogen‐free colony in accordance with guidelines set by University Health Network's Animal Care committee. Mice were genotyped by PCR as previously described.21 C57BL/6‐Tg (Tcra2D2,Tcrb2D2) Kuch1/J (2D2) transgenic mice with T‐cell receptor specificity for myelin oligodendrocyte glycoprotein (MOG) 35–55 were purchased from the Jackson Laboratory (Bar Harbor, ME) and bred with wild‐type C57BL/6 mice in our colony to obtain mice heterozygous for the 2D2 transgene. Mice were genotyped by flow cytometry as previously described.26
Bone‐marrow‐derived DC generation
Bone‐marrow‐derived dendritic cells (BMDCs) were generated as reported previously.27 Briefly, IFN‐β +/+ and IFN‐β −/− mice aged 6–12 weeks were killed, their femurs were harvested and flushed, and the resulting cells were passed through a 70‐μm filter. Progenitors were plated at 2 × 106 cells per plate in bacterial Petri dishes. Cells were cultured for 9 days in RPMI‐1640 medium supplemented with 10% fetal calf serum, 100 U penicillin and streptomycin, 2 mm l‐glutamine, 20 μm β‐mercaptoethanol (complete RPMI). Recombinant mouse granulocyte–macrophage colony‐stimulating factor (Peprotech, Rocky Hill, NJ) was added on days 0, 3, 6 and 8 at 40 ng/ml. On day 9, cells in suspension were aspirated, washed, counted and seeded at a density of 2 × 106/well in 24‐well tissue‐culture plates. BMDCs were matured by adding 1 μg/ml of lipopolysaccharide (LPS) (Escherichia coli, O55:B5; Sigma‐Aldrich, Oakville, ON, Canada) for 16 hr. When BMDCs were primed with MOG 35–55 peptide (McGill Sheldon Biotechnology Centre, Montreal, QC, Canada), it was at a concentration of 1 μg/ml for 2–3 hr on day 10 of DC culture. The LPS‐activated (or LPS‐activated, MOG‐primed) BMDCs were then washed three times with complete RPMI before use.
Dendritic cell isolation and activation
The IFN‐β +/+ or IFN‐β −/− mice, 6–8 weeks of age, were killed, and their spleens or inguinal LNs were harvested and digested with 1 μg/ml collagenase A (Roche Life Science, Oakville, ON, Canada) and 0·1 μg/ml DNase I (Roche) in PBS for 30 min at 37°. The reaction was stopped by incubating with 1 mm EDTA for 10 min at room temperature. Cell suspensions were subsequently passed through a 70‐μm filter and contaminating red blood cells were lysed by incubating in red blood cell lysis buffer (150 mm NH4Cl, 10 mm KHCO3, 0·1 mm Na2EDTA), for 5 min on ice. Cells were washed, resuspended in PBS and counted, then, as indicated, CD11c+ cells were isolated by magnetic separation (Miltenyi Biotec, Auburn, CA) as per the manufacturer's instructions. In other studies, unfractionated splenocytes were plated at 2 × 106 cells per well in 24‐well plates in complete RPMI medium. As indicated, CD11c+ DCs were activated with 1 μg/ml LPS, then identified by antibody staining and FACS analysis.
CD4+ T‐cell isolation and proliferation assays
Female 2D2 mice, 6–12 weeks of age, were killed, their spleens were harvested and CD4+ T cells were positively selected using a CD4+ selection kit (Miltenyi Biotec) as described previously.22 The resulting CD4+ T cells were labelled with Cell Proliferation Dye 450 (eBioscience, San Diego, CA), at a concentration of 1 μm, as per the manufacturer's instructions. Co‐cultures of 1 × 105 CD4+ 2D2 T cells with 5 × 104 IFN‐β +/+ or IFN‐β −/− CD11c+ DCs, isolated as described above, were established in individual wells of 96‐well round‐bottom culture plates, in complete RPMI medium. In experiments where naive DCs were treated with LPS, cells were plated at 5 × 104/well and incubated with 1 μg/ml LPS for 16 hr, then CD4+ 2D2 T cells were added. When MOG peptide was included in the cultures, it was added at a concentration of 1 μg/ml. After 48 or 72 hr, culture supernatants were harvested for cytokine analysis by ELISA, and cells were washed and stained for FACS analysis. Proliferation of CD4+ MHCII cells was quantified by cell proliferation dye dilution, using the unstimulated, untreated and unlabelled controls to gate on dividing versus undivided cells.
Fluorescence‐associated cell sorting
Cells to be analysed for surface markers were washed and resuspended in FACS buffer (PBS with 2% fetal calf serum). Non‐specific antibody binding was blocked using 10% mouse serum in FACS buffer for 10 min at room temperature. The following antibodies were used (all antibodies were from BioLegend, San Diego, CA, unless otherwise indicated): anti‐CD4‐BV650, anti‐CD3‐BV510, anti‐MHCII (IA/IE)‐FITC (eBioscience) or –BV650, anti‐CCR7‐phycoerythrin (eBioscience), anti‐CD80‐phycoerythrin‐Cy5 or ‐phycoerythrin (eBioscience), anti‐CD86‐FITC or –allophycocyanin, anti‐CD11c‐AlexaFluor 700, anti‐CD11b‐BV421, anti‐B220‐BV711 and anti‐CCR2‐AlexaFluor 647. Cells were stained for 30 min at 4° and then washed three times with FACS buffer. Cells were analysed using a Beckton Dickinson LSRII flow cytometer and appropriate isotype controls, and fluorescence minus one controls were used in each experiment to gauge non‐specific staining and to determine gates. Flow cytometry files were analysed using flowjo software (FlowJo, Ashland, OR).
Cytokine analysis (FlowCytomix and ELISA)
Supernatants from cell cultures were stored at −80°. To measure LPS‐induced cytokines in IFN‐β +/+ or IFN‐β −/− BMDCs, the FlowCytomix assay (flow‐cytometry based multiplex cytokine detection; eBioscience) was used as per the manufacturer's instructions. Cytokines measured included IL‐1α, IL‐1β, IL‐2, IL‐4, IL‐6, IL‐10, IL‐13, IL‐15, IL‐17, IL‐21, IL‐22, IL‐23, IL‐27, IFN‐γ, and tumour necrosis factor‐α (TNF‐α.). Ready‐Set‐Go ELISA kits (eBioscience) for IFN‐γ, IL‐6, IL‐17A, IL‐12p40, and IL‐23 were used as per the manufacturer's instructions to measure cytokine levels in DC‐CD4+ T‐cell co‐culture supernatants or from LPS‐stimulated BMDCs. Interferon‐β was detected using the LEGENDMAX Mouse IFN‐β ELISA kit (BioLegend).
Gene expression analysis
The BMDCs were generated from female IFN‐β +/+ (n = 3) and IFN‐β −/− (n = 3) mice as described above and either left untreated or treated with LPS (1 μg/ml) for 6 hr. Pooled BMDCs were then washed and RNA was extracted using the RNeasy isolation kit according to the manufacturer's instructions (Qiagen, Toronto, ON, Canada). RNA was then submitted to the UHN Microarray Facility, where 200 ng/sample was hybridized to the Illumina mouse whole genome WG‐6 v2 BeadChip, as per standard protocols. The WG‐6 v2 BeadChip contains probes for > 42 500 transcripts spanning the mouse genome, and includes probes identified from the NCBI RefSeq database, the mouse exonic evidence based oligonucleotide (MEEBO) probe set and protein coding sequences from the RIKEN FANTOM2 database. Data were analysed using Illumina's genomestudio software. Raw fluorescence intensities were background corrected and the resulting probe fluorescence intensities from LPS‐stimulated samples were compared with the untreated control for each mouse genotype. Fold change was calculated from the log2‐transformed data.
EAE and adoptive transfer experiments
Experimental autoimmune encephalomyelitis was induced in IFN‐β +/+ and IFN‐β −/− mice as previously described.21 Briefly, a subcutaneous injection of 50 μg of MOG peptide mixed with complete Freund's adjuvant (10 mg/ml Mycobacterium tuberculosis; Sigma Aldrich) was administered in the hind flank of mice on day 0, and 400 ng of pertussis toxin was given by intraperitoneal injection on days 0 and +2 relative to MOG. Disease progression was scored using a five‐point ascending scale: 0 = no signs of disease, 1 = limp tail, 2 = hind limb weakness, 3 = partial hind limb paralysis, 4 = full hind limb paralysis, 5 = moribund. For DC isolation and co‐culture with 2D2 CD4+ T cells, DCs were isolated from mice on day 3 of EAE, from the injection site draining LN, and CD11c+ cells were positively selected as described above. For adoptive transfer experiments, LPS‐activated, MOG‐primed IFN‐β +/+ and IFN‐β −/− BMDCs were generated as indicated above. The DCs were washed and labelled with Cell Vue Maroon dye (eBioscience) at a concentration of 1 μm, as per the manufacturer's instructions. Then, 2 × 106 labelled DCs were injected into IFN‐β +/+ and IFN‐β −/− mice on day 3 of EAE via the tail vein. Twenty‐four and 72 hr following injection, mice were killed, and their spleens, spinal cords and brains were removed and fixed in 10% formalin. After 24 hr, tissues were transferred into PBS, and imaged using a Xenogen IVIS (Xenogen, Caliper Life Science, Waltham, MA). The excitation was set at 640 nm and the emission filter used was 680 nm.
Results
Interferon‐β influences DC cytokine production
Our earlier studies identified that IFN‐β −/− mice are more susceptible to EAE than IFN‐β +/+ mice.21 Given that in MS DCs drive the Th1/Th17 T‐cell polarization associated with neuroinflammation, we undertook a series of experiments to determine whether the absence of IFN‐β might influence cytokine production by DCs, thereby affecting T‐cell polarization. Accordingly, we generated BMDCs from IFN‐β +/+ and IFN‐β −/− mice in vitro and stimulated these DCs with the Toll‐like receptor‐4 agonist, LPS, for 16 hr. Culture supernatants from these stimulated BMDCs were analysed for Th1/Th2/Th17/Th22 cytokines. Our data reveal increased IL‐6 and IL‐23 (Fig. 1a,b), decreased IL‐12p40, TNF‐α, IL‐27, IL‐10 and no IFN‐β production (Fig. 1c–g) from stimulated IFN‐β −/− BMDCs compared with stimulated IFN‐β +/+ BMDCs, which is suggestive of a pro‐Th17 environment generated by IFN‐β −/− DCs and a pro‐Th1/regulatory T‐cell environment generated by IFN‐β +/+ DCs.
Figure 1.
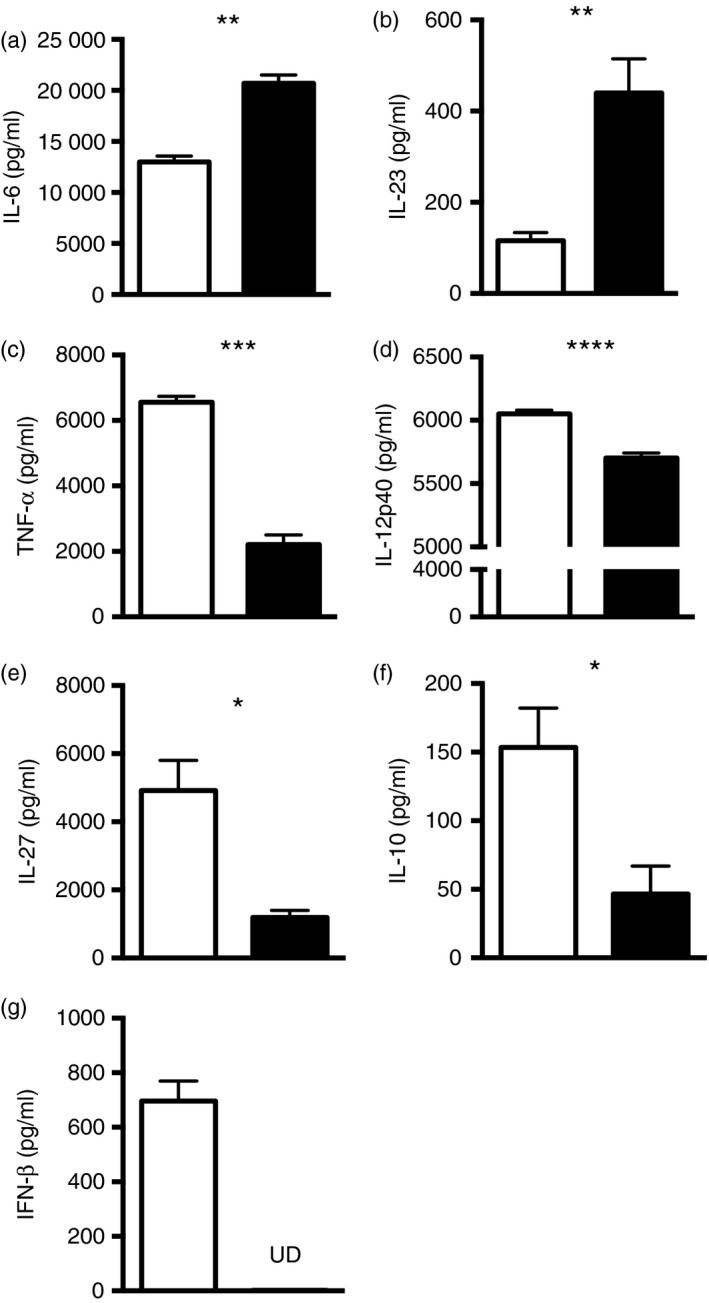
Activation of IFN‐β +/+ and IFN‐β −/− dendritic cells (DCs) leads to differential cytokine production. Bone marrow was isolated from the femurs of IFN‐β +/+ (open bars) or IFN‐β −/− (filled bars) mice and DCs were generated in vitro using granulocyte–macrophage colony‐stimulating factor (40 ng/ml). After 10 days in culture, DCs were harvested, stimulated with lipopolysaccharide (LPS) (1 μg/ml) for 16 hr, and the culture supernatants were assessed for cytokines by multiplex cytokine analysis or ELISA. Data for (a) interleukin‐6 (IL‐6), (b) IL‐23, (c) tumour necrosis factor‐α (TNF‐α, (d) IL‐12p40, (e) IL‐27, (f) IL‐10 and (g) interferon‐β (IFN‐β) are shown. UD = cytokine undetectable. Values are means ± SE. Significant differences were determined by Student's t‐test. *P < 0·05, **P < 0·01, ***P < 0·001, ****P < 0·0001. Data are representative of two independent experiments with three mice per group.
Interferon‐β affects DC‐mediated MOG‐transgenic CD4+ T‐cell proliferation
In our earlier publication we also provided evidence that DCs derived from IFN‐β −/− mice induce a greater MOG‐specific CD4 T‐cell proliferation, regardless of whether the CD4 T cells originated from IFN‐β +/+ or IFN‐β −/− mice, compared with DCs isolated from IFN‐β +/+ mice.21 To further investigate this effect of IFN‐β on DC‐mediated T‐cell proliferation, we examined DC : T‐cell co‐cultures using CD4+ T cells isolated from 2D2 transgenic mice. 2D2 CD4+ T cells are MOG‐specific, eliminating the need to generate antigen‐reactive T cells by inducing EAE. In Fig. 2 we provide evidence for greater proliferation when the 2D2 CD4+ T cells are cultured with LPS‐activated splenic CD11c+ DCs isolated from IFN‐β −/− mice compared with untreated CD11c+ DCs isolated from IFN‐β +/+ mice. Notably, although co‐culture of DCs and 2D2T cells with MOG peptide alone does increase proliferation of the T cells, Toll‐like receptor‐4 activation of the DCs by LPS enhances this proliferative response to a greater extent.
Figure 2.
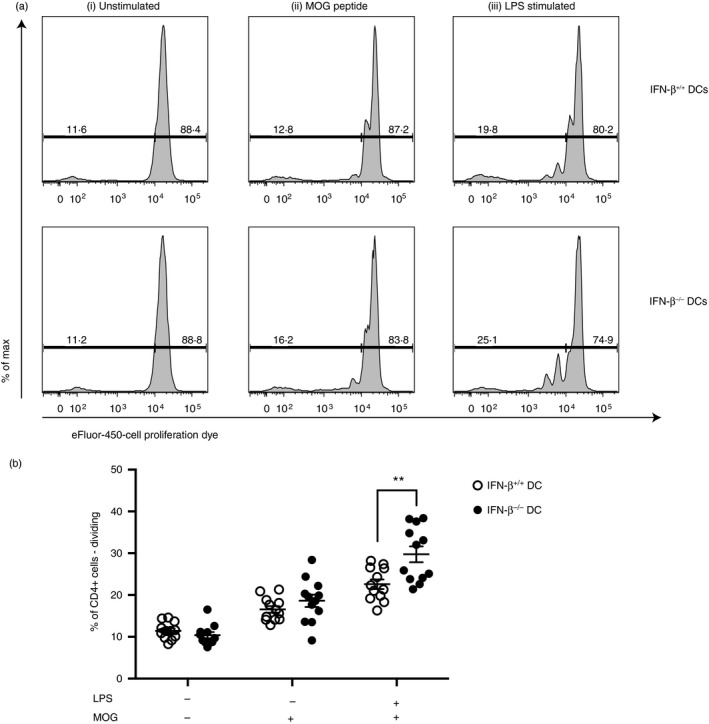
Dendritic cells (DCs) from IFN‐β −/− mice drive increased myelin oligodendrocyte glycoprotein (MOG)‐transgenic CD4+ T‐cell proliferation. CD11c+ DCs were isolated from the spleens of IFN‐β +/+ and IFN‐β −/− mice and either left untreated or treated with 1 μg/ml lipopolysaccharide (LPS) for 16 hr. MOG‐transgenic CD4+ T cells were isolated by positive selection and labelled with Cell Proliferation Dye (CPD). Then, 1 × 105 CD4+ T cells were cultured with 5 × 104 CD11c+ DCs. (a) Representative plots of CD4+ T‐cell dye dilution for each culture condition. 2D2 T cells were cultured with IFN‐β +/+ (top row) or IFN‐β −/− (bottom row) DCs. Culture conditions were as follows: (i) unstimulated DCs with 2D2 T cells, (ii) unstimulated DCs with 2D2 T cells in the presence of MOG peptide or (iii) LPS‐stimulated DCs with 2D2 T cells in the presence of MOG peptide for 48 hr. Following culture, cells were stained and CD4+ T‐cell proliferation was assessed. (b) Quantification of replicate samples of CD4+ T cells are shown. Horizontal bars denote means ± SE. Significant differences were measured using Student's t‐test. **P < 0·01. Data are representative of two independent experiments with four mice per group (three technical replicates per mouse).
Interferon‐β modulates co‐stimulatory molecule expression on DCs
As we have consistently observed a discriminating capacity for IFN‐β −/− CD11c+ DCs to enhance T‐cell proliferation, we investigated whether this might be a direct consequence of MHCII and co‐stimulatory molecule expression. At the outset we identified that a greater percentage of IFN‐β −/− CD11c+ DCs constitutively express CD80 (5·5%) compared with naive IFN‐β +/+ CD11c+ DCs (3·3%) (Fig. 3a). By contrast, we identified a modest, yet significant increase in the percentage of IFN‐β +/+ CD11c+ DCs expressing CD86 constitutively (5·4%), compared with IFN‐β −/− CD11c+ DCs (4·1%)(Fig. 3b). As anticipated, LPS treatment led to DC activation, indicated by increased percentages of CD80+ and CD86+ expressing CD11c+ DCs, and increased MHCII expression (Fig. 3c). Although LPS stimulation activated both IFN‐β +/+ and IFN‐β −/− DCs, the percentage of DCs expressing CD80, was significantly increased in IFN‐β −/− DCs (16%) compared with IFN‐β +/+ DCs (10·7%) (Fig. 3a). We observed a modest, yet significant increase in the percentage of CD86‐expressing DCs when derived from IFN‐β +/+ mice (43·8%) compared with IFN‐β −/− mice (35·5%), after LPS stimulation (Fig. 3b). When considering MHCII expression, we observed that > 95% of the DCs were positive for MHCII, whether this was constitutive expression or following LPS stimulation (data not shown). However, examination of MFI revealed that the level of MHCII expression was different between IFN‐β +/+ versus IFN‐β −/− splenic DCs. Specifically, in unstimulated DCs we found that MHCII expression was elevated on DCs from IFN‐β −/− mice compared with IFN‐β +/+ mice (Fig. 3c). Furthermore, LPS stimulation significantly increased MHCII expression to a greater extent on the IFN‐β −/− DCs compared with the IFN‐β +/+ DCs (Fig. 3c).
Figure 3.
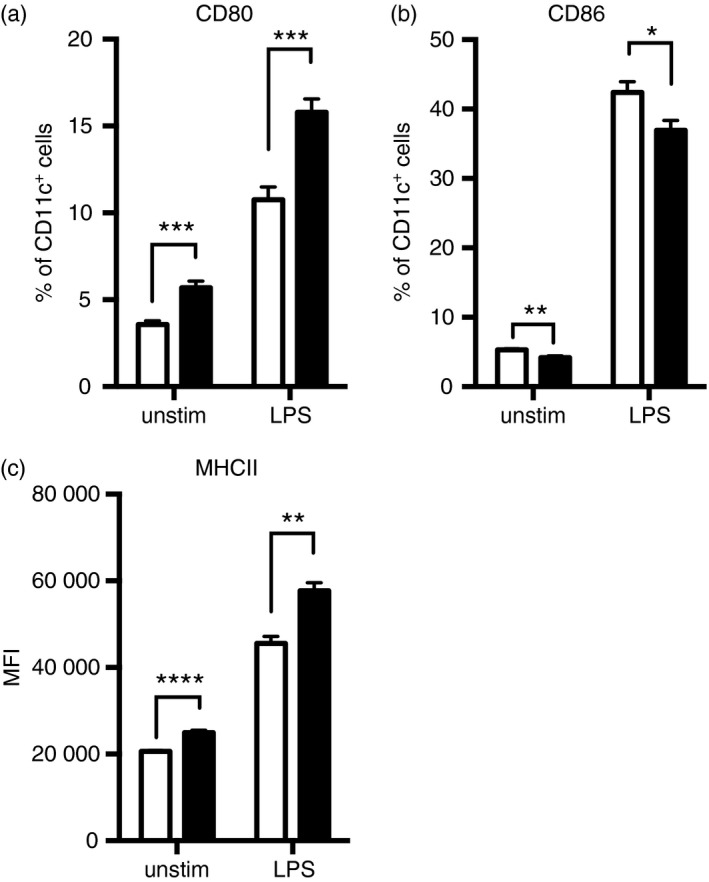
Lipopolysaccharide (LPS) stimulation modulates co‐stimulatory molecule and MHCII expression on splenic dendritic cells (DCs) from IFN‐β +/+ and IFN‐β −/− mice. Splenocytes were isolated from naive IFN‐β +/+ (open bars) and IFN‐β −/− (closed bars) mice and stimulated for 16 hr with LPS (1 μg/ml). Cells were stained with the relevant antibodies as described in the Materials and methods. The DCs were gated as CD45+ CD3− CD11c+. The percentage of cells expressing (a) CD80 and (b) CD86 and the mean fluorescence intensity of (c) MHCII are shown. Values are means ± SE. Significant differences were measured by the Mann–Whitney U‐test. *P < 0·05, **P < 0·01, ***P < 0·001, and ****P < 0·0001. Data are representative of at least three independent experiments with three mice per group and three technical replicates per mouse.
DCs from IFN‐β −/− mice with EAE induce enhanced MOG‐transgenic CD4+ T‐cell proliferation and drive Th17 polarization ex vivo
Activation of DCs using LPS may differ from activation of DCs in the context of EAE induction, as Toll‐like receptor agonists are known potent activators of DCs.28 When IFN‐β −/− and IFN‐β +/+ mice were induced to develop EAE, then their draining LNs harvested at 72 hr post induction and before symptom onset, we observed an increased percentage of CD80+ DCs and increased MHCII expression on DCs from IFN‐β −/− mice compared with the DCs from IFN‐β +/+ mice (Fig. 4a). Additionally, when co‐cultured with 2D2 T cells, these IFN‐β −/− DCs induced a greater proliferation of the T cells than their IFN‐β +/+ counterparts, yet this proliferative response by the 2D2 T cells required the presence of MOG peptide (Fig. 4b). Examination of the culture supernatants from these experiments showed that for the IFN‐β −/− DCs co‐cultured with 2D2 CD4+ T cells in the presence of MOG peptide, there were increased levels of IL‐6, IL‐17A, IL‐23 and IFN‐γ compared with the co‐cultures with IFN‐β +/+ DCs (Fig. 5). Interleukin‐12p40 levels detected were similar, regardless of whether IFN‐β −/− or IFN‐β +/+ DCs were present in the co‐cultures (Fig. 5).
Figure 4.
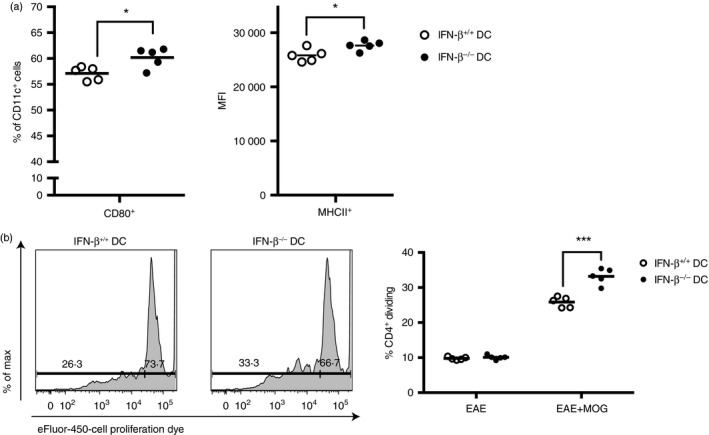
Dendritic cells (DCs) from IFN‐β −/− mice with experimental autoimmune encephalomyelitis (EAE) have increased CD80 and MHCII expression and drive increased proliferation of myelin oligodendrocyte glycoprotein (MOG) ‐transgenic CD4+ T cells. CD11c+ DCs were isolated from the draining lymph nodes of IFN‐β +/+ mice and IFN‐β −/− mice 72 hr after EAE induction. CD4+ T cells were collected by positive selection from the spleens of MOG‐transgenic (2D2) mice and labelled with Cell Proliferation Dye (CPD). Then, 1 × 105 CD4+ T cells were cultured with 5 × 104 CD11c+ DCs in the absence (EAE) or presence (EAE+MOG) of MOG peptide for 72 hr, when CD4+ T‐cell proliferation was assessed, as described in Materials and methods. (a) Cells were gated as MHCII+ CD11c+ and CD80+ (left hand side), and the MFI of MHCII expression on CD11c+ cells (right hand side) is shown. (b) Representative plots of CD4+ T‐cell dye dilution for 2D2 T cells cultured with IFN‐β +/+ or IFN‐β −/− DCs (left hand side) and quantification of replicate samples of CD4+ T cells (right hand side). Significant differences were measured by Student's t‐test. *P < 0·05, ***P < 0·001. Data are representative of two independent experiments with five technical replicates of three pooled mice per group. Horizontal bars denote means.
Figure 5.
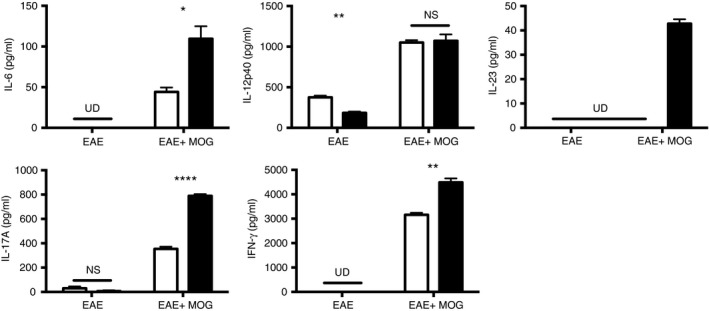
Activated IFN‐β −/− dendritic cells (DCs) produce pro‐T helper type 17 (Th17) cytokines and drive Th17 polarization ex vivo. CD11c+ DCs were isolated from the draining lymph nodes of IFN‐β +/+ (open bars) and IFN‐β −/− (filled bars) mice 72 hr after induction of experimental autoimmune encephalomyelitis (EAE). CD4+ T cells were collected by positive selection from the spleens of myelin oligodendrocyte glycoprotein (MOG) ‐transgenic (2D2) mice. Then, 1 × 105 CD4+ T cells were cultured with 5 × 104 CD11c+ DCs (EAE) or in the presence of MOG peptide (1 μg/ml) (EAE+MOG) for 72 hr. Cytokines [interleukin‐6 (IL‐6), IL‐12p40, IL‐23, IL‐17A interferon‐γ (IFN‐γ)] in culture supernatants were measured by ELISA. Values are means ± SE. Significant differences were measured by Student's t‐test. *P < 0·05, **P < 0·01, and ****P < 0·0001. UD = cytokine undetectable, ns = not significant. Data are representative of two independent experiments with three technical replicates from three pooled mice per group.
Interferon‐β modulates CCR7 expression on DCs influencing their migratory capacity
Dendritic cells that express CCR7 respond to gradients of CCL19 and CCL21, directing them to the T‐cell zones of lymphoid organs where they interact with and activate naive T cells. Moreover, DCs in CNS lesions have been shown to express CCR7.13 CCR2 expression has been extensively studied in EAE, with data indicating that CCR2−/− mice have fewer CNS‐infiltrating T cells and macrophages.5 We analysed the surface expression of CCR2 and CCR7 on BMDCs before and after 16 hr of LPS treatment and found that basal CCR2 expression was similar between IFN‐β +/+ and IFN‐β −/− BMDCs, and although CCR2 expression was increased after LPS stimulation, there was no significant difference in expression on the IFN‐β −/− DCs compared with the IFN‐β +/+ DCs (Fig. 6a). By contrast, basal CCR7 expression was significantly elevated on IFN‐β −/− BMDCs compared with IFN‐β +/+ BMDCs and was also increased to a greater level after LPS stimulation (Fig. 6a). Furthermore, we provide evidence that CCR7 expression is increased on DCs from the spleens and draining LNs of IFN‐β −/− mice with EAE (Fig. 6b). One of the proposed mechanisms of action of IFN‐β is to inhibit migration of DCs by modulation of CCR7 expression. This requires a functional signal transducer and activator of transcription 1 (STAT1), as studies have shown that CCR7 expression is not altered in response to IFN‐β treatment in STAT1‐deficient BMDCs.29 STAT1 activation is a key component of IFN‐β signalling that leads to the induction of IFN‐inducible gene expression.30 Not surprisingly, IFN‐β −/− macrophages have been shown to be deficient in their ability to activate STAT1 in response to LPS.31, 32 Cognizant that IFN‐β influences CCR7 expression, mediated by STAT1, we examined whether the effects of LPS on CCR7 expression might reflect differences in STAT1 expression between IFN‐β +/+ and IFN‐β −/− DCs. Basal levels of STAT1 gene expression were comparable between IFN‐β +/+ and IFN‐β −/− BMDCs, but we found that LPS stimulation increased STAT1 expression threefold in BMDCs from IFN‐β +/+ mice, yet had no effect on STAT1 gene expression in BMDCs from IFN‐β −/− mice (Fig. 6c).
Figure 6.
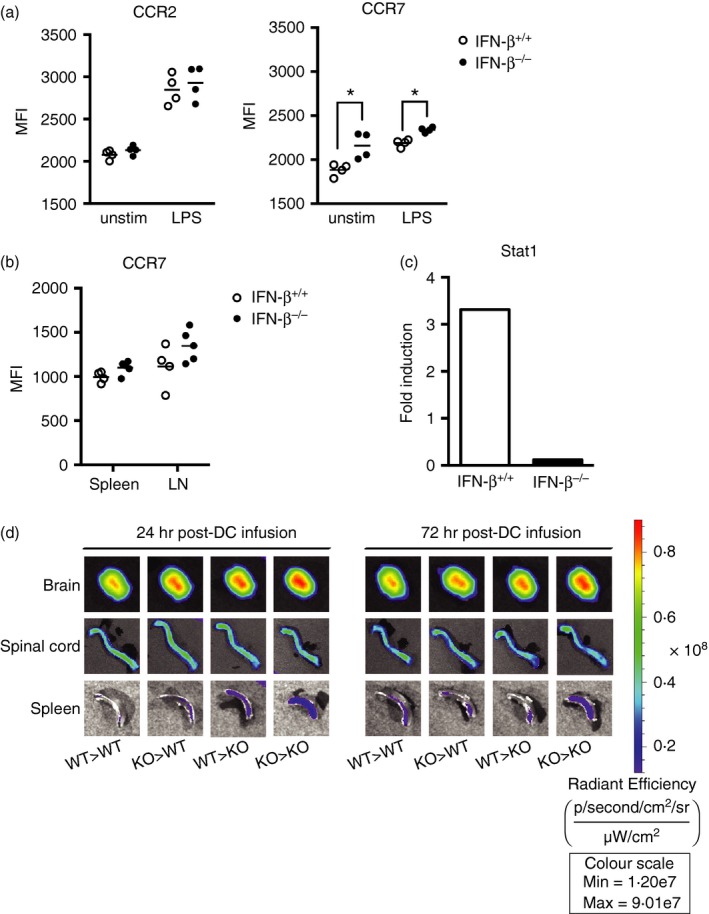
Interferon‐β (IFN‐β) affects CCR7 expression on dendritic cells (DCs), thereby contributing to their migratory capacity to the central nervous system. (a) Bone‐marrow‐derived DCs (BMDCs) (CD45+ CD11c+ CD11b+) from IFN‐β +/+ (n = 4) or IFN‐β −/− (n = 4) mice were either left untreated or stimulated with 1 μg/ml lipopolysaccharide (LPS) for 16 hr, then cell surface CCR2 and CCR7 expression was analysed. (b) CD11c+ splenic and lymph node DCs harvested from IFN‐β +/+ (n = 4) and IFN‐β −/− (n = 4) mice on day 9 after experimental autoimmune encephalomyelitis (EAE) induction were analysed for cell surface CCR7 expression. Horizontal bars denote means. Statistical differences were calculated using the Mann–Whitney U‐test. *P < 0·05. (c) BMDCs were generated from IFN‐β +/+ (n = 3) and IFN‐β −/− (n = 3) mice, as described in the Materials and methods, then treated for 6 hr with 1 μg/ml LPS. RNA was extracted, cDNA was prepared and gene expression for signal transducer and activator of transcription 1 was determined. (d) BMDCs generated from IFN‐β +/+ (n = 3) and IFN‐β −/− (n = 3) mice were LPS matured for 16 hr, pulsed with myelin oligodendrocyte glycoprotein (MOG) peptide (3 hr) and labelled with CellVue Maroon. DCs were adoptively transferred into IFN‐β −/− or IFN‐β +/+ recipient mice on day 3 of EAE and DC migration to brains, spinal cords and spleens was assessed at 24 and 72 hr after DC infusion using a Xenogen IVIS fluorescence imager. Images are representative of three mice per group, per time‐point. [Colour figure can be viewed at wileyonlinelibrary.com]
In a final series of experiments we examined the effects IFN‐β on DC migration to the CNS in the context of EAE. In vitro generated BMDCs, derived from IFN‐β +/+ and IFN‐β −/− mice were stimulated with LPS and MOG peptide, labelled with fluorescent CellVue Maroon, then introduced by intravenous injection into recipient mice, both IFN‐β +/+ and IFN‐β −/−, on day 3 post induction of EAE. As early as 24 and 72 hr post DC infusion, we detected the presence of the labelled DCs in the spleens, spinal cords and brains of all recipient mice, regardless of the source of the DCs (Fig. 6d). Notably, at 24 and 72 hr post‐infusion, none of these mice displayed any symptoms of EAE. At both 24 and 72 hr post‐infusion we observed a greater accumulation of DCs in the brains of IFN‐β −/− mice compared with the brains of IFN‐β +/+ mice. Additionally, the greatest accumulation of DCs in the brain was exhibited in IFN‐β −/− mice that were infused with IFN‐β −/− DCs. Analysis of the spinal cords of the IFN‐β +/+ and IFN‐β −/− mice, at both 24 and 72 hr post‐infusion, suggested little accumulation of DCs and no apparent difference whether the DCs were derived from IFN‐β +/+ or IFN‐β −/− mice, or whether they were infused into IFN‐β +/+ or IFN‐β −/− mice induced to develop EAE. Examination of spleens revealed that there was the greatest accumulation of DCs when IFN‐β −/− DCs were infused into IFN‐β −/− mice induced to develop EAE, at both time‐points.
Discussion
Dendritic cells represent an important component of the immune system, bridging the innate and adaptive immune responses: detecting and presenting antigens to T cells to induce an appropriate immune response and generate subsequent immunological memory. DCs may also present self‐antigens to T cells, effecting a breakdown in tolerance, leading to autoimmunity and detrimental immune responses against tissues and organs. As such, identifying the specific effects of disease‐modifying treatments in MS on DCs is important, as there is evidence that modifying DC function influences disease progression in EAE.6, 11, 14 As IFN‐β therapy remains an important treatment for MS, the molecular mechanisms involved in its beneficial effects related to DCs were the subject of these studies.
Patients with MS have higher levels of pro‐inflammatory cytokines in their serum and blood.33, 34 Treatment with IFN‐β alters cytokine profiles in EAE and in patients with MS, increasing regulatory or anti‐inflammatory cytokines,35 and decreasing pro‐inflammatory cytokines.36 DCs secrete pro‐ and anti‐inflammatory cytokines in MS and EAE, which guide T‐cell differentiation. Interleukin‐27 has been implicated in regulating Th17 responses in murine EAE experiments and in human CD4+ T cells.37, 38 There is evidence that IFN‐β treatment of both mouse and human DCs in vitro leads to production of both IL‐27 and IL‐10.35, 36, 39 Additionally, DCs generated from patients with MS that respond to IFN‐β therapy produce more IL‐27 in response to IFN‐β treatment, compared with non‐responders.35 In agreement, we identified that IFN‐β −/− DCs stimulated with LPS produced less TNF‐α, IL‐10 and IL‐27, associated with regulatory T cells, yet produced more IL‐6, less IL‐12p40 and more IL‐23, compared with IFN‐β +/+ DCs. The increase in production of IL‐6 by IFN‐β −/− DCs may contribute to polarization of CD4+ towards a Th17 cell lineage, as IL‐6 is required for Th17 cell differentiation and inhibits regulatory T‐cell polarization.40 Stimulation with LPS induced IFN‐β production from the IFN‐β +/+ BMDCs but not, as anticipated, from the IFN‐β −/− BMDCs, suggesting an autocrine effect of IFN‐β on the BMDC cytokine profile. Indeed, LPS stimulation of splenocytes or purified splenic DCs, leads to IFN‐β production (see Supplementary material, Fig. 1). These data are supportive of a role for IFN‐β regulating DC production of cytokines that affect CD4+ T‐cell polarization. These results align with our previous findings of increased CD4+ IL‐17A+ T cells in IFN‐β −/− mice, associated with exacerbated EAE.31, 32 We infer that in the presence of IFN‐β, activated DCs will limit CD4+ T‐cell polarization to pathogenic Th17 cells, thereby contributing to less severe EAE or MS.
A number of reports have indicated a role for IFN‐β in limiting IL‐17 production in MS and EAE,22, 23, 24, 36 however, recent work by Axtell and others indicated that IFN‐β could exacerbate Th17‐induced EAE.41 Herein we provide evidence that the absence of IFN‐β in DCs led to induction of pro‐inflammatory cytokines IL‐6, IL‐23, IL‐17 and IFN‐γ when cultured with 2D2 transgenic CD4+ T cells. The IFN‐β −/− DCs produced more of the Th17‐polarizing cytokines IL‐6 and IL‐23, which may drive the increased IL‐17 production in this 2D2 context. In earlier studies we showed that CD4+ T cells from IFN‐β −/− mice polarized more readily to the Th17 lineage21 and that IL‐6R and IL‐23R were both up‐regulated on activated IFN‐β −/− CD4+ T cells.22 This suggested a role for IFN‐β in T cells, influencing polarization toward the Th17 lineage and our current study suggests that IFN‐β may also regulate Th17‐polarizing cytokine production by DCs. We identified equivalent production of IL‐12p40, in IFN‐β +/+ and IFN‐β −/− DC : T‐cell co‐cultures, yet increased IFN‐γ in the IFN‐β −/− DC : T‐cell co‐cultures. Given the cytokine data we present in Fig. 5, we estimate that the ratio of Th1 : Th17 cells would be ~6 : 1 in the IFN‐β −/− DC co‐culture with 2D2 T cells, and ~7 : 1 in the IFN‐β +/+ DC co‐culture with 2D2 T cells. This may be a consequence of the increased capacity of IFN‐β −/− DCs to activate 2D2 CD4+ T cells and/or that these 2D2 T cells have been shown to produce large quantities of IFN‐γ in a recall response to MOG peptide.26 Notably, the DCs isolated from EAE mice were harvested on day 3 after disease induction, so we infer that it is unlikely that within this time period naive CD4+ T cells had been sufficiently activated and polarized to secrete cytokines that would, in turn, target these DCs in vivo. Taken together with our data indicating a role for IFN‐β in regulating expression of MHCII and CD80 on DCs, we postulate that IFN‐β may be required in DCs to limit interactions with CD4+ T cells that lead to their activation and polarization to the Th17 lineage.
One of the proposed mechanisms of IFN‐β action in MS is to regulate cell proliferation. In our earlier work, we provide evidence that IFN‐β −/− splenocytes are hyperproliferative in response to non‐specific stimuli.25 Interferon‐β exerts antiproliferative effects when added to peripheral blood monocyte cultures from patients with MS, inhibiting the proliferation of both CD4+ and CD8+ T cells.42 Given these inhibitory effects of IFN‐β on T‐cell proliferation, we postulated that IFN‐β may regulate the DC contribution to proliferation of T cells, herein supported by our data that DCs from IFN‐β −/− mice induce a greater proliferation of CD4+ T cells than DCs derived from IFN‐β +/+ mice (Fig. 2).
Circulating DCs in patients with MS have elevated levels of co‐stimulatory markers when compared with healthy controls,43 and patients with MS have more activated conventional DCs in their cerebrospinal fluid compared with their blood.44 Antigen‐presenting cells in the CNS have been implicated in re‐activation of T cells during EAE. Teige et al. provided evidence that IFN‐β down‐regulates the antigen‐presenting capacity of CNS glial cells, thereby potentially resulting in less efficient activation of autoreactive T cells in EAE.45 In the priming phase, in lymphoid tissues, we hypothesized that IFN‐β may function to regulate both proliferation and T helper cell polarization/activation, mediated by IFN‐β effects on DCs. In this study we found that treatment of splenic DCs with LPS led to their anticipated activation, detected as increased levels of MHCII, and an increase in the percentage of CD80+ and CD86+ DCs. Notably, we identified that LPS stimulation of IFN‐β −/− DCs resulted in enhanced levels of MHCII compared with IFN‐β +/+ DCs and increased the percentage of CD80+ DCs, in support of a heightened capacity for IFN‐β −/− DCs to activate T cells. The MFIs for CD80 and CD86 were comparable between IFN‐β +/+ and IFN‐β −/− DCs (data not shown), but we speculate that an increased percentage of cells expressing these markers would indicate an increased capacity to induce T‐cell proliferation. In the EAE setting, we identified a greater percentage of DCs expressing CD80 and increased expression of MHCII on CD11c+ DCs from the LNs of IFN‐β −/− mice compared with IFN‐β +/+ mice, whereas the expression of CD86 did not differ with IFN‐β status of the DCs (Fig. 4 and data not shown). Interestingly, plasmacytoid DCs from patients with MS treated with IFN‐β exhibit decreased expression of MHCII and CD86.46 Patients with MS that does not respond to IFN‐β treatment have increased expression of CD86 on their myeloid DCs, compared with patients whose MS responds to IFN‐β therapy.47 Given that IFN‐β therapy is only effective in about 30% of patients with MS, the variable response to IFN‐β therapy may reflect the variable responsiveness of different DC populations to IFN‐β, the subject of our ongoing investigations.
Migration of DCs to the CNS, orchestrated by chemokines that interact with CCR2 and CCR7, is critical for re‐activating T cells during EAE.6, 13, 14 Although we did identify an increase in CCR2 expression on BMDCs in response to LPS treatment, this was IFN‐β‐independent. Studies have identified the importance of the CCR7 ligands CCL19 and CCL21 for maintaining an inflammatory state in the CNS during EAE,8, 48 and there is evidence for CCR7+ cells accumulating in the CNS during MS.9, 49 We previously showed that LPS‐stimulated IFN‐β −/− bone‐marrow‐derived macrophages had lower STAT1 gene expression and were deficient in their ability to phosphorylate STAT1 compared with their IFN‐β +/+ counterparts.31 In other studies it has been shown that treatment of IFN‐β −/− macrophages with IFN‐β will induce STAT1 phosphorylation and restore IFN‐β‐inducible gene expression.32 Another study identified that IFN‐β treatment inhibited DC migration, through STAT1 transcriptional inhibition of CCR7.29 In our present study we provide evidence that IFN‐β −/− BMDCs are unable to induce STAT1 gene expression upon LPS stimulation, and although we did not analyse STAT1 phosphorylation in response to LPS treatment, given the findings of Sheikh et al.32 that LPS is unable to induce activation of STAT1 in IFN‐β −/− macrophages, we assume that a failure of STAT1 phosphorylation would also be observed in IFN‐β −/− BMDCs. We provide evidence that IFN‐β −/− BMDCs have increased surface expression of CCR7 when compared with IFN‐β +/+ BMDCs, whether naive (unstimulated) or in response to LPS stimulation. On day 9 after EAE induction, we observed a modest trend of increased CCR7 expression on DCs from the spleens and LNs of IFN‐β −/− mice compared with their IFN‐β +/+ counterparts, albeit not statistically significant (Fig. 6b). These findings are in agreement with the IFN‐β inhibition of CCR7 mediated by STAT1, described by Yen et al.,29 suggestive of a role for endogenous IFN‐β in the regulation of CCR7. Although CCR7−/− mice are not protected from EAE and develop disease comparable to CCR7+/+ mice, T‐cell responses appear to be initiated in the spleen, rather than the LNs of these mice. Of note, the Yen study did not address the migration of DCs or T cells to the CNS during disease, which may be altered in the absence of CCR7.50 Even though CCR7 does not appear to be necessary for induction of EAE, the findings that IFN‐β can inhibit its expression, in part by regulation of STAT1, suggest a role for IFN‐β in regulating cell migration in EAE. In our BMDC adoptive transfer experiment, we provide evidence that BMDCs generated from IFN‐β −/− mice rapidly migrate to the CNS of mice primed for EAE, in the absence of clinical scores. Our findings support the notion that one of the immunoregulatory roles of IFN‐β during neuroinflammation is to limit DC migration, in part mediated through STAT1 transcriptional inhibition of CCR7.
Taken together, our data highlight an important role for IFN‐β in regulating the interactions of T cells and DCs in the context of EAE. We show that IFN‐β is required to modulate the function of DCs in terms of antigen presentation and co‐stimulation of T cells, and regulates DC cytokine production that determines T‐cell lineage commitment. We point to a role for IFN‐β in regulating migration of DCs into lymphoid tissues and the CNS during EAE. Our findings of immunoregulatory properties of IFN‐β in DCs in the context of EAE provide further support for consideration of DC‐targeted therapies in MS.
Disclosure
The authors declare no conflict of interest.
Supporting information
Figure S1. IFN‐β is produced by splenocytes, splenic DCs and BMDCs in response to LPS stimulation.
Acknowledgements
LMP performed the experiments, LMP and ENF designed the experiments, analysed data, and prepared the manuscript. These studies were supported by a grant from the MS Society of Canada. ENF is a Tier 1 Canada Research Chair. LMP is a recipient of a Canadian Institutes of Health Research Banting and Best Doctoral Research Award.
References
- 1. McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol 2007; 8:913–9. [DOI] [PubMed] [Google Scholar]
- 2. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 2000; 47:707–17. [DOI] [PubMed] [Google Scholar]
- 3. Abdul‐Majid KB, Wefer J, Stadelmann C, Stefferl A, Lassmann H, Olsson T et al Comparing the pathogenesis of experimental autoimmune encephalomyelitis in CD4–/– and CD8–/– DBA/1 mice defines qualitative roles of different T cell subsets. J Neuroimmunol 2003; 141:10–9. [DOI] [PubMed] [Google Scholar]
- 4. Holman DW, Klein RS, Ransohoff RM. The blood–brain barrier, chemokines and multiple sclerosis. Biochim Biophys Acta 2011; 1812:220–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fife BT, Huffnagle GB, Kuziel WA, Karpus WJ. CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J Exp Med 2000; 192:899–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clarkson BD, Walker A, Harris MG, Rayasam A, Sandor M, Fabry Z. CCR2‐dependent dendritic cell accumulation in the central nervous system during early effector experimental autoimmune encephalomyelitis is essential for effector T cell restimulation in situ and disease progression. J Immunol 2015; 194:531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Simpson J, Rezaie P, Newcombe J, Cuzner ML, Male D, Woodroofe MN. Expression of the β‐chemokine receptors CCR2, CCR3 and CCR5 in multiple sclerosis central nervous system tissue. J Neuroimmunol 2000; 108:192–200. [DOI] [PubMed] [Google Scholar]
- 8. Alt C, Laschinger M, Engelhardt B. Functional expression of the lymphoid chemokines CCL19 (ELC) and CCL 21 (SLC) at the blood–brain barrier suggests their involvement in G‐protein‐dependent lymphocyte recruitment into the central nervous system during experimental autoimmune encephalomyelitis. Eur J Immunol 2002; 32:2133–44. [DOI] [PubMed] [Google Scholar]
- 9. Kivisäkk P, Mahad DJ, Callahan MK, Sikora K, Trebst C, Tucky B et al Expression of CCR7 in multiple sclerosis: implications for CNS immunity. Ann Neurol 2004; 55:627–38. [DOI] [PubMed] [Google Scholar]
- 10. Dardalhon V, Korn T, Kuchroo VK, Anderson AC. Role of Th1 and Th17 cells in organ‐specific autoimmunity. J Autoimmun 2008; 31:252–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Paterka M, Siffrin V, Voss JO, Werr J, Hoppmann N, Gollan R et al Gatekeeper role of brain antigen‐presenting CD11c+ cells in neuroinflammation. EMBO J 2016; 35:89–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bailey SL, Schreiner B, McMahon EJ, Miller SD. CNS myeloid DCs presenting endogenous myelin peptides ‘preferentially’ polarize CD4+ Th17 cells in relapsing EAE. Nat Immunol 2007; 8:172–80. [DOI] [PubMed] [Google Scholar]
- 13. Haines JL, Terwedow HA, Burgess K, Pericak‐Vance MA, Rimmler JB, Martin ER et al Linkage of the MHC to familial multiple sclerosis suggests genetic heterogeneity. The multiple sclerosis genetics group. Hum Mol Genet 1998; 7:1229–34. [DOI] [PubMed] [Google Scholar]
- 14. Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T et al Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 2005; 11:328–34. [DOI] [PubMed] [Google Scholar]
- 15. Zozulya AL, Ortler S, Lee J, Weidenfeller C, Sandor M, Wiendl H et al Intracerebral dendritic cells critically modulate encephalitogenic versus regulatory immune responses in the CNS. J Neurosci 2009; 29:140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Isaksson M, Ardesjö B, Rönnblom L, Kämpe O, Lassmann H, Eloranta M‐L et al Plasmacytoid DC promote priming of autoimmune Th17 cells and EAE. Eur J Immunol 2009; 39:2925–35. [DOI] [PubMed] [Google Scholar]
- 17. Yogev N, Frommer F, Lukas D, Kautz‐Neu K, Karram K, Ielo D et al Dendritic cells ameliorate autoimmunity in the CNS by controlling the homeostasis of PD‐1 receptor+ regulatory T cells. Immunity 2012; 37:264–75. [DOI] [PubMed] [Google Scholar]
- 18. Graber JJ, McGraw CA, Kimbrough D, Dhib‐Jalbut S. Overlapping and distinct mechanisms of action of multiple sclerosis therapies. Clin Neurol Neurosurg 2010; 112:583–91. [DOI] [PubMed] [Google Scholar]
- 19. Yu M, Nishiyama A, Trapp BD, Tuohy VK. Interferon‐β inhibits progression of relapsing–remitting experimental autoimmune encephalomyelitis. J Neuroimmunol 1996; 64:91–100. [DOI] [PubMed] [Google Scholar]
- 20. Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T et al IFN‐β gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol 2003; 170:4776–84. [DOI] [PubMed] [Google Scholar]
- 21. Galligan CL, Pennell LM, Murooka TT, Baig E, Majchrzak‐Kita B, Rahbar R et al Interferon‐β is a key regulator of proinflammatory events in experimental autoimmune encephalomyelitis. Mult Scler 2010; 16:1458–73. [DOI] [PubMed] [Google Scholar]
- 22. Pennell LM, Fish EN. Immunoregulatory effects of interferon‐β in suppression of Th17 cells. J Interferon Cytokine Res 2014; 34:330–41. [DOI] [PubMed] [Google Scholar]
- 23. Zhang X, Markovic‐Plese S. Interferon β inhibits the Th17 cell‐mediated autoimmune response in patients with relapsing–remitting multiple sclerosis. Clin Neurol Neurosurg 2010; 112:641–5. [DOI] [PubMed] [Google Scholar]
- 24. Martín‐Saavedra FM, Flores N, Dorado B, Eguiluz C, Bravo B, García‐Merino A et al β‐interferon unbalances the peripheral T cell proinflammatory response in experimental autoimmune encephalomyelitis. Mol Immunol 2007; 44:3597–607. [DOI] [PubMed] [Google Scholar]
- 25. Deonarain R, Verma A, Porter ACG, Gewert DR, Platanias LC, Fish EN. Critical roles for IFN‐β in lymphoid development, myelopoiesis, and tumor development: links to tumor necrosis factor α. Proc Natl Acad Sci USA 2003; 100:13453–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bettelli E, Pagany M, Weiner HL, Linington C, Sobel RA, Kuchroo VK. Myelin oligodendrocyte glycoprotein‐specific T cell receptor transgenic mice develop spontaneous autoimmune optic neuritis. J Exp Med 2003; 197:1073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lutz MB, Kukutsch N, Ogilvie AL, Rössner S, Koch F, Romani N et al An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 1999; 223:77–92. [DOI] [PubMed] [Google Scholar]
- 28. Zanoni I, Granucci F. Regulation of antigen uptake, migration, and lifespan of dendritic cell by Toll‐like receptors. J Mol Med 2010; 88:873–80. [DOI] [PubMed] [Google Scholar]
- 29. Yen J‐H, Kong W, Ganea D. IFN‐β inhibits dendritic cell migration through STAT‐1‐mediated transcriptional suppression of CCR7 and matrix metalloproteinase 9. J Immunol 2010; 184:3478–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Platanias LC. Mechanisms of type‐I‐ and type‐II‐interferon‐mediated signalling. Nat Rev Immunol 2005; 5:375–86. [DOI] [PubMed] [Google Scholar]
- 31. Thomas KE, Galligan CL, Newman RD, Fish EN, Vogel SN. Contribution of interferon‐β to the murine macrophage response to the toll‐like receptor 4 agonist, lipopolysaccharide. J Biol Chem 2006; 281:31119–30. [DOI] [PubMed] [Google Scholar]
- 32. Sheikh F, Dickensheets H, Gamero AM, Vogel SN, Donnelly RP. An essential role for IFN‐β in the induction of IFN‐stimulated gene expression by LPS in macrophages. J Leukoc Biol 2014; 96:591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martins TB, Rose JW, Jaskowski TD, Wilson AR, Husebye D, Seraj HS et al Analysis of proinflammatory and anti‐inflammatory cytokine serum concentrations in patients with multiple sclerosis by using a multiplexed immunoassay. Am J Clin Pathol 2011; 136:696–704. [DOI] [PubMed] [Google Scholar]
- 34. Romme Christensen J, Börnsen L, Hesse D, Krakauer M, Sørensen PS, Søndergaard HB et al Cellular sources of dysregulated cytokines in relapsing–remitting multiple sclerosis. J Neuroinflammation 2012; 9:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sweeney CM, Lonergan R, Basdeo SA, Kinsella K, Dungan LS, Higgins SC et al IL‐27 mediates the response to IFN‐β therapy in multiple sclerosis patients by inhibiting Th17 cells. Brain Behav Immun 2011; 25:1170–81. [DOI] [PubMed] [Google Scholar]
- 36. Ramgolam VS, Sha Y, Jin J, Zhang X, Markovic‐Plese S. IFN‐β inhibits human Th17 cell differentiation. J Immunol 2009; 183:5418–27. [DOI] [PubMed] [Google Scholar]
- 37. Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S et al Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17‐producing T cells. Nat Immunol 2006; 7:929–36. [DOI] [PubMed] [Google Scholar]
- 38. Murugaiyan G, Mittal A, Lopez‐Diego R, Maier LM, Anderson DE, Weiner HL. IL‐27 is a key regulator of IL‐10 and IL‐17 production by human CD4+ T cells. J Immunol 2009; 183:2435–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yen J‐H, Kong W, Hooper KM, Emig F, Rahbari KM, Kuo P‐C et al Differential effects of IFN‐β on IL‐12, IL‐23, and IL‐10 expression in TLR‐stimulated dendritic cells. J Leukoc Biol 2015; 98:689–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kimura A, Kishimoto T. IL‐6: regulator of Treg/Th17 balance. Eur J Immunol 2010; 40:1830–5. [DOI] [PubMed] [Google Scholar]
- 41. Axtell RC, de Jong BA, Boniface K, van der Voort LF, Bhat R, De Sarno P et al T helper type 1 and 17 cells determine efficacy of interferon‐β in multiple sclerosis and experimental encephalomyelitis. Nat Med 2010; 16:406–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Noronha A, Toscas A, Jensen MA. Interferon β decreases T cell activation and interferon γ production in multiple sclerosis. J Neuroimmunol 1993; 46:145–53. [DOI] [PubMed] [Google Scholar]
- 43. Karni A, Abraham M, Monsonego A, Cai G, Freeman GJ, Hafler D et al Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol 2006; 177:4196–202. [DOI] [PubMed] [Google Scholar]
- 44. Pashenkov M, Huang YM, Kostulas V, Haglund M, Söderström M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain 2001; 124:480–92. [DOI] [PubMed] [Google Scholar]
- 45. Teige I, Liu Y, Issazadeh‐Navikas S. IFN‐β inhibits T cell activation capacity of central nervous system APCs. J Immunol 2006; 177:3542–53. [DOI] [PubMed] [Google Scholar]
- 46. Lande R, Gafa V, Serafini B, Giacomini E, Visconti A, Remoli ME et al Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon‐β . J Neuropathol Exp Neurol 2008; 67:388–401. [DOI] [PubMed] [Google Scholar]
- 47. Comabella M, Lünemann JD, Río J, Sánchez A, López C, Julià E et al A type I interferon signature in monocytes is associated with poor response to interferon‐β in multiple sclerosis. Brain 2009; 132:3353–65. [DOI] [PubMed] [Google Scholar]
- 48. Columba‐Cabezas S, Serafini B, Ambrosini E, Aloisi F. Lymphoid chemokines CCL19 and CCL21 are expressed in the central nervous system during experimental autoimmune encephalomyelitis: implications for the maintenance of chronic neuroinflammation. Brain Pathol 2003; 13:38–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M et al CCL19 is constitutively expressed in the CNS, up‐regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol 2007; 190:72–9. [DOI] [PubMed] [Google Scholar]
- 50. Pahuja A, Maki RA, Hevezi PA, Chen A, Verge GM, Lechner SM et al Experimental autoimmune encephalomyelitis develops in CC chemokine receptor 7‐deficient mice with altered T‐cell responses. Scand J Immunol 2006; 64:361–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. IFN‐β is produced by splenocytes, splenic DCs and BMDCs in response to LPS stimulation.