

Abstract
The use of monoclonal antibodies (mAbs) for the manufacture of innovator and biosimilar biotherapeutics has increased tremendously in recent years. From a structural perspective, mAbs have high disulfide bond content, and the correct disulfide connectivity is required for proper folding and to maintain their biological activity. Therefore, disulfide linkage mapping is an important component of mAB characterization for ensuring drug safety and efficacy. The native disulfide linkage patterns of all four subclasses of IgG antibodies have been well established since the late 1960s. Among these IgG subtypes, disulfide mediated isoforms have been identified for IgG2 and IgG4, and to a lesser extent in IgG1, which is the most studied IgG subclass. However, no studies have been carried out so far to investigate whether different IgG3 isoforms exist due to alternative disulfide connectivity. In an effort to investigate the presence of disulfide-mediated isoforms in IgG3, we employed a bottom-up mass spectrometry approach to accurately determine the disulfide bond linkages in endogenous human IgG3 monoclonal antibody and our results show that no such alternative disulfide bonds exist. While many antibody-based drugs are developed around IgG1, IgG3 represents a new, and in some cases, more desirable drug candidate. Our data represent the first demonstration that alternative disulfide bond arrangements are not present in endogenous IgG3; and therefore, they should not be present in recombinant forms used as antibody-based therapeutics.
Keywords: Disulfide bond, Monoclonal Antibody, Electron Transfer Dissociation, Liquid Chromatography-Mass Spectrometry
Introduction
Human IgG3 monoclonal antibody is the most efficient IgG subclass in mediating effector functions, followed by IgG1, IgG2, and IgG4, respectively.1,2 IgG3 displays the highest complement dependent cytotoxicity (CDC) and comparable antibody dependent cell-mediated cytotoxicity (ADCC) to IgG1, making it an ideal antibody drug candidate. Despite these ideal drug qualities and the rapidly growing use of monoclonal antibodies as biotherapeutics against various diseases, IgG3 is the only IgG subclass that has not yet been used for the production of antibody-based drugs. This is mainly due to its short half-life of seven days, compared to 21 days for the other IgG's.3 This short half-life of IgG3 is generally attributed to its long hinge region of 62 amino acid residues compared to 12 and 15 residues for the other IgG subclasses, making IgG3 more susceptible to proteolysis.4 However, a recent report by Stapleton et al showed that the short half-life is primarily due to the presence of arginine at position 435 (R435) of the IgG3 heavy chain, as opposed to histidine (H435) for the other IgG subclasses.5 Mutation of the IgG3 heavy chain arginine 435 to histidine (H435-IgG3) extends the IgG3 half-life. This finding by Stapleton et al has energized interest in the production of IgG3-based biotherapeutics.
Consequently, timely studies on the structure and properties of endogenous IgG3 are now an urgent priority. For example, Plomp et al recently identified three O-glycosylation sites (each having about 10% site occupancy) in the hinge region of endogenous IgG3 samples from six donors.6 In a complementary line of work, we take on the challenge of investigating whether IgG3 is similar to the other IgG's in displaying endogenous isoforms resulting from alternative disulfide connectivity.
Disulfide bonds are vital post-translational modifications in therapeutic proteins as they play a key role in mediating protein folding, stability and biological function.7-9 The disulfide bond patterns of the four IgG subclasses (IgG1, IgG2, IgG3, and IgG4) were established in the late 1960s and early 1970s by Milstein et al. using diagonal paper electrophoresis and Edman degradation.10-14 In addition to these classical IgG disulfide connections, alternative (non-classical) disulfide bonds have been identified in the constant regions of some IgG subclasses, leading to their disulfide-mediated structural isoforms. For example, in addition to the classical IgG4 structure with inter-chain disulfide bonds in the hinge region, IgG4 also forms intra-chain disulfide bonds in the hinge region, thereby forming an isoform that consists of two half molecules.15,16 Additionally, both native and recombinant IgG2 antibodies have been shown to have two disulfide-mediated isoforms in addition to the classical IgG2 structure.17-19 Furthermore, one report observed a trace amount of alternative intra-chain IgG1 disulfide bonds in the hinge region in addition to its conventional inter-chain disulfide bonds.16 However, there is currently no study determining whether IgG3 also contains disulfide-mediated IgG3 isoforms. This lack of information is likely because IgG3 has been overlooked as a promising drug candidate, due to its short half-life, which is also reflected by the lack of IgG3-based drugs in the market. With the discovery of H435-IgG3 which has comparable half-life to IgG1, IgG2, and IgG4, it is important to confirm the classical IgG3 disulfide bond connectivity and to determine whether or not disulfide-mediated isoforms exist in endogenous IgG3. Data from such a study would facilitate future drug development work based on the IgG3 scaffold, because it would provide a blueprint for the appropriate disulfide bonding profile for recombinant IgG3-based therapeutics.
Herein, we use liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) to experimentally characterize the disulfide bond connectivity of IgG3 for the first time. Two batches of native (endogenous) IgG3 were obtained from two different sources and extensive disulfide bond characterization was done by expanding upon our previously published Extracted Ion Chromatogram/Electron Transfer Dissociation (XIC/ETD) approach for rapid disulfide bond analysis in proteins.20 With the combination of both the ETD-based method and further analysis of collision induced dissociation (CID) data, all of the disulfide bonds in the constant region of the protein were accounted for. Using these techniques, we confirmed the classical disulfide bond pattern in the constant region of endogenous IgG3 antibodies and showed that unlike IgG2 and IgG4, which have well established conformational isoforms due to alternative disulfide bonds in their constant regions, endogenous IgG3 does not have any alternative disulfide bonds in its constant region and therefore does not have disulfide-mediated isoforms.
Experimental Section
Materials and Reagents
Tris(hydroxylmethyl)aminomethane (Trizma) base, guanidine hydrochloride, sodium acetate anhydrous, acetonitrile, N-Ethylmaleimide, and Gamma-globulins from human serum were purchased from Sigma-Aldrich (St. Louis, MO). High capacity Protein A resin, sodium phosphate, glycine hydrochloride, sodium chloride, calcium chloride dihydrate, and optima grade formic acid were from Fisher Scientific (Pittsburgh, PA). Native human IgG3 (with λ light chain) was purchased from Fitzgerald (Acton, MA), mouse anti-human IgG3 antibody was purchased from Invitrogen (ThermoFisher Scientific, Grand Island, NY), sequencing grade trypsin was acquired from Promega (Madison, WI), and Protein G resin was prepared in-house.
Isolation of IgG3 from human Gamma-globulins
Gamma-globulins from human serum were used as a source of serum IgG. IgG3 was isolated by sequential affinity purification using Protein A and Protein G. Protein A binds to IgG1, IgG2 and IgG4, and Protein G binds to all IgG subclasses. About 100 mg of gamma globulins was re-suspended in 20 mM sodium phosphate, 150 mM sodium chloride, pH 7.0 (equilibration buffer) and passed over a Protein A affinity column (5 ml bed volume of Pierce High-Capacity Protein A Ultralink resin) to capture all IgG subclasses except IgG3. The flow through from Protein A column (containing IgG3) was collected and the IgGs retained in the column were eluted with 0.1 M glycine hydrochloride, pH 2.5 (elution buffer). The column was re-equilibrated with phosphate buffer and the flow-through was reloaded onto the column. The procedure was repeated to remove any residual IgG subclasses other than IgG3. To further purify the IgG3, the final flow-through from the Protein A column was loaded onto Protein G column (5 ml bed volume of Protein G resin), washed thoroughly with equilibration buffer (20× column volume), eluted with elution buffer, and immediately neutralized. IgG3 was dialyzed against the equilibration buffer, concentrated using Amicon ultra-centrifugal filters (30 kDa cutoff), and stored at -20 °C. The isolated IgG3 was checked by SDS-PAGE and validated by Western blot using a mouse anti-human IgG3 antibody.
Proteolysis
To prevent disulfide bond shuffling, IgG3 samples were prepared under non-reducing conditions following a protocol modified from reference 16. About 100 μg IgG3 at a concentration of 1 μg/μL in 20 mM phosphate buffer (pH 7.0) was buffer exchanged using a 10 kDa molecular weight cut-off filter (Millipore, Billerica, MA) into 100 mM acetate buffer (pH 6.5) containing 7 M guanidine hydrochloride and 10 mM N-ethylmaleimide (NEM). The sample was incubated at 37 °C for two hours to allow for denaturation and capping of any free cysteine residues. After denaturation and alkylation, excess NEM and guanidine hydrochloride were removed by subjecting the samples to centrifugal filtration using a 10 kDa molecular weight cutoff filter, and reconstituted to a final concentration of 0.8 μg/μL in 100 mM Tris buffer (pH 7.0) containing 1 mM calcium chloride. Trypsin was added at an enzyme-to-protein ratio of 1:10 (w/w) and incubated for 15 hours at 37 °C. Tryptic digestion was stopped by adding 1% formic acid. The digested IgG3 samples were diluted with water to a final concentration of 0.6 μg/μL and aliquots were stored at -20 °C until analysis. For the purpose of reproducibility, samples from the same IgG3 source (Sigma-Aldrich or Fitzgerald) were digested on two different days and each digested sample was run at least two times on different days using the same experimental procedure as described in the LC-MS Analysis section.
LC-MS Analysis
Digested IgG3 samples were analyzed using reversed phase HPLC (Waters Acquity, Milford, MA) coupled with a LTQ Orbitrap Velos Pro hybrid mass spectrometer equipped with ETD (Thermo Scientific, San Jose, CA). A solution of 5 μL of the tryptic digest was injected onto a C18 Aquasil Gold column (100 × 1 mm i.d, 175 Å, Thermo Scientific, San Jose, CA). The mobile phase A was 99.9% water with 0.1% formic acid; and mobile phase B was 99.9% acetonitrile with 0.1% formic acid. After sample injection, the tryptic peptides were eluted from the column at a flow rate of 50 μL/min using the following gradient: Mobile phase B, initially held at 2% for 5 min, was increased to 35% in 55 min, and then ramped to 60% in 15 min, followed by a 10 minute isocratic elution at 95% B and re-equilibration.
Data acquisition was done in the data-dependent scan mode. After a survey MS scan from m/z 400 to 2000 in the Orbitrap mass analyzer at a resolution of 30,000 at m/z 400, the top 5 ions were sequentially selected for ETD (or CID, during CID experiments) in the LTQ mass analyzer. ETD and CID experiments were performed separately in different runs. For ETD experiments, charge state dependent ETD time and supplemental activation were enabled in order to enhance ETD efficiency. The ion-ion reaction time was maintained at 100 ms. For CID experiments, the activation time was set at 10 ms, and the normalization collision energy was 35%. The dynamic exclusion window and isolation width were set at 2 min and 2 Da, respectively, for both CID and ETD experiments. All data were collected in the positive ion mode with the ESI source spray voltage of 3.0 kV and capillary temperature of 250 °C. The data were acquired and analyzed using Xcalibur 2.7 software (ThermoElectron Corp, San Jose, CA)
Results and Discussion
Disulfide Analysis Approach
Most disulfide assignments were done manually using an augmented version of a method reported elsewhere.20 A schematic representation of the disulfide mapping approach used to verify expected (classical) and alternative IgG3 disulfide bonds is shown in Figures 1A and 1B, respectively. To verify the classical disulfide linkage pattern, extracted ion chromatograms (XIC's) are constructed from ETD data based on the m/z values of two Cys-containing peptides that are expected to be linked through a disulfide bond (e.g. peptides P1 and P2, shown in Figure 1A). The XIC's of the two peptides are then compared to quickly verify whether the expected disulfide bond is present. If peaks having the same retention time (RT) are identified (such as the highlighted peaks in the figure) the peptides are preliminarily assigned to be disulfide-bonded partners, and the corresponding ETD spectrum is inspected. ETD preferentially cleaves disulfide bonds and produces intense peaks for each bonded peptide,20,21 so the ETD spectrum is interrogated to determine whether intense marker ion peaks for peptides P1 and P2 are present, along with c and z fragment ions from each bonded chain and the intact disulfide bonded peptide. If all these ions are present, the identity of the disulfide-linked peptides is assigned. Additionally, the assignment is quickly validated by matching the precursor ion mass to the theoretical mass of the dipeptide (the sum of the masses of the two Cys-containing peptides minus 2 Da).
Figure 1.
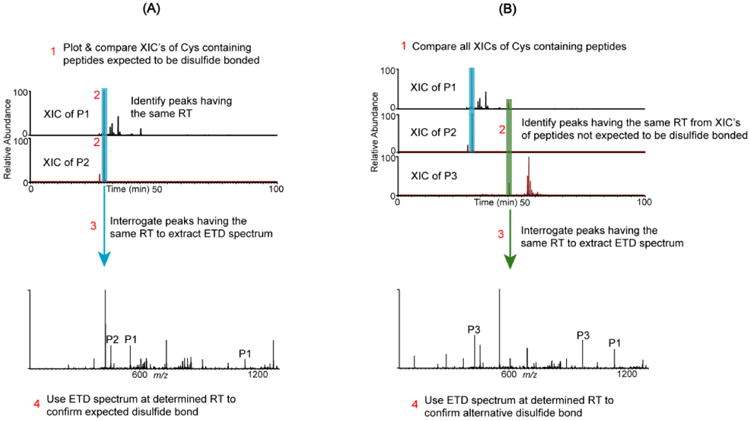
Schematic representation of the disulfide mapping approach for expected and alternative disulfides. (A) Assignment of an expected disulfide bond between Cys-containing peptides P1 and P2. Step 1: Plot XIC's for each peptide. Step 2: Identify peaks with the same retention time. Step 3: Extract the corresponding ETD spectrum, which confirms the disulfide bond. Step 4: Verify that marker ions of each chain (P1 & P2 peaks) and c and z ions from both chains are present in the ETD spectrum. (B) Alternative disulfide bonds are verified by the following: Step 1: Plot and compare the XIC's of all Cys-containing peptides. Step 2: If peaks with the same RT are identified in the XIC's of peptides that are not expected to be disulfide bonded, then steps 3 and 4 in (A) are used to verify whether the two peptides are bonded by an alternative disulfide bond.
After confirming the presence of the disulfide linked peptides, alternatively connected disulfide-linked peptides are also searched for in a similar manner (Figure 1B). For example, in addition to the correct disulfide bond between P1 and P2, if peptide P1 is also alternatively linked to peptide P3, the XIC's of both peptides would have peaks where the P1-P3 dipeptide (alternative disulfide bonded dipeptide) eluted. Therefore, by comparing the XIC's for peptides P1 and P3, and following the procedure described in the previous paragraph for identifying disulfide bonds between two peptides, the alternative disulfide bond between peptide P1 and peptide P3 could be identified, if it were present. Hence, to rapidly search for alternatively disulfide linked peptides, the XIC's of all Cys-containing peptides were compared, asking the question: Are there any peaks that show up at the same retention time in at least two chromatograms, which could be aberrant disulfide linked peptides? When such peaks are identifiable, their ETD spectra are interrogated, as described above, to determine if the ion responsible for the peak is an aberrant disulfide-bonded peptide.
Assignment of Expected (Classical) IgG3 Disulfide bonds
The classical disulfide bond structure of IgG3 is shown in Figure 2A. The comprehensive structure consists of 12 domains, two heavy chains and two light chains, with each chain having a variable and constant region. There are a total of 50 Cys residues that form 25 disulfide bonds: 21 in the constant regions and four in the variable regions. Because the IgG3 samples used in this study were isolated from human serum, the amino acid sequences of the variable regions were unknown; therefore, only the disulfide bonds in the constant regions were mapped. Additionally, disulfide bonds for both the lambda and kappa light chains were mapped. The expected tryptic disulfide bonded peptides from the constant region are shown in Figure 2B. We verified the presence of these expected disulfide bonds prior to investigating disulfide bond variants. For example, Figure 3 shows XIC's and ETD data that support the assignment of the disulfide bonded peptide in the CH2 domain of the Fitzgerald IgG3 antibody. The XIC's of the CH2-1 (TPEVTCVVVDVSHEDPEVQFK) and CH2-2 (CK) Cys-containing peptides were plotted by searching ETD data in the m/z range of 1178-1181 (which encompasses the CH2-1 theoretical m/z of 1179 in the plus two charge state) and 248-251 (which encompasses the CH2-2 theoretical m/z 250 in the plus one charge state), respectively. This resulted in XIC's with intense peaks at the retention time of 37.5 min for both the CH2-1 peptide (Figure 3a) and the CH2-2 peptide (Figure 3b). The presence of peaks at the same retention time in both XIC's suggests that the two peptides are potential disulfide bonded partners. To verify this, the ETD spectrum of the ion that eluted at 37.5 was extracted (Figure 3c). The marker ion peaks for the CH2-1 and the CH2-2 peptides (at m/z 1179 and 250, respectively) were conspicuously present in the ETD spectrum, and c and z ions from the CH2-1 peptide, as well as c and z ions containing the intact disulfide bond (labeled in red) were identified, thereby confirming that the two peptides are indeed disulfide bonded. Additionally, using high resolution data, the monoisotopic molecular mass of the precursor ion was determined to be within 3 ppm of the theoretical mass of the dipeptide. Hence the CH2 domain disulfide bond was assigned. The CH3, kappa and lambda constant light chain (CL), and the kappa and lambda heavy chain-light chain (HC-LC) domain disulfide bonds were assigned in a similar manner, and these results are shown in Supplementary Figures 1 to 5.
Figure 2.
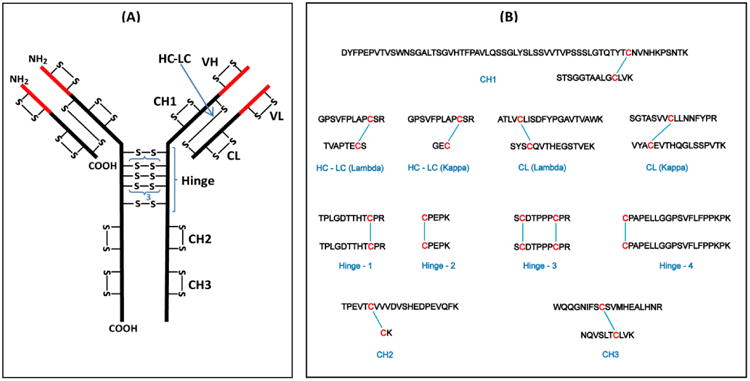
(A) Structure of a typical human IgG3 antibody showing the disulfide bond pattern. There are a total of 50 Cys residues and 25 disulfide bonds (–S–S–). The red parts are the variable (V) regions and the black parts are the constant (C) regions. H and L indicate the heavy and light chains, respectively; VL and CL are domains of the light chain; VH, CH1, CH2, and CH3 are domains of the heavy chain. The hinge region has a 15-residue segment that is repeated three times. (B) Expected tryptic dipeptides from human IgG3 constant region.
Figure 3.
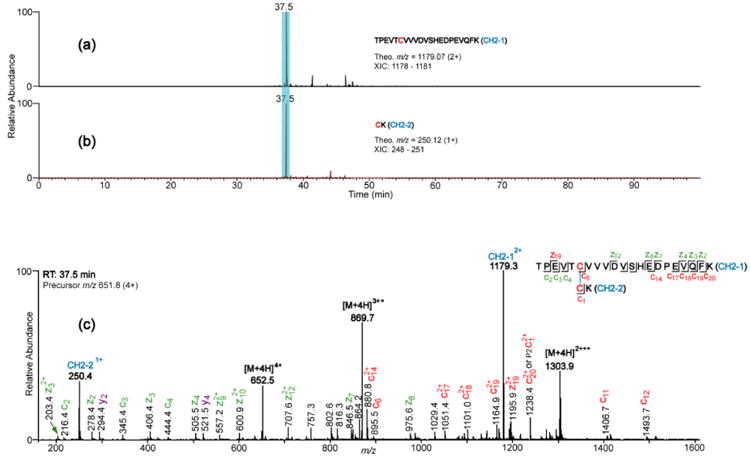
XIC's and ETD spectrum showing assignment of the CH2 domain disulfide bond. (a) and (b) are XIC's of peptides TPEVTCVVVDVSHEDPEVQFK and CK, respectively; clearly showing the RT of the dipeptide (highlighted). The peptide sequence, theoretical m/z, and the m/z range that was used to plot the XIC's are shown in the inset. (c) ETD spectrum of the CH2 domain dipeptide. Marker ion peaks resulting from the cleavage of the disulfide bond are labeled in blue; product ions (c/z ions) not containing the disulfide bond are labeled in green; product ions containing the disulfide bond are labeled in red; unexpected product ions (b/y ions) are labeled in purple.
Assignment of disulfide bonds between identical Cys-containing peptides
Proteolytic digestion of IgG antibodies usually produce dipeptides with identical disulfide bonded chains originating from the hinge regions of the antibodies. This happens because all IgG antibodies have inter-chain disulfide bonds in their hinge regions that link identical sequences of the heavy chains. For instance, IgG3 has four tryptic dipeptides (Hinge-1, Hinge-2, Hinge-3, and Hinge-4) that have identical disulfide bonded chains. Our previously published method for assigning disulfide bonds does not address this possibility; therefore, we have improved the method to account for these species. Because the disulfide bonded peptides are identical, it may seem that the XIC data would not be useful for identifying them, since plotting the same XIC data twice results in two identical chromatograms. Nonetheless, we determined that the XIC's do, in fact, become useful if the charge states of the peptide marker ions are different for the two XIC's. For example, Figure 4 shows the assignment of the Hinge-1 disulfide bond between two identical tryptic peptides (TPLGDTTHTCPR disulfide-linked to TPLGDTTHTCPR). Although the disulfide bonded peptides are identical, by using charge states of 1 and 2 (corresponding to m/z 1298.6 and 649.8, respectively) distinct XIC's were obtained (Figure. 4a and 4b), and the peaks in the two XIC's with the same retention time correspond to the disulfide bonded dimers. The ETD spectrum of the XIC peak at RT 22.1 min (Figure 4c) shows the marker ion peaks at m/z 649 and 1298, along with c and z product ion peaks with and without the disulfide bond, labeled in red and green respectively. Additionally, the observed monoisotopic mass of the precursor ion mass was within 2 ppm of the theoretical mass of the expected Hinge-1 dipeptide, further confirming the assignment. Another example of the assignment of disulfide bonded peptides, where both peptides are identical, is shown in Supplementary Figure 6; it confirms the Hinge-4 assignment.
Figure 4.
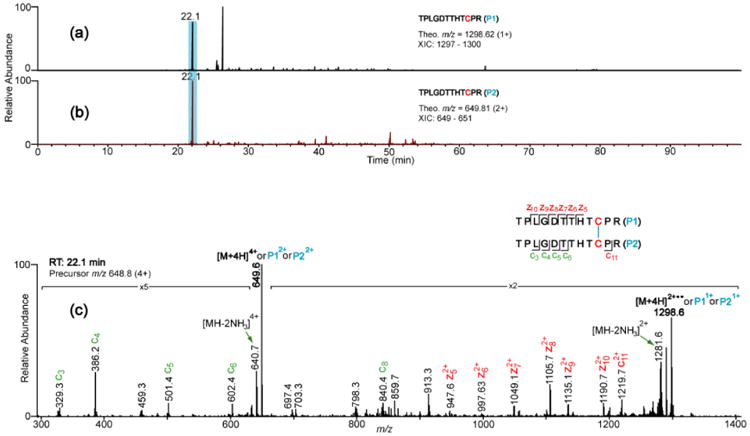
Representative XIC's (a and b) and ETD spectrum (c) that support the assignment of the Hinge-1 disulfide, which has identical Cys-containing peptides. Since the peptides are identical, the XIC's for different charge states (+1 and +2) were plotted so as to avoid plotting the same XIC twice. Details about the product ion colors are given in Figure 3.
Disulfide bonds identified using CID data
Three expected disulfide bonded peptides (in the Hinge-2, Hinge-3, and CH1 regions) were not readily assigned using the XIC/ETD method. The Hinge-2 dipeptide is small (CPEPK bonded to CPEPK), and the highest charge state identified for the dipeptide was two, which is not sufficient for efficient ETD fragmentation. Additionally, the presence of two proline residues prevented ETD cleavage before and after the proline residues, limiting the formation of c and z ions. The Hinge-3 dipeptide, which was also not rapidly detected using the XIC/ETD method, contains two SCDTPPPCPR tryptic peptides connected by two disulfide bonds. In this case, ETD data was acquired on the dipeptide, but the ETD spectrum showed only charged reduced species and no peptide marker ions. Therefore, the XIC's of the disulfide bonded partners were not useful for assigning this dipeptide. The disulfide bonded peptide in the CH1 domain also was not identified by the XIC/ETD method because XIC's of these tryptic peptides did not show marker ions for the two peptide partners in the same ETD spectrum. The absence of these ions could be due to the large size of one of the tryptic peptides. The CH1-2 tryptic peptide is 63 amino acids long (without any missed cleavage) and the Cys residue is 11 residues from the C-terminus, leaving 52 amino acids after the disulfide bond. It is possible that because the portion of the peptide is very long, it could fold around the disulfide bond, thereby preventing efficient transfer of the ETD reagent ion to the disulfide bond and consequent cleavage of the bond by ETD. A shorter CH1 domain dipeptide may be obtainable using a different enzyme; in that case, the XIC/ETD method may be used to assign the disulfide bond.
These three disulfide linked peptides were assigned using a complementary strategy: Searching for the in-tact disulfide bonded peptides in the high resolution MS data first, followed by confirmation of the species using CID data, a procedure reported by Go et al.22 A prediction table containing the theoretical masses and m/z's of the dipeptides at different charge states was generated, and the XIC of each in-tact dipeptide was constructed from the total ion chromatogram. High resolution MS data and the corresponding CID spectra were used to assign the dipeptides. For instance, Figure 5 shows the assignment of the CH1 domain disulfide bond using this approach. The XIC of the ion m/z 1130 was plotted (Figure 5A) and the corresponding high resolution mass spectrum at 53.3 min is shown in the insert. It contains five peaks at m/z 879, 989, 1130, 1319, and 1582, which correspond to the theoretical m/z's of the CH1 domain dipeptide at charge states of 9+, 8+, 7+, 6+, and 5+, respectively. Figure 5B shows the CID spectrum of the ion, m/z 1130, eluting at 53.3 min. Abundant b and y ions resulting from the fragmentation of both CH1 bonded peptides are present, and they are used to unequivocally confirm the CH1 disulfide bonded peptide. The CID spectra showing the assignments of the Hinge-2 and Hinge-3 disulfide bonded peptides are shown in Supplementary Figures 7 and 8, respectively.
Figure 5.

XIC and CID spectrum supporting the assignment of the CH1 domain disulfide bond. (A) XIC of the CH1 domain tryptic dipeptide at m/z 1130 (7+). The insert shows the full mass spectrum at 53.3 min. (B) CID spectrum of the 1130.86 ion (7+), supporting the assignment of the CH1 domain disulfide bond. Product ions (b/y) that contain the disulfide bond are labeled in red; product ions that do not contain the disulfide bond are labeled in green.
Overall, by combining two different MS-based approaches for assigning disulfide-linked peptides,20, 22 we identified all the expected disulfide bonds in the constant region of endogenous IgG3 from two different sources. A summary of the disulfide bond assignments using the XIC/ETD method is shown in Table 1, and those identified using high resolution and CID data are shown in Supplementary Table 1.
Table 1.
Summary of Disulfide Bond Assignments in Fitzgerald and Sigma IgG3 mAbs Using XIC/ETD Data.
| Fitzgerald IgG3 | Sigma IgG3 | |||||
|---|---|---|---|---|---|---|
|
|
||||||
| Cys-Containing Tryptic peptides | Position | Peptide XIC m/z (Theo) | RT | Peptide XIC m/z (Expt) | RT | Peptide XIC m/z (Expt) |
| GPSVFPLAPCSR | HC-LC | 1230.6 | 33.9 | 1230.8 | 34.2 | 1230.3 |
| GEC | Kappa | 308.1 | 33.9 | 308.2 | 34.2 | 308.1 |
|
| ||||||
| GPSVFPLAPCSR | HC-LC | 1230.6 | 34.9 | 1229.7 | 35.1 | 1229.7 |
| TVAPTECS | Lambda | 807.4 | 34.9 | 807.4 | 35.1 | 807.4 |
|
| ||||||
| ATLVCLISDFYPGAVTVAWK | CL | 1077.62+ | 50.0 | 1077.42+ | 50.3 | 1077.42+ |
| SYSCQVTHEGSTVEK | Lambda | 827.92+ | 50.0 | 827.62+ | 50.3 | 827.72+ |
|
| ||||||
| SGTASVVCLLNNFYPR | CL | 870.92+ | 44.4 | 870.92+ | 44.6 | 870.82+ |
| VYACEVTHQGLSSPVTK | Kappa | 910.02+ | 44.4 | 909.82+ | 44.6 | 909.82+ |
|
| ||||||
| TPLGDTTHTCPR | Hinge-1 | 1298.6 | 22.1 | 1298.6 | 22.6 | 1298.8 |
| TPLGDTTHTCPR | 649.82+ | 22.1 | 649.62+ | 22.6 | 649.62+ | |
|
| ||||||
| CPAPELLGGPSVFLFPPKPK | Hinge-4 | 1047.62+ | 53.4 | 1047.82+ | 53.7 | 1047.82+ |
| CPAPELLGGPSVFLFPPKPK | 698.73+ | 53.4 | 698.9 | 53.7 | 698.83+ | |
|
| ||||||
| TPEVTCVVVDVSHEDPEVQFK | CH2 | 1179.12+ | 37.5 | 1179.32+ | 37.8 | 1179.62+ |
| CK | 250.1 | 37.5 | 250.4 | 37.8 | 250.1 | |
|
| ||||||
| WQQGNIFSCSVMHEALHNR | CH3 | 1129.02+ | 41.0 | 1128.92+ | 41.2 | 1129.22+ |
| NQVSLTCLVK | 1104.6 | 41.0 | 1104.7 | 41.2 | 1104.6 | |
Unless otherwise stated, all product ions are in the plus one charge state. Precursor ions ranged from the +2 to +6 charge states.
Assignment of Disulfide Bond Variants (Alternative disulfide bonds)
After verification that all of the classical IgG3 disulfide bonds were present, we investigated the presence of alternative disulfide bonds. The same approaches used to identify the classical disulfide bonds were used to search for alternative disulfide bonds. For the dipeptides that were identified using the XIC/ETD method, the XIC's of all the Cys-containing peptides were aligned and compared (Figure 6). In addition to the peaks that revealed the expected disulfide bonds (highlighted in blue), three sets of low abundant peaks in the XIC's of peptides that are not expected to be disulfide bonded were identified to have the same retention time (peaks at 19.2, 38.1 and 41.0 mins), suggesting the possible presence of alternative disulfide bonds. To determine if the ions generating these peaks are aberrant disulfide bonded peptides, the corresponding ETD spectra were extracted and studied. None was found to correspond to disulfide bonded peptides. For example, the XIC's of CPAPELLGGPSVFLFPPKPK (Hinge-4, P2, m/z 698.72 at charge state of three) and WQQGNIFSCSVMHEALHNR (CH3-1, m/z 1129.02 at charge state of two), which are not expected to be disulfide bonded, both have peaks at 19.2 min, suggesting that they are potentially linked by an alternative disulfide bond. The MS data that generated the ETD spectrum of the ion that eluted at 19.2 min was interrogated. Immediately, this peak was confirmed not to be an aberrantly disulfide bonded peptide because the precursor ion mass that generated this peak was from a 1267.6 Da ion, which does not match the theoretical mass of 4347.2 for disulfide bonded CPAPELLGGPSVFLFPPKPK and WQQGNIFSCSVMHEALHNR. Hence, there was no alternative bond between these two peptides. After further interrogation, this particular peptide was assigned as the Hinge-2 dipeptide with a non-specific N-ethylmaleimide alkylation (Theoretical mass of 1267.6). The MS data for the peaks at 38.1 and 41.0 min were interrogated in a similar manner, and they were also found not be related to any alternatively linked disulfide bonded peptides. Overall, no alternative disulfide bonded peptides were found between any cys-containing peptides that are not expected to be disulfide bonded.
Figure 6.
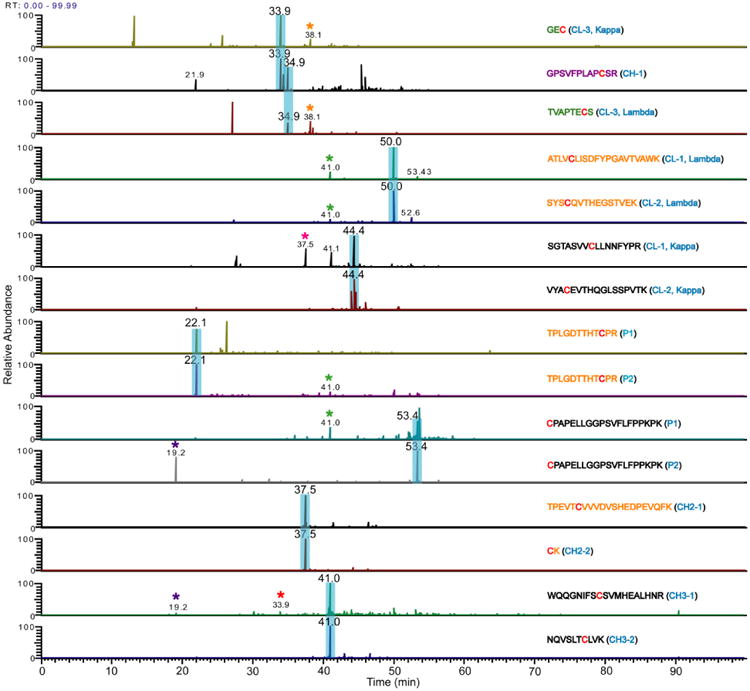
XIC's of Cys-containing peptides identified in Table 1. Adjacent Cys-containing peptides of the same color (orange or black) are disulfide bonded, and the peaks in their XIC's that lead to their identification are highlighted in blue. The CH1 Cys-containing peptide (purple) is disulfide bonded to the third Cys residue of the kappa and lambda light chains (green). Peaks with the same retention time in XIC's of peptides that are not expected to be disulfide bonded are indicated by asterisks. Each of these peaks was interrogated to determine whether or not it was from an aberrant disulfide-linked peptide. In each case, the peaks were verified to be from another source, confirming that no aberrant disulfide-linked peptides are present.
For the CH1, Hinge-2, and Hinge-3 domain disulfide bonds (see Figure 2B for the tryptic disulfide bonded peptides), which were identified using high resolution (MS1) and CID data, a prediction table containing the masses and m/z values of plausible alternative disulfide bonds between these Cys-containing peptides and other Cys-containing peptides in close proximity was constructed, as described previously.22 This table includes six plausible four-peptide disulfide-linked chains involving two SCDTPPPCPR peptides and three other Cys-containing peptides in close proximity. The calculated m/z's were searched using high resolution MS and CID data, and no alternative disulfide linked peptides were identified in either IgG3 sample using this approach.
In summary, no disulfide bond variants were detected, even though they were searched for using two different search strategies. It is theoretically possible that disulfide bond variants exist for IgG3 and remained undetected, but if that is the case, we expect these variants to be in very low abundance, perhaps less than 2% of the protein. This estimate is based on substantial prior work we have completed using these methods and mapping disulfide bond variants in HIV-1 Envelope proteins.20, 22-26 In one case, the MS methods described here were able to detect an aberrant disulfide bonded isoform that was present in substantially less than 5% of the total protein population.26
Conclusion
We carried out an extensive disulfide bond analysis of two endogenous human IgG3 samples to experimentally verify the presence of the classical IgG3 disulfide bonds and to investigate the possibility of disulfide-mediated isoforms resulting from alternative disulfide bonds. All expected disulfide bonded peptides in the constant regions of the IgG3 samples from two different sources were unambiguously assigned. Both the kappa and lambda forms were fully characterized in each protein. Although disulfide-mediated isoforms have been identified for IgG2 and IgG4 antibodies, and to a lesser extent in IgG1, the data presented herein show that there are no alternative disulfide bonded peptides within the constant region of native IgG3 antibodies, indicating that endogenous IgG3 antibodies do not have disulfide-mediated isoforms. The data presented herein provide the first benchmark for a complete native IgG3 disulfide bonding profile, and the analytical approach described herein can be readily applied to recombinant IgG3 antibodies or any other IgG.
Supplementary Material
Acknowledgments
This manuscript is dedicated to Professor Craig E. Lunte, who was a wonderful leader and friend of the bioanalytical community. The work was supported by NIH grant number R01AI094797 and R01GM103547 to Heather Desaire and R01GM090080 to Thomas Tolbert.
References
- 1.Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, Daeron M. Blood. 2009;113:3716–3725. doi: 10.1182/blood-2008-09-179754. [DOI] [PubMed] [Google Scholar]
- 2.Hogarth PM, Pietersz GA. Nat Rev Drug Discov. 2012;11:311–331. doi: 10.1038/nrd2909. [DOI] [PubMed] [Google Scholar]
- 3.Morell A, Terry WD, Waldmann TA. J Clin Invest. 1970;49:673–680. doi: 10.1172/JCI106279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jefferis R. Expert opinion on biological therapy. 2007;7:1401–1413. doi: 10.1517/14712598.7.9.1401. [DOI] [PubMed] [Google Scholar]
- 5.Stapleton NM, Andersen JT, Stemerding AM, Bjarnarson SP, Verheul RC, Gerritsen J, Zhao Y, Kleijer M, Sandlie I, de Haas M, Jonsdottir I, van der Schoot CE, Vidarsson G. Nat Commun. 2011;2:599. doi: 10.1038/ncomms1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Plomp R, Dekkers G, Rombouts Y, Visser R, Koeleman CAM, Kammeijer GSM, Jansen BC, Rispens T, Hensbergen PJ, Vidarsson G, Wuhrer M. Mol Cell Proteomics. 2015;14:1373–1384. doi: 10.1074/mcp.M114.047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abkevich VI, Shakhnovich EI. J Mol Biol. 2000;300:975–985. doi: 10.1006/jmbi.2000.3893. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Chou CP, Moo-Young M. Biotechnol Adv. 2011;29:923–929. doi: 10.1016/j.biotechadv.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 9.Betz SF. Prot Sci. 1993;2:1551–1558. doi: 10.1002/pro.5560021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milstein C, Frangion B. Biochem J. 1971;121:217–225. doi: 10.1042/bj1210217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangione B, Milstein C. J Mol Biol. 1968;33:893–906. doi: 10.1016/0022-2836(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 12.Frangione B, Milstein C. Nature. 1967;216:939–941. doi: 10.1038/216939b0. [DOI] [PubMed] [Google Scholar]
- 13.Pink JR, Milstein C. Nature. 1967;216:941–942. doi: 10.1038/216941a0. [DOI] [PubMed] [Google Scholar]
- 14.Pink JR, Milstein C. Nature. 1967;214:92–94. doi: 10.1038/214092a0. [DOI] [PubMed] [Google Scholar]
- 15.Schuurman J, Perdok GJ, Gorter AD, Aalberse RC. Mol Immunol. 2001;38:1–8. doi: 10.1016/s0161-5890(01)00050-5. [DOI] [PubMed] [Google Scholar]
- 16.Bloom JW, Madanat MS, Marriott D, Wong T, Chan SY. Prot Sci. 1997;6:407–415. doi: 10.1002/pro.5560060217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, Fodor S, Kelner DN, Flynn GC, Liu YD, Bondarenko PV, Ricci MS, Dillon TM, Balland A. J Biol Chem. 2008;283:16194–16205. doi: 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez T, Guo A, Allen MJ, Han M, Pace D, Jones J, Gillespie R, Ketchem RR, Zhang Y, Balland A. Biochemistry. 2008;47:7496–7508. doi: 10.1021/bi800576c. [DOI] [PubMed] [Google Scholar]
- 19.Zhang A, Fang J, Chou RYT, Bondarenko PV, Zhang Z. Biochemistry. 2015;54:1956–1962. doi: 10.1021/bi5015216. [DOI] [PubMed] [Google Scholar]
- 20.Clark DF, Go EP, Desaire H. Anal Chem. 2013;85:1192–1199. doi: 10.1021/ac303124w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu SL, Jiang H, Lu Q, Dai S, Hancock W, Karger BL. Anal Chem. 2009;81:112–122. doi: 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Go EP, Hua D, Desaire H. J Proteome Res. 2014;13:4012–4027. doi: 10.1021/pr5003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Go EP, Zhang Y, Menon S, Desaire H. J Proteome Res. 2011;10:578–591. doi: 10.1021/pr100764a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kassa A, Dey AK, Sarkar P, Labranche C, Go EP, Clark DF, Sun Y, Nandi A, Hartog K, Desaire H, Montefiori D, Carfi A, Srivastava IK, Barnett SW. Plos One. 2013 doi: 10.1371/hournal.pone.0076139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ringe RP, Yasmeen A, Ozorowski G, Go EP, Pritchard LK, Guttman M, Ketas TA, Cottrell CA, Wilson IA, Sanders RW, Cupo A, Crispin M, Lee KK, Desaire H, Ward AB, Klasse PJ, Moore JP. J Virol. 2015;89(23):12189–12210. doi: 10.1128/JVI.01768-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Go EP, Cupo A, Ringe RP, Pugach P, Moore JP, Desaire H. J Virol. 2016;90(6):2884–2894. doi: 10.1128/JVI.01953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.