

Abstract
The major codon preference model suggests that codons read by tRNAs in high concentrations are preferentially utilized in highly expressed genes. However, the identity of the optimal codons differs between species although the forces driving such changes are poorly understood. We suggest that these questions can be tackled by placing codon usage studies in a phylogenetic framework and that bacterial genomes with extreme nucleotide composition biases provide informative model systems. Switches in the background substitution biases from GC to AT have occurred in Gardnerella vaginalis (GC = 32%), and from AT to GC in Lactobacillus delbrueckii (GC = 62%) and Lactobacillus fermentum (GC = 63%). We show that despite the large effects on codon usage patterns by these switches, all three species evolve under selection on synonymous sites. In G. vaginalis, the dramatic codon frequency changes coincide with shifts of optimal codons. In contrast, the optimal codons have not shifted in the two Lactobacillus genomes despite an increased fraction of GC-ending codons. We suggest that all three species are in different phases of an on-going shift of optimal codons, and attribute the difference to a stronger background substitution bias and/or longer time since the switch in G. vaginalis. We show that comparative and correlative methods for optimal codon identification yield conflicting results for genomes in flux and discuss possible reasons for the mispredictions. We conclude that switches in the direction of the background substitution biases can drive major shifts in codon preference patterns even under sustained selection on synonymous codon sites.
Keywords: codon usage, mutation bias, Lactobacillus, Bifidobacterium, optimal codon
Introduction
More than 30 years ago it was discovered that synonymous codons are not used randomly (Grantham et�al. 1980, 1981). In bacteria, codon usage patterns differ between genomes, as reflected in GC content values at third codon positions that range from 20% to 80% (Muto and Osawa 1987). Codon frequencies also differ between genes within the same genome due to selection for translational efficiency and accuracy (reviewed in Andersson and Kurland 1990; Bulmer 1991; Sorensen and Pedersen 1991; Akashi 1994; Novoa and Ribas de Pouplana 2012). Horizontal gene transfer events (Moszer et�al. 1999; Ochman et�al. 2000), strand-specific mutation biases (Lobry 1996; McLean et�al. 1997) and biased gene conversion (Lassalle et�al. 2015) also contribute to gene-specific differences in codon frequencies.
Early work done in the classical model organisms Escherichia coli, Bacillus subtilis and Saccharomyces cerevisiae showed that the frequencies of preferred codons in the highly expressed genes (Bennetzen and Hall 1982; Gouy and Gautier 1982; Grosjean and Fiers 1982; Shields and Sharp 1987; Bulmer 1991), correlated with the abundance of the corresponding isoacceptor tRNAs (Ikemura 1981, 1985; Bulmer 1987). Furthermore, the level of codon bias in each individual gene correlated with its expression level (Ikemura 1985; Kanaya et�al. 1999; Ghaemmaghammi et�al. 2003; Goetz and Fugelsang 2005). These observations formed the basis for the so-called major codon preference model, which suggests that selection for codon bias is a growth optimization strategy (Kurland 1987; Andersson and Kurland 1990). Consistently, it has been shown that rapidly growing bacterial species have more tRNA genes and show a stronger influence of selection on codon bias than slowly growing bacteria (Rocha 2004).
The preferred codons in E. coli, also designated the “optimal” codons (Ikemura 1981), include both AT- and GC-ending codons. In two-codon boxes read by a single tRNA, C-ending codons are generally favored over U-ending codons (Sharp et�al. 2005), due to a more efficient interaction with the G in the first anticodon position of the tRNA. However, in four- and six-codon boxes read by multiple tRNAs the identities of the preferred codons differ between species due to differences in isoacceptor tRNA concentrations (Grantham et�al. 1980; Sharp et�al. 2005). For example, GC-ending codons are preferred in Drosophila (Shields et�al. 1988; Vicario et�al. 2007) whereas AT-ending codons are preferred in Saccharomyces (Bennetzen and Hall 1982). Bacterial genomes display the whole spectrum of patterns, with subsets of optimal codons that are unique for each species (Hershberg and Petrov 2009). This raises a conundrum: if selection favors a stable co-adaptive relationship between codon frequencies and the concentrations of the corresponding isoacceptor tRNAs, how can the identity of optimal codons change over evolutionary time, leading to diverse sets of preferred codons in different species?
Three hypotheses have been proposed to explain switches of optimal codons, and they differ in the role that selection is thought to play. The first hypothesis suggests that prolonged periods of weak selection on translational efficiency are needed to generate diversity in codon preference patterns (reviewed in Sharp et�al. 2010). This hypothesis is inspired by the Sewall Wright’s shifting balance theory of evolution (Wright 1977), which introduces drift to explain shifts in codon usage without invoking changes in the mutational spectrum. Previous studies found evidence for selection on translational in 25% to almost 100% of the species examined, depending on the methodology used (dos Reis et�al. 2004; Carbone et�al. 2005; Sharp et�al. 2005; Supek et�al. 2010). However, even when signatures of selection are detectable, these might result from long-term selective constraints that could have been interspersed by shorter periods during which selection for codon bias was lost due to population bottlenecks, host-adaptation or other lifestyle changes. It is hypothesized that periods of weak or no selection randomized codon usage patterns. The re-imposition of selective constraints would then introduce a new co-adaptive stage during which different sets of codons and isoacceptor tRNAs increase in abundance and become selectively favored. According to this model, the choice of optimal codons is the result of a frozen accident and as such is unpredictable. Here, we refer to this model as the “relaxed selection” hypothesis.
The second hypothesis stresses the importance of the mutation pressure and suggests that a dramatic change in global GC content may lead to a large reduction of previously preferred codons and introduce new codons in high abundance (Shields 1990). The term “mutation “is here used in the broad sense to include factors other than single nucleotide substitutions such as biased gene conversion, which can also change genomic GC contents (Hershberg and Petrov 2010; Hildebrand et�al. 2010). According to this model, optimal codon choice follows changes in global GC content and is thus predictable. Importantly, loss of selection would not be required for a shift of codon preference patterns. We refer to this model the “mutation-driven” hypothesis.
The third hypothesis is a variant of the “mutation-driven” hypothesis and suggests that horizontal gene transfer events may mediate changes in codon frequencies by introducing genes with a different codon bias (Hershberg and Petrov 2008). If high expression levels of the recently acquired genes are essential for survival of the bacterial cells, tRNA isoacceptor concentrations might change to fit the codon bias of the foreign genes, after which codon frequencies in all other genes will be adjusted to match the new tRNA expression levels. Thus, also in this model optimal codon shifts would occur without weakened selection.
Although the three hypotheses have been available for some time, no attempts have been made to assess their importance as it is difficult to collect an appropriate data set to differentiate them. A broad study of more than 700 genomes, of which 675 were bacterial genomes, showed that the GC-richness of the optimal codons correlate with the global GC content values, indirectly supporting mutation-driven hypotheses (Hershberg and Petrov 2009). However, the strengths of selection acting on the individual genomes were not estimated and no studies of optimal codon switches in phylogenetically related bacterial species with different GC contents were performed to validate the hypothesis.
Lactic acid bacteria provide good model systems for studies of codon usage patterns. They comprise a wide range of phylogenetically disparate species that ferment carbohydrates into lactic acid as a major end product, including genera such as Lactobacillus, Enterococcus, Lactococcus, Streptococcus and Bifidobacterium (Klein et�al. 1998). These bacteria share many biochemical and physiological properties, and they tend to inhabit ecological niches that are rich in carbohydrates (Stiles and Holzapfel 1997). Lactobacillus species evolve under strong AT-bias, whereas Bifidobacterium species evolve under strong GC-bias. Within genera, most species conform to the characteristic codon usage patterns. However, a few outlier species within each genus have substantially higher or lower genomic GC contents than the majority, which provides an opportunity to test hypotheses about how shifts in the mutational spectrum have affected the choice of optimal codons. Lactobacillus species grow rapidly in nutrient-rich habitats and it has been shown that codon usage patterns are influenced by selection for translational efficiency (Nayak 2012).
In this study, we have examined whether the altered genomic GC content in the outlier species of Lactobacillus and Bifidobacterium have induced switches of optimal codons in the highly expressed genes. We refer to the changes in genomic GC content as changes in the direction of the background substitution patterns, irrespectively of whether these changes are caused by single nucleotide mutations or recombination events. We define optimal codons as codons that evolve under selection for translational efficiency in the highly expressed genes, and abundant codons as codons used at high frequency in the majority of genes. We discuss the results in the context of the proposed hypotheses for how major codon preference patterns change over evolutionary time.
Materials and Methods
Bacterial Genomes
Complete genomes of Lactobacillus species as of 1 January 2015 were retrieved from the National Center for Biotechnology Information (NCBI) and additional L. kunkeei genomes were added from (Ellegaard et�al. 2015; Tamarit et�al. 2015). The Bifidobacterium data set comprised the diversity of species with complete genomes deposited at NCBI as of May 2014 and was completed with B. asteroids and B. coryneforme genomes from (Ellegaard et�al. 2015). All Lactobacillus genomes used for the initial phylogeny are shown in supplementary fig. S1, Supplementary Material online. All Lactobacillus and Bifidobacterium genomes used for the codon usage analyses are listed in supplementary table S1, Supplementary Material online.
Phylogenetic Analyses
For each Lactobacillus genome, all annotated proteins shorter than 50 amino acids were filtered out, and an all-against-all BLAST comparison was done using an E-value cutoff of 1e-05 (Altschul et�al. 1990). The Lactobacillus proteome was classified into protein families using OrthoMcl, using an inflation parameter value of 1.5 (Li et�al. 2003). Of these, 54 protein families contained a single protein from each one of the 135 taxa. The 54 single-copy panorthologs were individually aligned with Mafft-linsi (Katoh et�al. 2002, 2005), trimmed for all positions with over 50% gaps with trimAl (Capella-Gutierrez et�al. 2009), and concatenated using a custom perl script. The phylogeny was inferred using RAxML (Randomized Axelerated Maximum Likelihood) with the PROTCATLG model and 100 bootstrap pseudoreplicates (Stamatakis 2006). A reduced data set of 34 genomes from the Lactobacillaceae and Leuconostocaceae families was selected for codon usage analysis. The 54 single-copy panorthologs from the reduced set of taxa were aligned with Probcons (Do et�al. 2005) and trimmed with BMGE (Criscuolo and Gribaldo 2010) with default parameters. A tree was inferred using RAxML with the PROTGAMMALG model and 100 bootstrap pseudoreplicates. The Bifidobacterium data set was treated similarly: OrthoMcl was first used to detect 400 single-copy panorthologs, which were then aligned with Mafft-linsi (Katoh et�al. 2005), trimmed for positions with over 50% gaps, and concatenated with local perl scripts. A tree was then reconstructed with RAxML as before.
Codon Usage Analysis and Genome Statistics
Genome statistics, including GC content, GC3s and Nc, and correspondence analyses were calculated with the aid of the software CodonW (Peden 1999). The Nc values were calculated based on the GC3s values by the method defined by Wright, as Ncexpect = 2 + GC3s + 29/(GC3s + (1−GC3s)2) (Wright 1990; Chen 2013). The codon usage index (CAI) was calculated by CAI and cusp function from EMBOSS package (Rice et�al. 2000). The relative synonymous codon usage (RSCU) values were calculated using the program GCUA (General Codon Usage Analysis) (McInerney 1998). The strength of selected codon usage bias was estimated from the S index, which is used as a proxy for translational selection on individual genomes (Sharp et�al. 2005, 2010). The number of tRNA genes and the inference of anti-codons were made with the aid of tRNAscan-SE 1.3.1 (Lowe and Eddy 1997). Other statistics, including length of CDS and nucleotide position within the genome, were calculated from Genbank or annotation files using Perl and R scripts.
The genes in each data set were categorized into highly expressed and all genes (Sharp et�al. 2005). The highly expressed genes were defined as in (Sharp et�al. 2005), and included genes for translation elongation factor Tu, Ts and G, and 37 large ribosomal proteins, including rplA-rplF, rplI-rplT and rpsB-rpsT, whereas the all genes data set included all genes in the genome. Codons used significantly more or less frequently in the highly expressed gene data set compared with the whole genome data set (chi-squared test, cutoff P = 0.01) were defined as optimal (+) and nonoptimal (−) codons according to the Ribosomal Protein (RP) method. Optimal codons were also predicted by the correlative test (Hershberg and Petrov 2009). In this test, the Nc value for each gene was plotted against the RSCU value for each codon and, for each amino acid, and the codons showing the strongest negative correlation with high significance (P < 0.05/number of codons in the codon family) were inferred to represent the optimal codons. For G. vaginalis, L. delbrueckii and L. fermentum, we also identified optimal codons by testing the correlation between the RSCU values and Nc′ values, with Nc′ values calculated using the ENCprime package (Novembre 2002).
For the Akashi test (1994), we extracted single copy panorthologs genes from (Ellegaard et�al. 2015; Tamarit et�al. 2015). The extracted data set included 400 genes from the Bifidobacterium species and 302 genes from Lactobacillus species. Amino acid sequence alignments were built with MAFFT-linsi (Katoh et�al. 2002), and then backtranslated to nucleotide sequence alignments. For the identification of conserved and variable sites, we used A. phenanthrenivorans as the reference species for G. vaginalis and S. pyogenes as the reference species for L. delbrueckii and L. fermentum. Conserved sites were defined as codon sites that code for the same amino acid as the sequence in the reference genome, and variable sites as codon sites in the alignment that code for different amino acids. For the implementation of the Akashi’s test (1994), we used the procedure described on the website “http://drummond.openwetware.org/Akashi’s_Test.html”. It is suggested that the Akahi’s test is implemented using the Mantel–Haenszel test in the open-source statistical package R. However, we realized that the test in R is not appropriate because it does not distinguish positive from negative signs. Instead, we followed the procedure exactly as detailed on the website.
Species-Specific Genes
The species-specific proteins were obtained by analysing the output of the bifidobacterial orthoMcl reconstruction (Ellegaard et�al. 2015), and the 135-genomes Lactobacillus orthoMcl reconstruction. The species-specific genes were defined as the singletons in these reconstructions, plus all proteins present in clusters with no other species from the ingroup. These proteins were used as queries in BLASTP searches against the Non-redundant database (NR), using an E-value cutoff of 1e-03. All genes yielding more hits to other species within the ingroup than to foreign genera within the best 50 hits were discarded. Hits from the same species as the query were filtered out as self hits, as were also hits from closely related species with similar GC content, such as the Lactobacillus species L. panis, L. oris, L. vaginalis, L. antri, L. frumenti and L. pontis, in the case of L. reuteri (Vogel et�al. 1994; Felis and Dellaglio 2007); L. equicursoris in the case of L. delbrueckii (Morita et�al. 2010) and L. hakayitensis in the case of L. salivarius (Morita et�al. 2007).
In the initial search, the species-specific genes of G. vaginalis yielded numerous hits to Chlamydia trachomatis. These hits originated from sequencing projects published by the Sanger Institute on 10 March 2015 in NCBI. The samples were claimed to represent C. trachomatis genomes, but contained several thousand contigs and several thousand genes. Phylogenetic inferences based on all recruited BLAST hits showed that the identified C. trachomatis sequences clustered inside the G. vaginalis clade, and that they were never represented by more than one or two sequences. In order to assess whether these C. trachomatis genomes were contaminated with G. vaginalis, we blasted the contigs of seven genomes against all 119 Chlamydia complete genomes (of which 88 belong to C. trachomatis) and 4 G. vaginalis complete genomes found in NCBI at 20 November 2015. The seven genomes had between 3 and 1,110 contigs with best BLASTn hits to the Gardnerella rather than the Chlamydia genomes. Therefore, we concluded that these hits came from metagenomes formed by contaminations or co-infections with G. vaginalis, and were filtered out as self-hits. The next best 250 hits were retrieved with the aid of a tBLASTn search (E < 1e-05). The sequences of the hits were retrieved and their GC3s values were calculated using CodonW (Peden 1999).
Results
Switches in GC Content in Lactobacillus and Bifidobacterium Species
To study how shifts in GC content have affected codon preference patterns, we selected 14 species from the family Bifidobacteriaceae and 34 species from the family Lactobacillaceae for an in-depth analysis (supplementary table S1, Supplementary Material online). The data sets were selected so as to represent the genetic diversity of species for which complete genome sequence data were available in the public database as of 1 January 2015 (supplementary fig. S1, Supplementary Material online). Genome sizes for the taxa included in the Lactobacillus data set ranged from 1.3 Mb in Lactobacillus sanfranciscensis to 3.0 Mb in Lactobacillus plantarum and Lactobacillus rhamnosus and in the Bifidobacterium data set from 1.6 Mb in Gardnerella vaginalis to 2.8 Mb in Bifidobacterium longum (supplementary table S2, Supplementary Material online). The selected genomes cover the whole spectrum of base composition variation in bacteria, as reflected in GC content values that span from <20% to >80% at third codon synonymous sites (GC3s) in the individual genomes (fig. 1).
Fig. 1.—
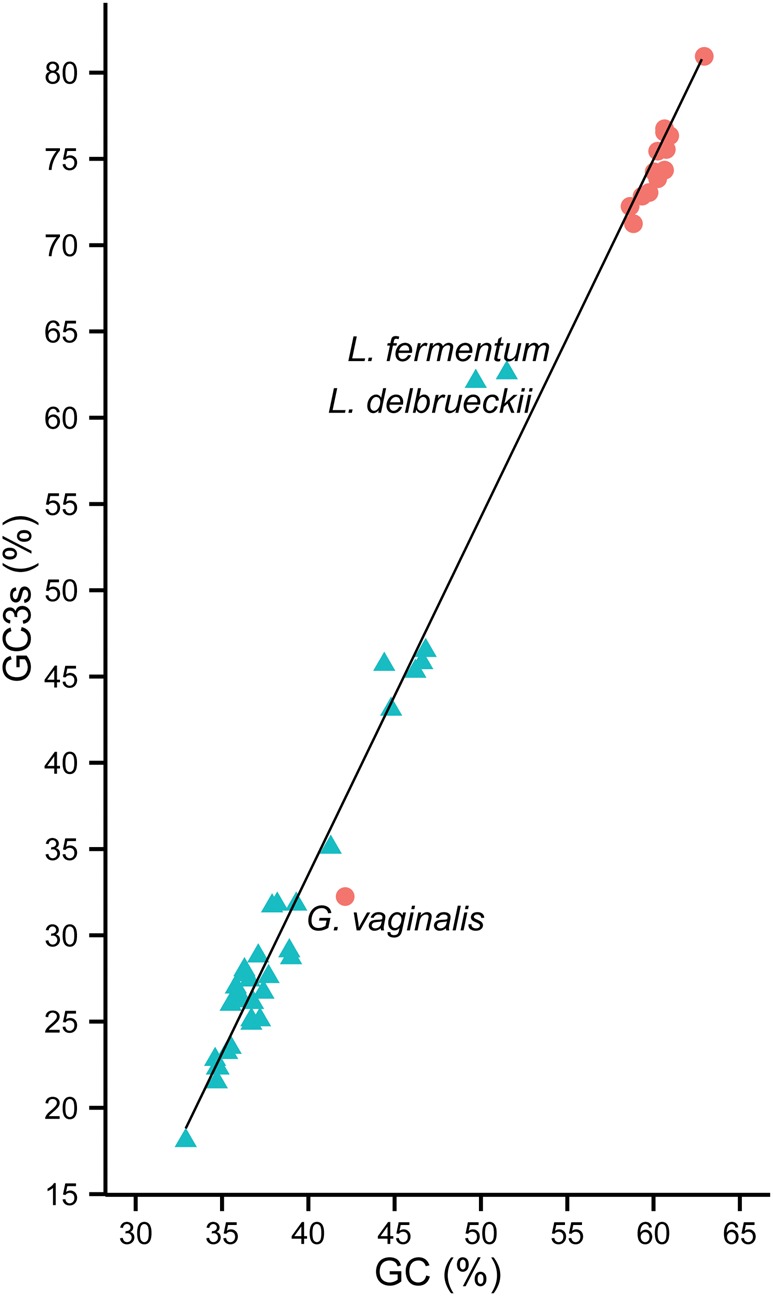
Variation in genomic GC content values in Lactobacillus and Bifidobacterium. The GC content at third codon synonymous sites (GC3s) is plotted against the overall genomic GC content (GC) for Lactobacillus (blue) and Bifidobacterium (red). The individual species are listed in supplementary table S1, Supplementary Material online, and their GC content at third codon synonymous sites in supplementary table S2, Supplementary Material online. Three species with a markedly deviating GC content, Gardnerella vaginalis, Lactobacillus fermentum and L. delbrueckii, are highlighted.
Importantly for the aim of this study, three genomes have GC content values that deviate drastically from the values of the group that they belong to, indicating that the data set is a good representation of intra- and inter-phyla variation of GC content values in bacterial genomes. The genes in the Gardnerella vaginalis genome have a mean GC3s value of only 32% as compared with 75% in the other bifidobacterial species, and the genes in the Lactobacillus fermentum and Lactobacillus delbrueckii genomes have mean GC3s values of 62–63%, as compared with 29% in the other Lactobacillus species. It is proposed that mutations are universally biased towards AT (Hershberg and Petrov 2008), and other factors contribute to a bias towards GC, such as gene conversion (Lassalle et�al. 2015), influence of an error-prone alpha subunit of DNA polymerase III (Wu et�al. 2014) and selection for GC-ending codons (Ran et�al. 2014). Indeed, we confirmed a mutation bias towards AT in recent nucleotide substitutions in G. vaginalis and, comparatively, a much higher rate of recent nucleotide substitutions contributing to GC richness in L. fermentum and L. delbrueckii (supplementary text S1, Supplementary Material online). However, none of the other factors, such as biased gene conversion or selection for GC-ending codons, provided a good fit for the increase in GC content in L. delbrueckii and L. fermentum (supplementary text S1, Supplementary Material online).
To place the Lactobacillus species with a deviating GC-content in their evolutionary context, we inferred a genome phylogeny based on a concatenated protein alignment of 54 single copy orthologs using maximum likelihood methods (fig. 2A). The tree topology showed that the two Lactobacillus species with higher GC-content than the others, L. fermentum and L. delbrueckii, are not sister species, as has also been shown elsewhere (Felis and Dellaglio 2007; Lukjancenko et�al. 2012; Ellegaard et�al. 2015). Rather, L. fermentum with a GC3s value of 62% clustered with Lactobacillus reuteri with a GC3s value of only 29%. Likewise, the analysis showed that L. delbrueckii with a GC3s value of 62% is embedded in a clade of Lactobacillus species with GC3s values of 22–30%. Less dramatic increases in GC content were mapped on the node to Lactobacillus plantarum, Lactobacillus brevis and Lactobacillus buchneri, and on the node to Lactobacillus casei and Lactobacillus rhamnosus, all species of which have GC3s values of 43–46%. It is noteworthy that L. buchneri is a sister species to Lactobacillus sanfranciscensis and Lactobacillus apinorum with GC3s values of only 21.5–22.8%. This suggests that the increase in GC-content in the ancestor of L. buchneri, L. brevis and L. plantarum was followed by a reversal in the direction of the background substitution bias towards AT in one of the descending lineages.
Fig. 2.—

Phylogenetic relationships and genome features of Lactobacillus species. The maximum likelihood phylogeny was inferred from a concatenated alignment of 54 single-copy orthologs present in all species. The species were selected so as to represent the diversity of Lactobacillus based on the phylogeny shown in supplementary fig. S1, Supplementary Material online. Numbers at nodes refer to bootstrap support values, and branch lengths correspond to the substitution rate. Accession numbers for all Lactobacillus strains and outgroup taxa are detailed in supplementary table S1, Supplementary Material online. A schematic figure illustrates the variation in GC contents, selective constraints (S-value), genome sizes and number of tRNAs in the species selected for an in-depth analysis. The color’s saturation represents the numeric values for the genomic statistics.
A phylogeny of the Bifidobacterium species indicated that G. vaginalis with a GC3s value of 32% is an early diverging species within one of the two bifidobacterial clades (fig. 2B) which otherwise encompasses species with GC3s values >70% (supplementary table S2, Supplementary Material online) (Ellegaard et�al. 2015). This is in line with previous phylogenetic inferences, which also placed G. vaginalis within the genus Bifidobacterium (Miyake et�al. 1998; Munoz et�al. 2011). We conclude that the direction of the background substitution bias has switched several times in the genus Lactobacillus, and at least once in the genus Bifidobacterium, resulting in genomes with drastically different GC content values compared with their most closely related species.
Selection on Codon Usage in Genomes with Altered Genomic GC Contents
To study the intra-species variation in synonymous codon usage for the genomes in our data set, we used two different statistical approaches. We illustrate the general trends in our data sets with Lactobacillus salivarius and Bifidobacterium bifidum, which have GC3s values of 18% and 81%, respectively. These two species are here used as the “archetype species” of Lactobacillus and Bifidobacterium. With “archetype species” we mean those species displaying the canonical codon usage pattern of the majority of species in their respective families, in contrast to the species in which the genomic G + C content have recently shifted.
First, we conducted a correspondence analysis (fig. 3), which is a multivariate statistical method that generates a series of orthogonal axes that display the variation in the data set (Peden 1999). The first axis accounted for 10–25% of the variation, and the second axis for 5–10% of the variation in both groups of bacteria (supplementary table S3, Supplementary Material online). In the archetype species L. salivarius (fig. 3A) and B. bifidum (fig. 3B), axis one correlated with the codon adaptation index (CAI), confirming a strong influence of selective constraints on synonymous codon usage. In all species of Bifidobacterium, axis one also correlated with the GC3s values (supplementary figs. S2–S4, Supplementary Material online), suggesting that both mutation and selection have driven codon usage patterns towards GC-rich codons, and that the resulting gradient in GC content among genes explains most of the intra-specific variation in codon usage patterns. In Lactobacillus, we observed a much larger variation in correlation statistics among species (supplementary table S3, Supplementary Material online). For the majority of Lactobacillus species, axis one correlated with the CAI values but not with the GC3s values, and in a few species no correlation was observed to either of these two factors in the first two axes (supplementary figs. S5–S7, Supplementary Material online).
Fig. 3.—

Correspondence analysis of codon usage frequency variation. The two main axes produced the correspondence analyses are shown for (A) Lactobacillus salivarius, (B) Bifidobacterium bifidum, (C) L. delbrueckii, (D) Gardnerella vaginalis, (E) L. fermentum and (F) L. reuteri. Genes for ribosomal proteins and elongation factors are shown in black. Species-specific genes are shown in red. The variation (v) and correlation (r) value for the correspondence analysis are shown in the bottom of each plot.
Having established the intra-genomic variation in species with the canonical codon usage patterns, we turned to the species in which the direction of the background substitution bias has been altered. Despite a much higher overall A + T content in G. vaginalis, axis one correlated with both CAI and GC3s values, as in the other bifidobacteria (fig. 3D). In L. delbrueckii (fig. 3C) and L. fermentum (fig. 3E), axis one correlated with GC3s and axis two with the CAI values. The three species showed a rabbit-ear shape of the plots, in which the ribosomal protein genes were located in one ear and several of the species-specific genes were located in the other ear. The segregation of these two groups of genes along the second axes correlated with the CAI values. Thus, all three genomes displayed the characteristic signs of selection.
For an intuitively more appealing visualization of the patterns, we estimated the effective number of codons (Nc) for all species in our data set (fig. 4 and supplementary fig. S8, Supplementary Material online). The archetype species L. salivarius and B. bifidum showed the expected Nc values for the given GC3s values, and the spread of the dots was quite narrow (fig. 4A and B). The ribosomal protein genes were situated at the lower ends of the cloud of dots, as expected for genes with a codon bias that is shaped by selective constraints.
Fig. 4.—

The effective number of codons (Nc) used in each gene for a range of G + C content values at third codon synonymous sites (GC3s). The correlation between the Nc and the GC3s values are shown for (A) Lactobacillus salivarius, (B) Bifidobacterium bifidum, (C) L. delbrueckii, (D) Gardnerella vaginalis, (E) L. fermentum and (F) L. reuteri. Genes for ribosomal proteins and elongation factors are shown in black. Species-specific genes are shown in red. The curve represents the null hypothesis that Nc values are determined solely by the mutational bias.
The Nc plots for L. delbrueckii (fig. 4C) and L. fermentum (fig. 4E) were strikingly different from that of L. salivarius (fig. 4A) and L. reuterii (fig. 4F) in that the genes showed a broader variation in GC3s values, ranging from 20% to 80%, consistent with GC3s explaining most of the variation among genes in the correspondence analyses. The majority of genes were located at the right half of the plot with GC3s values >50%, whereas the ribosomal protein genes presented GC3s values of ∼50% and Nc values <40 in both species (fig. 4C and E). If selection had been relaxed, then we would have expected the ribosomal protein genes to show similarly high GC3s values as the majority of genes. The similarity in codon usage patterns in the two species is rather indicative of convergence due to selection for the previously used subset of AT-rich codons. However, the GC3s values of the ribosomal protein genes is slightly higher in L. fermentum than in L. delbrueckii, which might indicate a faster rate of evolution, weaker selective constraints or longer time because the switch of the background substitution bias. Genes with GC3s values <40% and Nc values >40 in L. fermentum and L. delbrueckii encoded restriction-modification systems, integrases, glycosyltransferases and ABC transporters (supplementary table S4, Supplementary Material online), suggesting that they represent horizontal gene transfers. The Nc plot for G. vaginalis also displayed a quite large spread of the GC3s values, and was more similar to the Nc plots for the archetype Lactobacillus species than to the Nc plot of the archetype Bifidobacterium species (fig. 4D). The species-specific genes in G. vaginalis showed homology to species that are part of the healthy or opportunistic microbiota of the vaginal tract, such as Neisseria gonorrhoeae, Atopobium vaginae and Lactobacillus iners, supporting gene exchange within this niche (supplementary table S4, Supplementary Material online). Many species-specific genes were co-located with genes that showed similarity to A. vaginae and L. iners in chromosomal segments with low GC3s values (supplementary table S4, Supplementary Material online). Thus, in addition to selective constraints on highly expressed genes, codon usage patterns in the three GC-shifted genomes have also been influenced by horizontal gene transfer events.
In order to quantify the relative strength of selection, we calculated the S indexes for the genomes in this study (supplementary table S2, Supplementary Material online). The S index is inferred from the relative use of C-ending codons for Asn, Ile, Phe, Tyr (Sharp et�al. 2005), which form a standard G:C basepair with the tRNA at the first anticodon position and are generally considered to be translationally optimal, although this may depend on species-specific tRNA modifications. In a previous study of 80 bacterial genomes, the S index ranged from −0.88 to 2.65, and >0.2 were chosen as a cutoff for translational selection (Sharp et�al. 2005). The S indexes in the Lactobacillus genomes also showed a broad range of variation, from −0.35 to 1.80, and the estimates were largely concordant with the species-specific variation in indications of selection on synonymous codon choice in the correspondence analyses. The highest S values in our data set, 1.7–1.8, were estimated for L. kunkeei, suggesting selective constraints on synonymous sites in the highly expressed genes of this species. On the other extreme, the S value in Lactobacillus mellis and Lactobacillus mellifer were ∼−0.3, implying that translational selection on codon usage patterns in these species is not detectable by this method.
Importantly, L. delbrueckii and L. fermentum have S values of 1.2 and 1.3, respectively, as did also L. reuteri. Thus, despite the difference in GC content in the close relatives L. fermentum and L. reuteri, both species have evolved under selective constraints on the highly expressed genes. In Bifidobacterium, the S index ranged from 0.81 to 1.38, including G. vaginalis with an S index of 1.03. We conclude that the codon preference patterns in the highly expressed genes of all three species with GC shifted genomes have been influenced by selective constraints on synonymous codon sites.
Inferences of Optimal Codon Identities
To identify optimal codons, we contrasted the relative synonymous codon usage (RSCU) values for a data set consisting of 37 ribosomal protein genes and three other highly expressed genes (the RP data set) (figs. 5–7) with the RSCU values of all genes in a data set consisting of all protein coding genes in the whole genomes (the WG data set) of Bifidobacterium and Lactobacillus (supplementary figs. S9–S11, Supplementary Material online). We defined optimal codons as codons that were significantly more abundant in the RP than in the WG data set, irrespectively of their actual frequencies. Thus, it should be noted that both highly abundant (major) and lowly abundant (minor) codons might be considered optimal. We refer to the identification of optimal codons using this approach as the RP method.
Fig. 5.—
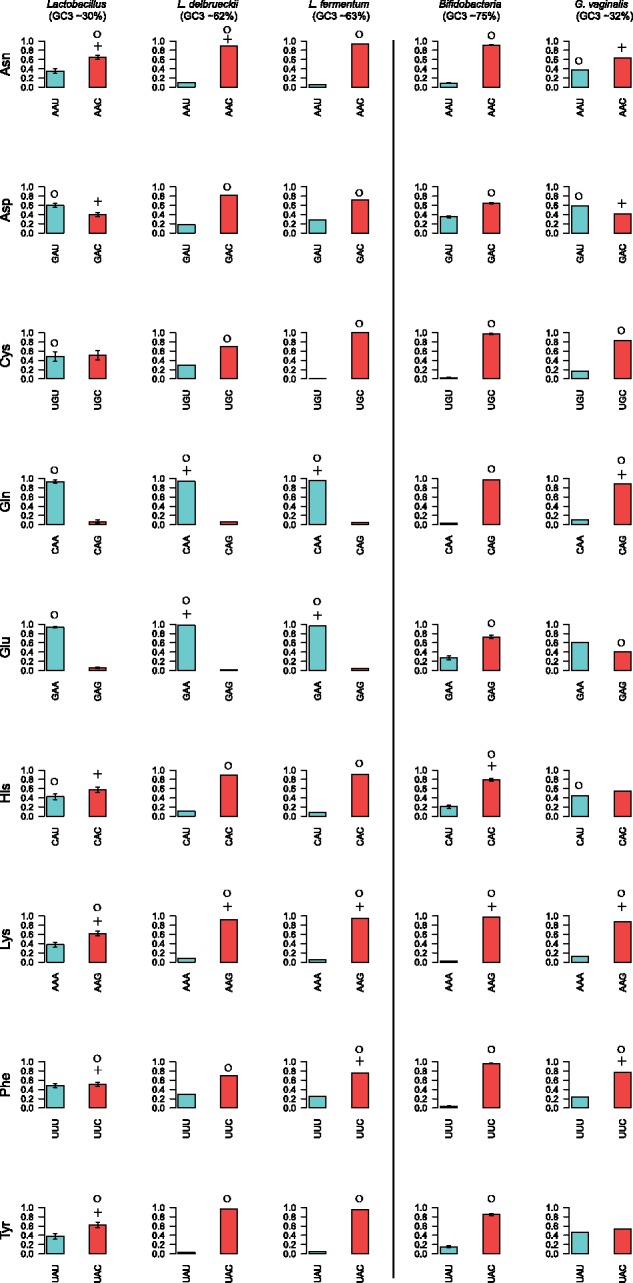
Relative fraction of codons for amino acids encoded by two codons. The figure shows the relative fraction of codons in each two-codon box for the highly expressed genes of Lactobacillus (to the left) and Bifidobacterium (to the right), estimated as the average frequency over the species included in the analysis, excluding the GC-shifted genomes. The error bar shows the standard error for each codon. Blue and red colors refer to AU- and GC-ending codons, respectively. The optimal codons predicted by RP method are marked with the symbol “+”, and optimal codons predicted by correlative method are marked with the symbol “o”. Codons were defined as “optimal” in Lactobacillus and Bifidobacterium if predicted to be optimal in > 40% of the species.
Fig. 6.—

Relative fraction of codons for amino acids encoded by four codons. The figure shows the relative fraction of codons in each four-codon box for the highly expressed genes of Lactobacillus (to the left) and Bifidobacterium (to the right), estimated as the average frequency over the species included in the analysis, excluding the GC-shifted genomes. The error bar shows the standard error for each codon. Blue and red colors refer to AU- and GC-ending codons, respectively. The optimal codons predicted by RP method are marked with the symbol “+”, and optimal codons predicted by correlative method are marked with the symbol “o”. Codons were defined as “optimal” in Lactobacillus and Bifidobacterium if predicted to be optimal in > 40% of the species.
Fig. 7.—

Relative fraction of codons for amino acids encoded by three and six codons. The figure shows the relative fraction of codons in each three- and six-codon box in the highly expressed genes of Lactobacillus (to the left) and Bifidobacterium (to the right), estimated as the average frequency over the species included in the analysis, excluding the GC-shifted genomes. The error bar shows the standard error for each codon. Blue and red colors refer to AU- and GC-ending codons, respectively. The optimal codons predicted by RP method are marked with the symbol “+”, and optimal codons predicted by correlative method are marked with the symbol “o”. Codons were defined as “optimal” in Lactobacillus and Bifidobacterium if predicted to be optimal in > 40% of the species.
To begin, we examined the use of codons in two-codon boxes read by a single tRNA (fig. 5 and supplementary fig. S9, Supplementary Material online). In these families, the codons with the best codon–anticodon interactions are expected to represent the optimal codons. Overall, the 2-fold degenerate codon sites were fairly robust to changes in the background substitution biases, with the exception of CAA/CAG and GAA/GAG for Gln and Glu that co-varied with the genomic base composition patterns.
Next, we examined whether the identity of the optimal codons in the three-, four- (fig. 6 and supplementary fig. S10, Supplementary Material online) and six-codon boxes (fig. 7 and supplementary fig. S11, Supplementary Material online) read by multiple tRNAs have changed following the switches in the direction of the background substitution biases. In these codon families, the codons read by the tRNA isoacceptors in highest concentration are expected to represent the optimal codons. Since tRNA concentrations are adaptable, the three-, four- and six-codon families are expected to be more sensitive to biases in the direction of the background substitutions. We therefore investigated optimal codon choice in more detail for codon families read by multiple tRNAs.
Optimal Codons after Switches in the Direction of the Background Substitution Bias from AT to GC
In the Lactobacillus species with high genomic AT content values, 75% of all codons end in A or U. Optimal codons were mostly inferred to be U-ending (e.g., GUU, ACU, GCU, CGU, GGU), and A-ending (e.g., CCA and UCA) (summarized in figs. 6 and 7; shown for each individual species in fig. 8), suggesting that selection has acted on codons favored by the AT background substitution bias.
Fig. 8.—

Heatmap of Lactobacillaceae RSCU values comparing optimal codons predicted with the RP method and the correlative method. Each panel represents log(RSCU) values calculated for each codon in Lactobacillaceae genomes. The upper panel shows log(RSCU) values for the RP data set, with “+” and “−” symbols indicating codons used significantly more and less than in the WG data set, respectively. Thus, “+”represents optimal codons detected by the RP method. The lower panel shows log(RSCU) values for the WG data set, with “o” symbols indicating codons detected as optimal by the correlative method.
Following the switch in the direction of the background substitution bias in L. delbrueckii and L. fermentum, the abundance of GC-ending codons has increased, but more so in the WG than in the RP gene data set. In effect, the large majority of codons predicted to be optimal by the RP method corresponded to the previously used AU-ending codons, such as, e.g., GUU, CCA, ACU, GCU, CGU, and UCA, which have RSCU values of > 2.5 in one or both species. Only one shift in optimal codon identity towards a more GC-rich codon, UUG for Leucine, was identified in L. delbrueckii. We conclude that the old set of optimal codons was still mostly inferred to be optimal in L. delbrueckii and L. fermentum, although the overall frequency of the GC-ending codons has increased in both the WG and the RP data set following the switch in the direction of the background substitution bias.
These results were surprising because a previous publication claimed that none of the optimal codons ends in A or T in L. delbrueckii or L. fermentum (Nayak 2012). The latter study used a slightly different set of putatively highly expressed genes for the analysis, which might have influenced the results. To test this concern, we contrasted the codon usage patterns of genes with the highest and lowest 10% CAI values. However, also in this data set the AU-ending codons were over-represented in the genes with the highest CAI values (P < 0.05, chi squared test). In fact, our analyses have shown that selection for a subset of AU-ending codons in L. delbrueckii and L. fermentum in the highly expressed genes became even more apparent after the shift in the background substitution bias because of the increasing abundance of GC-ending codons in all other genes.
Optimal Codons after Switches in the Direction of the Background Substitution Bias from GC to AT
In the Bifidobacterium species with high genomic GC content values, the majority of codons end in G or C. Optimal codons in these species were mostly C-ending (e.g., ACC, UCC, CGC, GGC, GUC) (summarized in figs. 6 and 7; shown for each individual species in fig. 9). This suggests that selection for translational efficiency has acted on codons favored by the GC background substitution bias. Thus, the identity of the optimal codons differed in Lactobacillus and Bifidobacterium, but was in both cases a subset of those generated by the background substitution bias.
Fig. 9.—

Heatmap of Bifidobacteriaceae RSCU values comparing optimal codons predicted with the RP method and the correlative method. Each panel represents log(RSCU) values calculated for each codon in the Bifidobacteriaceae genomes. The upper panel shows log(RSCU) values for the RP data set, with “+” and “−” symbols indicating codons used significantly more and less than in the WG data set, respectively. Thus, “+”represents optimal codons detected by the RP method. The lower panel shows log(RSCU) values for the WG data set, with “o” symbols indicating codons detected as optimal by the correlative method.
Consistent with a change in the direction of the background substitution bias towards AT in G. vaginalis, the frequency of AU-rich codons have increased 2- to 5-fold in the WG gene data set, whereas the previously preferred C-ending codons have been reduced >2-fold in most codon boxes. This is also true for genes in the RP data set, as a result of which the AU-ending codons are equally or more abundant than the C-ending codons. For example, the RSCU value for the codon CCA for Proline is 5 times higher in the RP gene data set in G. vaginalis than in the other Bifidobacterium species. Furthermore, the codon CCA is used significantly more frequently in the RP gene data set than in the WG gene data set in G. vaginalis, providing an example of how a previous minor codon has become an optimal codon after the shift in the background substitution bias. The increased usage of the CCA codon has been associated with a dramatic decrease in the usage of the codon CCC and loss of the isoacceptor tRNA with the anticodon GGG, recognizing CCC.
However, despite the dramatic increase in the abundance of the AU-ending codons in G. vaginalis, the relative differences in codon usage frequencies between genes in the RP and the WG data set have mostly remained. In effect, the C-ending codons were still mostly inferred to be the optimal codons although their total abundances may have decreased below those of the U-ending codons. For example, the codon ACC for Threonine has decreased 2-fold whereas the codon ACU has increased 4-fold in frequency such that the two codons are now equally abundant in the RP gene data set of G. vaginalis. Yet, it is still the old codon ACC that is inferred to be the optimal codon because it is used significantly more frequently in the RP gene data set. Another counter-intuitive example of codon bias was observed for Alanine. The frequency of the codon GCU for Alanine has increased in G. vaginalis, such that it is 4- to 5-fold more abundant than GCC, yet the RP method suggests that the minor codon GCC is the optimal codon for Alanine, because GCC was inferred to be used significantly more frequently in the RP gene data set. In most other Bifidobacterium species the minor codon GCU was inferred to be the optimal codon for Alanine, because the major codon CGC was highly abundant in both the RP and the WG data set. Overall, the RP method predicted the old set of GC-rich codons to represent the optimal codons in G. vaginalis. Thus, codons used at low frequencies were occasionally inferred to represent the optimal codons both before and after the shift of the background substitution bias.
Predicting Optimal Codons with the Correlative Method
Another approach to predict optimal codons is the so-called correlative method (Hershberg and Petrov 2009). This method identifies optimal codons as those codons that show a statistical difference in frequency between lowly and highly biased genes. The gene bias is estimated from the effective number of codons Nc, or a version thereof, Nc′, that corrects for the background GC content (Hershberg and Petrov 2009). A comparison of optimal codons identified by the correlative method (figs. 6–9) confirmed an overall preference for U-ending codons in the AT-rich Lactobacillus genomes, and for GC-ending codons in the GC-rich Bifidobacterium genomes. Thus, both methods predicted similar sets of optimal codons for the majority of Lactobacillus and Bifidobacterium species.
The similarity in predictive power of the two methods for the archetype Lactobacillus and Bifidobacterium species contrasted with converse predictions of optimal codons in the three genomes that have recently experienced a shift in the direction of the background substitution bias (fig. 10). Thus, the optimal codons were mostly predicted to be G- and C-ending in L. delbrueckii and L. fermentum, but A- and U-ending in G. vaginalis. The Nc and Nc′ methods yielded similar predictions of optimal codons in the two Lactobacillus species (fig. 10). However, the Nc′ method predicted four G- and C-ending codons to be optimal in G. vaginalis that were not identified as optimal by the Nc method, three of which were predicted to be the optimal codons by the RP method. Vice versa, five A- and U-ending codons were predicted to be optimal solely by the Nc method, none of which were predicted to be optimal by the RP method. Intriguingly, CGU for Arginine and GGU for Glycine were predicted by all methods to represent the optimal codons in G. vaginalis.
Fig. 10.—

Comparison of optimal codon identities in Lactobacillus delbrueckii, L. fermentum and Gardnerella vaginalis with the RP and the correlative methods for codons in three-, four-, and six-codon boxes. The upper rows in each panel shows the log(RSCU) values for the RP data set, with “+” and “−” symbols indicating codons used significantly more and less than in the WG data set, respectively. Thus, “+”represents optimal codons detected by the RP method. The lower rows in each panel shows the log(RSCU) values for the WG data set, with “o” symbols indicating codons detected as optimal by the correlative method, based on correlations to the Nc and Nc′ values, respectively. The row at the bottom of the upper panel, labeled HP, shows the results of a previous study of L. fermentum (Hershberg and Petrov 2009), with the “#” symbols indicating codons detected as optimal by the correlative method.
Thus, the correlation method indicated that shifts in optimal codon choices have occurred in most four- and six-codon families in all three species with shifted AT/GC background substitution biases, whereas the RP method predicted no or only a few changes of optimal codons.
Optimal Codons and Conserved Amino Acids
In an attempt to distinguish the optimal codon predictions made by the RP and correlative methods in the GC-shifted genomes, we tested whether any of the predicted optimal codons evolve under selection for translational accuracy as inferred from the so-called Akashi’s test (1994). This test is based on a comparison of codon usage patterns at sites that code for conserved versus nonconserved amino acids. We defined conserved codons as sites that code for the same amino acid in pairwise gene alignments of the species to be tested and a reference species, selected from one of the outgroup taxa in the phylogenies of Lactobacillus and Bifidobacterium, respectively.
We first performed the test for two large gene data sets, one consisting of 400 orthologs in Bifidobacterium and another consisting of 302 orthologs in Lactobacillus. The results provided indications for an association between optimal codons and conserved amino acid sites in the archetype genome as well as in the GC-shifted genomes (supplementary table S5, Supplementary Material online). However, the association was observed irrespectively of the method used for optimal codon predictions, with the exception of the RP-predicted optimal codons in L. fermentum. Since the ribosomal protein genes have a higher fraction of conserved sites (supplementary table S6, Supplementary Material online) as well as of optimal codons, we were concerned that gene expression levels might have influenced the results.
To test for an association between optimal codons and conserved amino acid sites for genes with similar expression levels, we repeated the test for data sets consisting only of the ribosomal protein genes. For these data sets the correlation between optimal codons and conserved amino acid sites were abolished in all genomes, again irrespectively of the method used to predict optimal codons (supplementary table S5, Supplementary Material online). However, this does not mean that all codons behaved similarly. For example, there was a tendency for codons read by a single tRNA and suggested by both methods to be optimal to be used more frequently at the conserved sites. There was also a tendency for optimal codons predicted by the RP method in families read by multiple tRNAs to be preferentially utilized at the conserved sites of the GC-shifted genomes. Interestingly, the codon CGU for Arginine was used more frequently at the conserved sites in all genomes irrespectively of their GC content, indicating that CGU evolves under selection for translational accuracy or under some other selective constraints that are not affected by GC content.
Placing Shifts in Optimal Codons in a Phylogenetic Framework
The identification of optimal codons builds on the biological assumption that the most frequently used codons in the highly expressed genes (i.e., the major codons) also correspond to the codons favored in translation (i.e., the optimal codons). Thus, we reasoned that comparisons of the major codons in the highly expressed genes of G. vaginalis, L. delbrueckii and L. fermentum to the major codons in species that belong to the same phylogenetic group, respectively, should reveal identity shifts in optimal codons. We performed such a comparison of codon usage patterns in orthologous genes across phylogenetically related genomes as a complement to the comparisons across different genes within the same genome, as in the RP and correlative methods. The results of these phylogenetic comparisons suggested that the major codons have not changed in the two Lactobacillus genomes, whereas they have been altered in G. vaginalis (figs. 6 and 7). Taken together, we suggest that the optimal codons have shifted in G. vaginalis, but not yet in the two Lactobacillus genomes.
Discussion
The aim of this study was to investigate whether a strong background substitution bias alone can drive shifts in optimal codons. The novelty of our study is that we have placed species with an altered direction of the background substitution bias in a phylogenetic framework, which has enabled us to study the evolutionary process whereby optimal codons change in identity. Importantly, the results suggest that the major codon preference patterns in the highly expressed genes in G. vaginalis have changed following a switch in direction of the background substitution bias, which supports mutation-driven hypotheses to explain shifts in optimal codons.
We have used two bioinformatics methods, the RP and the correlative method, to identify optimal codons in the genomes under study. For the majority of genomes, the two methods predicted similar optimal codons, suggesting that GC-ending codons are selectively favored in the GC-rich bifidobacterial genomes whereas AU-ending codons are favored in the AT-rich lactobacilli genomes. These results are consistent with a broader study of 675 bacterial genomes, which also indicated that selection for optimal codons follows, rather than counters, the background substitution bias (Hershberg and Petrov 2009). Surprisingly, the two methods yielded conflicting results regarding the identification of optimal codons in the three species in which the background substitution bias has switched. Thus, the RP method indicated that the optimal codons remain un-shifted in all three genomes, whereas the correlative method suggested that the optimal codons have shifted in all genomes. In effect, the major codon for a particular amino acid was not necessarily predicted to be the optimal codon by either method. Thus, how to define an optimal codon merits further discussion (Wang et�al. 2011; Hershberg and Petrov 2012).
The RP method is based on the hypothesis that the codons favored by selection are those used significantly more frequently in the highly expressed genes than in the lowly expressed genes. Experimental data on tRNA expression levels from E. coli and other classical bacterial model systems support the assumptions of the RP method and it thus has a strong biological foundation (Ikemura 1985; Bulmer 1987; Sorensen and Pedersen 1991). By extrapolation to other species for which no experimental data is available, the ribosomal protein genes and a few other translation genes are therefore normally used as a proxy for high expression levels. However, it has been suggested that there might be selective constraints operating on the ribosomal protein genes not related to translational efficiency, which could lead to biased results (Hershberg and Petrov 2012). As we have shown here, the identity of the codons predicted to be optimal in the RP gene data set are mostly different in the GC-rich and GC-poor genomes, which argues against the influence of universal constraints on the RP genes that may bias the results. We do not know why the RP method has problems with GC-shifted genomes, but one hypothesis is that this method recognizes selective patterns that were established prior to the shift and may persist for some time even after the shift.
The correlation method on the other hand identifies optimal codons as those codons that show a statistical difference in frequency between lowly and highly biased genes, with the latter defined as the genes with the lowest Nc or Ncc. If the ribosomal protein genes represent the most highly biased genes, as demonstrated in our study, we would expect the two methods to yield similar results, as was indeed observed for the majority of genomes studies here as well as for 658 out of 675 taxonomically diverse genomes examined previously (Hershberg and Petrov 2009). However, the correlation method classifies as putatively optimal codons any set of codons with a biased usage profile; thus, it may mispredict a codon to be optimal if it is rapidly changing in frequency due to a recent switch in the background substitution bias. Broad-scale studies of optimal codons in genomes that are changing in GC content may suffer from either of these two types of mispredictions, and it may even be difficult to recognize that there is a problem if the analyses are based on single genomes or multiple, unrelated genomes.
We have taken a different approach to identify shifts of optimal codons. Placing studies of codon usage patterns in a phylogenetic framework, as performed here, could help identify taxa in which the codon usage pattern is in the process of changing, thus guiding the interpretation of the results. By comparing codon usage patterns in the highly expressed genes to the patterns in the orthologs of their closest relatives, we were able not only to identify shifts in the identity of optimal codons but also to dissect the contribution of mutation and selection to these processes.
The mutation-driven hypothesis states that the background substitution bias alone can drive changes of the preferred set of codons even without an intervening period during which selection is lost (Shields 1990). The stronger the background substitution biases the higher the likelihood that the optimal codons will shift identity. It is estimated that shifts in codon preference patterns will occur if the mutation rate from GC to AT is sufficiently different from the mutation rate from AT to GC (Shields 1990). Based on their genomic GC content values, which is 32% in G. vaginalis and 62–63% in L. fermentum and L. delbrueckii, we hypothesized that the background substitution biases in these three species should be high enough to drive changes in major codons even under sustained selection for translational selection.
Consistent with these expectations, we noted massive changes in codon abundances in all three species in accordance with the new directions of the background substitution biases. However, L. delbrueckii and L. fermentum used the same major codons in the RP gene data set as most other Lactobacillus species, which suggests that the identity of the optimal codons have not yet shifted. The similarity in major codon preference patterns for all members of the Lactobacillus indicates that L. fermentum and L. delbrueckii are in a pre-shift phase in which codon abundances are starting to change, but selection is still acting on the old set of optimal codons. In contrast, we found that the strong AT background substitution bias in G. vaginalis has induced a shift in the identity of the most highly abundant codons in the RP gene data set as compared with the codon preference patterns in these genes in the other Bifidobacterium species. Thus, G. vaginalis seems to be in a post-shift phase in which selection is favoring a new set of optimal codons.
The relaxed selection hypothesis suggests that a shift in major codons may occur if the selective constraints are lost or reduced, after which a new set of codons is favored when selection resumes (Wright 1977; Sharp et�al. 2010). However, we found no indications of relaxed selection in species with an altered direction of the GC-bias. Rather, the selective constraints on codon usage remained consistently strong in all species with an altered background substitution bias, as inferred from their high S values. Nor did we find any indications of loss of selection in their most closely related sister taxa. Importantly, the Lactobacillus species with low S values (S < 0.3), did not share a common ancestor with the species in which the background substitution bias has been altered. However, it should be cautioned that the S value reflects long-term evolution (Sharp et�al. 2010) and relaxed selection would not immediately erase all codon usage bias from the highly expressed genes. Thus, it cannot be excluded that selected codon usage bias has started to decay recently. However, it is unlikely that the increased use of A- and U-ending codon in 4- and 6-fold codon boxes in the highly expressed genes of G. vaginalis is due to loss of selection because the frequencies of C-ending codons in two-codon boxes has remained high.
The reasons for why the three species are currently at different stages of a shift in optimal codons could be that a longer time has elapsed because the switch of the background substitution bias or that there is a stronger bias from GC to AT in G. vaginalis than from AT to GC in Lactobacillus. Consistent with the latter explanation is that recent mutations showed a strong trend towards AT with 84% of GC-changing SNPs in G. vaginalis, whereas the mutations towards GC in L. fermentum and L. delbrueckii were 65% and close to 50% of AT-changing single nucleotide substitutions, respectively. Hypothetically, the optimal codons in L. fermentum and L. delbrueckii will never completely shift to match the new G + C content of the two genomes.
Finally, it should be emphasized that the composition and expression of the isoacceptor tRNAs will have an impact on the ability of a species to adopt novel optimal codons. Comparative analyses of closely related Escherichia species showed that the optimal codons matched the ancestral rather than the extant tRNA population (Withers et�al. 2006) suggesting that that the isoacceptor tRNA pool adapts only slowly to changes in codon preference patterns (Iriarte et�al. 2013). Measurement of tRNA expression levels in the three species in which the direction of the background substitution bias has recently been reversed is an interesting avenue for future research.
Conclusions
The results of this study suggests that shifts in the direction of the background substitution bias can drive changes in optimal codon identities without an intervening period in which selection is lost. The likelihood for a shift in optimal codon identity depends on the strength of the background substitution bias, the number of isoacceptor-tRNAs and how easily these tRNAs will adapt to the changes in codon frequencies. Overall, we found that two-codon families read by a single tRNA were fairly robust to changes in the mutational biases (Higgs and Ran 2008; Ran and Higgs 2010), whereas four- and six-codon boxes read by multiple tRNAs were highly sensitive to the AT/GC background substitution biases.
Capturing genomes in this veering phase, during which new optimal codons rise and ancestrally optimal codons decline in frequency, may provide clues about the order in which individual codon families respond to an altered direction of the background substitution bias or to changes in the expression levels or gene copy numbers of the isoacceptor-tRNAs. Based on the result of our study, and consistent with the model from Hershberg and Petrov (2009), we suggest the following order of events leading to a shift of optimal codons in the codon families read by multiple tRNAs: (1) a change in the direction of the background substitution bias initiates the process, (2) the new background substitution bias leads to altered codon usage patterns in all genes, after which (3) gene copy numbers and expression levels of the tRNA isoacceptors adjust to the new codon usage patterns, and finally (4) codon preference patterns and tRNA expression levels are fine-tuned to the new patterns.
Supplementary Material
Supplementary figures S1–S11, tables S1–S6 and text S1 are available at Genome Biology and Evolution online (http://www.gbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
We are thankful to two anonymous reviewers, and especially to the editor, for constructive comments on the article. We thank Kirsten Ellegaard for help regarding bifidobacterial phylogenetics. This work was supported by the Swedish Research Council (349-2007-8732, 621-2014-4460) and the Knut and Alice Wallenberg Foundation (2011.0148, 2012.0075) to S.G.E.A., and by an European Union grant from the Marie Curie ITN SYMBIOMICS (264774) to D.T.
Literature Cited
- Akashi H. 1994. Synonymous codon usage in Drosophila melanogaster: natural selection and translational accuracy. Genetics 136:927–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ.. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Andersson SG, Kurland CG.. 1990. Codon preferences in free-living microorganisms. Microbiol Rev. 54:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennetzen JL, Hall BD.. 1982. Codon selection in yeast. J Biol Chem. 257:3026–3031. [PubMed] [Google Scholar]
- Bulmer M. 1987. Co-evolution of codon usage and transfer RNA abundance. Nature 325:827–730. [DOI] [PubMed] [Google Scholar]
- Bulmer M. 1991. The selection-mutation-drift theory of synonymous codon usage. Genetics 129:897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T.. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone A, Kepes F, Zinovyev A.. 2005. Codon bias signatures, organization of microorganisms in codon space, and lifestyle. Mol Biol Evol. 22:547–561. [DOI] [PubMed] [Google Scholar]
- Chen Y. 2013. A comparison of synonymous codon usage bias patterns in DNA and RNA virus genomes: quantifying the relative importance of mutational pressure and natural selection. Biomed Res Int. 2013:406342.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criscuolo A, Gribaldo S.. 2010. BMGE (Block Mapping and Gathering with Entropy): a new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol Biol. 10:210.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MS, Brudno M, Batzoglou S.. 2005. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res. 15:330–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L.. 2004. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res. 32:5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard KM, et al. 2015. Extensive intra-phylotype diversity in lactobacilli and bifidobacteria from the honeybee gut. BMC Genomics 16:284.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felis GE, Dellaglio F.. 2007. Taxonomy of Lactobacilli and Bifidobacteria. Curr Issues Intest Microbiol. 8:44–61. [PubMed] [Google Scholar]
- Ghaemmaghammi S, et al. 2003. Global analysis of protein expression in yeast. Nature 425:737–741. [DOI] [PubMed] [Google Scholar]
- Goetz RM, Fugelsang A.. 2005. Correlation of codon ibas measures with mRNA levels: analysis of transcriptome data from Escherichia coli. Biochem Biophys Res Commun. 327:4–7. [DOI] [PubMed] [Google Scholar]
- Gouy M, Gautier C.. 1982. Codon usage in bacteria: correlation with gene expressivity. Nucleic Acids Res. 10:7055–7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Jacobzone M, Mercier R.. 1981. Codon catalog usage is a genome strategy modulated for gene expressivity. Nucleic Acids Res. 10:r43–r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grantham R, Gautier C, Gouy M, Mercier R, Pave A.. 1980. Codon catalog usage and the genome hypothesis. Nucleic Acids Res. 8:r49–r62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H, Fiers W.. 1982. Preferential codon usage in prokaryotic genes: the optimal codon-anticodon interaction energy and the selective codon usage in efficiently expressed genes. Gene 18:199–209. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2008. Selection on codon bias. Annu Rev Genet. 42:287–299. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2009. General rules for optimal codon choice. PLoS Genet. 5:e1000556.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2010. Evidence that mutation is universally biased towards AT in bacteria. PLoS Genet. 6:e1001115.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA.. 2012. On the limitations of using ribosomal genes as references for the study of codon usage: A rebuttal. PLoS One 7:e49060.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgs PG, Ran W.. 2008. Coevolution of codon usage and tRNA genes leads to alternative stable states of biased codon usage. Mol Biol Evol. 25:2279–2291. [DOI] [PubMed] [Google Scholar]
- Hildebrand F, Meyer A, Eyre-Walker A.. 2010. Evidence of selection upon genomic GC-content in bacteria. PLoS Genet. 6:e1001107.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikemura T. 1981. Correlation between the abundance of Escherichia coli transfer RNAs and the occurrence of the respective codons in its protein genes. J Mol Biol. 146:1–21. [DOI] [PubMed] [Google Scholar]
- Ikemura T. 1985. Codon usage and tRNA content in unicellular and multicellular organisms. Mol Biol Evol. 2:13–34. [DOI] [PubMed] [Google Scholar]
- Iriarte A, Baraibar JD, Romero H, Castro-Sowinski S, Musto H.. 2013. Evolution of optimal codon choices in the family Enterobacteriaceae. Microbiology 159:555–564. [DOI] [PubMed] [Google Scholar]
- Kanaya S, Yamada Y, Kudo Y, Ikemura T.. 1999. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 238:143–155. [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T.. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Misawa K, Kuma K, Miyata T.. 2002. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein G, Pack A, Bonaparte C, Reuter G.. 1998. Taxonomy and physiology of probiotic lactic acid bacteria. Int J Food Microbiol. 41:103–125. [DOI] [PubMed] [Google Scholar]
- Kurland CG. 1987. Strategies for efficiency and accuracy in gene expression. 1. The major codon preference. A growth optimization strategy. Trends Biochem Sci. 12:126–128. [Google Scholar]
- Lassalle F, et al. 2015. GC-content evolution in bacterial genomes: the biased gene conversion hypothesis expands. PLoS Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Stoeckert CJ Jr., Roos DS.. 2003. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13:2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobry JR. 1996. Assymetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 13:660–665. [DOI] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR.. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukjancenko O, Ussery DW, Wassenar TM.. 2012. Comparative genomics of Bifidobacterium, Lactobacillus and related probiotic genera. Microb Ecol. 63:651–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInerney JO. 1998. GCUA: general codon usage analysis. Bioinformatics 14:372–373. [DOI] [PubMed] [Google Scholar]
- McLean MJ, Devine KM, Wolfe KH.. 1997. Base composition skews, replication orientation, and gene orientation in 12 prokaryotic genomes. J Mol Evol. 47:691–696. [DOI] [PubMed] [Google Scholar]
- Miyake T, Watanabe K, Watanabe T, Oyaizu H.. 1998. Phylogenetic analysis of the genus Bifidobacterium and related genera based on 16S rDNA sequences. Microbiol Immunol. 42:661–667. [DOI] [PubMed] [Google Scholar]
- Morita H, et al. 2007. Lactobacillus hayakitensis sp. nov., isolated from intestines of healthy thoroughbreds. Int J Syst Evol Microbiol. 57:2836–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita H, et al. 2010. Lactobacillus equicursoris sp. nov., isolated from the faeces of a thoroughbred racehorse. Int J Syst Evol Microbiol. 60:109–112. [DOI] [PubMed] [Google Scholar]
- Moszer I, Rocha EPC, Danchin A.. 1999. Codon usage and lateral gene transfer in Bacillus subtilis. Curr Opin Microbiol. 2:524–528. [DOI] [PubMed] [Google Scholar]
- Munoz JA, et al. 2011. Novel probiotic Bifidobacterium longum subsp. infantis CECT 7210 strain active against rotavirus infections. Appl Environ Microbiol. 77:8775–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto A, Osawa S.. 1987. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc Natl Acad Sci U S A. 84:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak KC. 2012. Comparative study on factors influencing the codon and amino acid usage in Lactobacillus sakei 23K and 13 other lactobacilli. Mol Biol Rep. 39:535–545. [DOI] [PubMed] [Google Scholar]
- Novembre JA. 2002. Accounting for background nucleotide composition when measuring codon usage bias. Mol Biol Evol. 19:1390–1394. [DOI] [PubMed] [Google Scholar]
- Novoa EM, Pavon-Eternod M, Pan T, Ribas de Pouplana L.. 2012. A role for tRNA modifications in genome structure and codon usage. Cell 149:202–213. [DOI] [PubMed] [Google Scholar]
- Novoa EM, Ribas de Pouplana L.. 2012. Speeding with control: codon usage, tRNAs, and ribosomes. Trends Genet. 28:574–581. [DOI] [PubMed] [Google Scholar]
- Ochman H, Lawrence JH, Groisman EA.. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304. [DOI] [PubMed] [Google Scholar]
- Peden JF. 1999. Analysis of codon usage Department of Genetics, Vol Doctor of Philosophy, University of Nottingham. [Google Scholar]
- Ran W, Higgs PG.. 2010. The influence of codon-anticodon interactions and modified bases on codon usage bias in bacteria. Mol Biol Evol. 27:2129–2140. [DOI] [PubMed] [Google Scholar]
- Rice P, Longden I, Bleasby A.. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet. 16:276–277. [DOI] [PubMed] [Google Scholar]
- Rocha EP. 2004. Codon usage bias from tRNA’s point of view: redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 14:2279–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Bailes E, Grocock RJ, Peden JF, Sockett RE.. 2005. Variation in the strength of selected codon usage bias among bacteria. Nucleic Acids Res. 33:1141–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Emery LR, Zeng K.. 2010. Forces that influence the evolution of codon bias. Philos Trans R Soc Lond B Biol Sci. 365:1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Li WH.. 1986. An evolutionary perspective on synonymous codon usage in unicellular organisms. J Mol Evol. 24:28–38. [DOI] [PubMed] [Google Scholar]
- Shields DC. 1990. Switches in species-specific codon preferences: the influence of mutation biases. J Mol Evol. 31:71–80. [DOI] [PubMed] [Google Scholar]
- Shields DC, Sharp PM.. 1987. Synonymous codon usage in Bacillus subtilis reflects both translational selection and mutational biases. Nuclec Acids Res. 15:8023–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields DC, Sharp PM, Higgins DG, Wright F.. 1988. “Silent” sites in Drosophila genes are not neutral: evidence of selection among synonymous codons. Mol Biol Evol. 5:704–716. [DOI] [PubMed] [Google Scholar]
- Sorensen MA, Pedersen S.. 1991. Absolute in vivo translation rates of individual codons in Escherichia coli. The two glutamic acid codons GAA and GAG are translated with a threefold difference in rate. J Mol Biol. 222:265–280. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stiles ME, Holzapfel WH.. 1997. Lactic acid bacteria of foods and their current taxonomy. Int J Food Microbiol. 36:1–29. [DOI] [PubMed] [Google Scholar]
- Supek F, Skunca N, Repar J, Vlahovicek K, Smuc T.. 2010. Translational selection is ubiquitous in prokaryotes. PLoS Genet. 6(6):e1001004.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamarit D, et al. 2015. Functionally structured genomes in Lactobacillus kunkeei colonizing the honey crop and food products of honeybees and stingless bees. Genome Biol Evol. 7:1455–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicario S, Moriyama EN, Powell JR.. 2007. Codon usage in twelve species of Drosophila. BMC Evol Biol. 7:226.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel RF, et al. 1994. Identification of lactobacilli from sourdough and description of Lactobacillus pontis sp. nov. Int J Syst Bacteriol. 44:223–229. [DOI] [PubMed] [Google Scholar]
- Wang B, et al. 2011. Optimal codon identities in bacteria: Implications from the conflicting results of two different methods. PLoS One 6:e22714.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers M, Wernisch L, doe Reis M.. 2006. Archaeology and evolution of transfer RNA genes in the Escherichia coli genome. RNA 12:933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright F. 1990. The ‘effective number of codons’ used in a gene. Gene 87:23–29. [DOI] [PubMed] [Google Scholar]
- Wright S. 1977. Evolution and the genetics of population. University of Chicago Press. [Google Scholar]
- Wu H, Fang Y, Yu J, Zhang Z.. 2014. The quest for a unified view of bacterial land colonization. ISME J. 8:1358–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.