

This review by Conlon and Manley focuses on RNA processing in neurodegeneration. It describes how neurons are particularly vulnerable to disruption of RNA-binding proteins (RBPs), leading to a wide range of neurological and neurodegenerative diseases.
Keywords: disease mechanisms, neurodegeneration, RNA-binding proteins
Abstract
Neurodegeneration is a leading cause of death in the developed world and a natural, albeit unfortunate, consequence of longer-lived populations. Despite great demand for therapeutic intervention, it is often the case that these diseases are insufficiently understood at the basic molecular level. What little is known has prompted much hopeful speculation about a generalized mechanistic thread that ties these disparate conditions together at the subcellular level and can be exploited for broad curative benefit. In this review, we discuss a prominent theory supported by genetic and pathological changes in an array of neurodegenerative diseases: that neurons are particularly vulnerable to disruption of RNA-binding protein dosage and dynamics. Here we synthesize the progress made at the clinical, genetic, and biophysical levels and conclude that this perspective offers the most parsimonious explanation for these mysterious diseases. Where appropriate, we highlight the reciprocal benefits of cross-disciplinary collaboration between disease specialists and RNA biologists as we envision a future in which neurodegeneration declines and our understanding of the broad importance of RNA processing deepens.
Alternative transcript processing is an effective mechanism to generate functional diversity from a fixed volume of genetic information. Such types of justifications are often called on to rationalize the lack of correlation between the number of genes in an organism's genome and its perceived complexity. Nowhere is this more pertinent than in studies of the human brain, the most cellularly and functionally complex organ among all living things. While much research has focused on how cis-acting elements within alternatively processed transcripts provide instructions for the multitude of forms that they can take, the mechanisms through which these signals impart different meaning in a context-dependent manner depends heavily on their trans-acting effectors: RNA-binding proteins (RBPs). RBPs seldom act alone but rather form extensive protein–protein and protein–RNA interactions in an immense number of permutations that allow for spatial and temporal control of gene expression in response to a range of stimuli. Given the scope and intricacy of functions that the human central nervous system must regulate, it is thus not surprising that disruption of precise RBP stoichiometry through mutations and pathological events leads to a wide range of neurological and neurodegenerative diseases. In this review, we discuss recent findings and recurrent questions regarding the widening role of RBPs in human neurodegenerative diseases. For further insight into this topic, we refer the reader to a number of excellent related reviews (Cooper et al. 2009; Vanderweyde et al. 2013; Nussbacher et al. 2015; Cookson 2017; De Conti et al. 2017).
RNA metabolism in the brain—a heightened demand
The human brain contains a remarkable number of cells, currently estimated at 170 billion, with an ∼1:1 ratio of neurons:nonneurons (glia) (Azevedo et al. 2009). Neurons, the targets of neurodegeneration, come in countless varieties, differing in position (Fig. 1), size, neurotransmission, and electrophysiological properties yet are united by several key features such as their polarization, ability to form networks with other neurons, and enormous energy demands (Herculano-Houzel 2012; Sharpee 2014).
Figure 1.

Overview of diseases and genes covered. Top-down schematic of neurodegeneration illustrating the tissue-wide phenomena and the genetic and molecular changes that occur at the subcellular level. (Left) Approximate sites of pathology and degenerating neurons in diseases covered in this review, including amyotrophic lateral sclerosis (ALS), spinocerebellar ataxia type 36 (SCA36), spinal muscular atrophy (SMA), frontotemporal dementia (FTD), Huntington's disease (HD), Huntington's disease-like 2 (HDL2), and myotonic dystrophy type 1 (DM1) and DM2. (Right) Mutations at the DNA result in a variety of changes at the level of RNA processing, such as repression of cryptic splicing (i), formation of exonic splicing silencers (ii), formation of hairpin structures that trigger RNA silencing pathways and sequester proteins (iii), formation of stable G-quadruplex (G-Q) structures that can form aggregates with RBPs (iv), mutations in RBPs that affect their RNA processing functions (v), and formation of aggregation-prone polyglutamine (polyQ) and dipeptide repeat (DPR) proteins (vi,vii). Genes harboring mutations are categorized as being RBP-encoding genes (TARDBP, FUS, hnRNP A1/A2B1, EWSR1, and MATR3 in ALS), noncoding expansions (C9ORF72 in ALS–FTD, FMR1 in fragile X syndrome and fragile X-associated tremor/ataxia syndrome, NOP56 in SCA36, and DMPK and CNBP in DM1 and DM2, respectively) or coding expansions (HTT in HD, JPH3 in HDL2, ATXN2 in spinocerebellar ataxia type 2 and ALS, and AR in X-linked spinal and bulbar muscular atrophy). Nucleotide repeat units for each noncoding and coding expansion are in quotations.
While our analysis focuses primarily on genetically determined neurodegeneration, the greatest risk factor for neurodegeneration is normal aging (Bishop et al. 2010). This underscores the point that neurodegeneration is a gradual, age-dependent process that may exploit another unique feature of neurons, which is their persistent metabolic activity across long life spans (Magistretti and Allaman 2015). The molecular mechanisms put forward for neurodegenerative diseases must always strive to explain how accumulative damage due to inherited/germline mutations can manifest decades later in highly specific neuronal loss. Likewise, an important aspect of faithful disease modeling is the consideration of how different genetic variants lead to earlier onset and faster progression, as the factors that result in more serious disease may be of therapeutic interest as well as informative of the diseases themselves (Van Damme et al. 2017).
Alternative mRNA processing greatly increases the dimensions of gene expression beyond the on/off duality through splicing, polyadenylation, targeted localization, and post-transcriptional silencing, and the cells in the brain take great advantage of these different strategies to diversify their repertoires. For example, it has been shown that the brain undergoes particularly high levels of alternative splicing relative to other human tissues and also tends to follow more distinctive patterns (Yeo et al. 2004). Analogous types of analyses have similarly revealed that brain tissues are unique in alternative polyadenylation choices (Zhang et al. 2005). In recent years, more classes of RNAs, including microRNAs (miRNAs) (Wang et al. 2012), enhancer RNAs (eRNAs) (Kim et al. 2010), and long intergenic noncoding RNAs (lincRNAs) (Sauvageau et al. 2013; Wu et al. 2013), have been demonstrated to contribute to the fine-tuning of gene expression in the brain that specifies its distinctive proteome and capacity for adaptive responses. While splicing makes up the majority of our discussion, in reality, it is impossible to consolidate the many ways that RNA and, by association, RBPs make neurons unique.
The outcomes of alternative splicing can have important biological relevance in the brain. This can include, for example, determining isoform expression patterns of receptors and channels critical to neurotransmission (Grabowski and Black 2001). Sometimes, these brain-specfic events are regulated by binding of brain-restricted splicing factors such as NOVA (Jensen et al. 2000) or neural polypyrimidine tract-binding protein (nPTB) (Coutinho-Mansfield et al. 2007; Licatalosi et al. 2012). However, for many specific neuronal splicing events and for splicing events in general, splice site choices rely on the concerted action of many indispensable factors that are ubiquitously expressed (Chan and Black 1997; Chou et al. 1999). Thus, it is seen that alternative RNA processing is driven by the competition inherent in the dynamic concentrations of RBPs with respect to other protein factors and the abundance of high-affinity sequences that they bind (Dreyfuss et al. 1993; Chen and Manley 2009).
Early clinical examples
Disease has been one of our biggest teachers when it comes to the importance of RBPs in defining neural identity. Just as classical ablation studies have enabled researchers to map the contributions of different brain regions, naturally occurring human encephalopathies have unlocked molecular signatures of the brain's function.
Paraneoplastic syndromes
The neuronal RBPs Hu and Nova-1/2 were discovered as the targets of high-titer antibodies present in the sera of cancer patients with paraneoplastic neurologic disorders (PNDs). These proteins represent the misexpressed pathological culprits behind paraneoplastic encephalomyelitis/sensory neuronopathy (PEM/SN) and paraneoplastic opsoclonus–myoclonus ataxia (POMA), respectively (Darnell 1996). PNDs arise due to ectopic expression of brain-specific proteins in tumors; for instance, in small cell lung cancer (PEM/SN and POMA) and breast and fallopian cancer (POMA) (Yang et al. 1998). The abnormal expression of these brain-restricted proteins causes the immune system to mount an attack on these antigens, thus, in the case of POMA, releasing autoantibodies that attack Nova-1 in the regions of the brain and spinal cord where they are expressed in a highly restricted manner (Zhou et al. 2014). The generation of Nova-1-null mice highlighted the tissue-specific relevance of this protein, as the animals exhibited progressive motor dysfunction in their short life spans before dying within 10 d postnatally and, importantly, displayed defects in specific splicing events critical to the affected cells and developmental period (Jensen et al. 2000). Subsequently, Nova-1 was the first protein to be subjected to CLIP (cross-linking immiunoprecipitation) analysis. CLIP enables profiling of the RNAs associated with a protein in vivo and has been instrumental in much of the research that we discuss here. For Nova-1, this effort led to the striking observation that three-quarters and one-third of Nova-1 splicing targets function in neuronal synapses and inhibition, respectively (Ule et al. 2003).
The main symptoms of POMA are indicative of lower motor dysfunction (Buckanovich et al. 1993). However, it has been noted that symptoms of POMA sometimes include atrophy of the cerebellum, a region not expressing Nova-1. Furthermore, POMA antisera are reactive against regions of the brain outside the limits of Nova-1 expression. These inconsistencies led to the discovery of a highly similar protein, Nova-2 (Yang et al. 1998). Subsequent CLIP analysis of Nova-2 demonstrated a role in both brain-specific splicing and alternative polyadenylation (Licatalosi et al. 2008). Decreased Nova activity has been found in brains from a pooled cohort of frontotemporal dementia (FTD) and Alzheimer's disease patients (Tollervey et al. 2011b).
Spinal muscular atrophy (SMA)
SMA is a juvenile-onset neurodegenerative disorder marked by the loss of lower motor neurons (MNs) from the anterior horn of the spinal cord. In its most extreme form, it is the most common monogenic cause of infant mortality (Prior 2010). The disease is caused by homozygous loss-of-function (LOF) mutations in the gene SMN1, which, together with the nearly identical SMN2 gene (unique to humans), encodes the protein survival of MN (SMN), a protein chaperone for spliceosomal factor biogenesis (Pellizzoni 2007; So et al. 2016). While SMN is not an RBP itself, its interaction with gemin proteins is critical for the efficient assembly of Sm proteins and small nuclear RNAs (snRNAs) into snRNPs (Kolb et al. 2007). SMN thus resides upstream of the proper functioning of many RBPs by orchestrating the splicing process in which they participate.
While SMN in an essential protein in organisms from Schizosaccharomyces pombe to humans, its range of suboptimal levels in patients is the defining factor in the spectrum of differentially severe forms of SMA. This has prompted much inquiry into why MNs are seemingly more susceptible than other cells to low SMN levels (Kolb et al. 2007; Simone et al. 2016; Tu et al. 2017).
SMA is classified into three types based on age of onset and disease severity (Simone et al. 2016). The different clinical manifestations reflect the overall amounts of SMN protein (Lefebvre et al. 1997), which are determined by the mutation (deletion vs. partial LOF) in SMN1 (Tu et al. 2017), SMN2 gene copy number (Taylor et al. 1998; Harada et al. 2002), and the amount of full-length SMN2 produced (Fig. 1). Full-length SMN2 represents the minority of total SMN2 produced due to a translationally silent single-nucleotide difference distinguishing exon 7 of SMN1 from SMN2. This polymorphism generates a novel exonic splicing silencer in the latter (or perhaps also disrupts a splicing enhancer in the former) (Cartegni and Krainer 2002) that is bound repressively by the RBPs hnRNP A1 and Sam68 (Kashima and Manley 2003; Pedrotti et al. 2010). MNs have been shown to express lower levels of exon 7-containing SMN2 than other cells in the spinal cord, which is negatively reinforced in an SMN1-deficient context where splicing is most compromised (Ruggiu et al. 2012). Development of antisense oligonucleotides (ASOs) that promote exon 7 inclusion, increase full-length SMN protein levels (Hua et al. 2007), and can rescue severe SMA mice when injected neonatally (Hua et al. 2011) thus represents a major therapeutic breakthrough. This technology led to the development of Biogen's Spinraza, the first Food and Drug Administration-approved therapy for SMA (Ottesen 2017), and has increased the hope that ASOs may be successful at mitigating other neurodegenerative diseases.
A key question when disease-linked changes in gene expression arise from mutations affecting a transcription/RNA processing factor is the identity of the critical gene targets that lead to disease, and this is certainly true with SMN. A number of studies (Zhang et al. 2008; Lotti et al. 2012; Doktor et al. 2017) have pointed to defects in splicing of a class of introns known as U11/U12 introns. These introns represent <1% of all introns and differ at their consensus 5′ donor and 3′ acceptor sites, requiring a spliceosome with distinct snRNPs for intron removal. Splicing of U11/U12 introns is less efficient than splicing of standard introns, which is thought to be a rate-limiting factor in the expression of genes that contain a U11/U12 intron (Turunen et al. 2013). Interestingly, a rare class of genes with more than one U11/U12 intron is the voltage-gated ion channel superfamily of genes (Wu and Krainer 1999), which themselves are mutated in neurodegenerative and neuromuscular disease (Andavan and Lemmens-Gruber 2011), suggesting that selectively compromised splicing of these transcripts may have disease relevance. Recently, it was shown in mice that SMN deficiency leads to intron retention, particularly among U11/U12 introns, which can be reversed by increasing SMN levels (Jangi et al. 2017). While the affected genes were not similar in function, their pre-mRNA structures were, as they tended to have weaker 5′ and 3′ splice sites, be GC-enriched, and contain more R-loop structures to which SMN has been independently been shown to be recruited (Zhao et al. 2016; Jangi et al. 2017).
Efforts are ongoing to determine whether there is indeed a select repertoire of splicing changes associated with SMN deficiency from which one can rationalize disease features or whether SMN depletion leads to global splicing disruption that manifests differently depending on the splicing factors in a given cell type. While these mechanistic questions still remain, the evidence that SMN is indeed critical for MN survival builds. As was shown in SMA and control patient-derived induced pluripotent stem cell (iPSC) MNs, even among this cell population, there is variability in SMN levels in individual cells, and those with the lowest levels were the fastest to die even when they were derived from healthy controls. Furthermore, while increasing SMN levels promoted survival of iPSC-derived MNs in the face of stress, the same was not true for cells differentiated into cortical neurons (Rodriguez-Muela et al. 2017). While the special function of SMN in MNs remains a mystery, it is noteworthy that loss of nuclear Gems, a hallmark of SMA, has also been observed in amyotrophic lateral sclerosis (ALS) (Shan et al. 2010; Yamazaki et al. 2012; Tsuiji et al. 2013), raising the possibility that SMN disruption may occur indirectly in this adult-onset MN disease. snRNP assembly/splicing is not the only process of mRNA biogenesis in which SMN has been implicated. Evidence for a role in axonal mRNA transport provides an intriguing link to pathology, as the degeneration of axon terminals at neuromuscular junctions is a disease hallmark (Dombert et al. 2014). MNs have remarkable architecture, with a large cell body and extremely long axons that are very far removed from the nucleus and soma. Axons undergo many changes during development and in response to stimuli and injury that require responses at the level of protein expression, with high spatial and temporal resolution; this is enabled by local protein translation (Jung et al. 2012). SMN has been shown to localize to axons in a complex with several mRNA-binding proteins, including FMRP (Piazzon et al. 2008), HuD (Fallini et al. 2011), and hnRNP R (Dombert et al. 2014). Additionally, compartmentalized siRNA-mediated knockdown of SMN was observed to cause differential effects in the somatodendritic and axonal compartments, with the latter displaying down-regulation of genes involved in axonal growth (Saal et al. 2014). Decreased SMN at axon terminals might lead to loss of important gene regulation at the synapse, and it will be interesting to see whether this impinges on early clinical changes to the motor circuitry in MNs and other cells (Mentis et al. 2011).
Mutations affecting RBPs in cis
ALS
SMA is an excellent example of an RNA processing-centric mechanism, as disease is demonstrably controlled by SMN levels and is thus a straightforward case of LOF. Several points differentiate ALS and SMA: First, while both diseases have some systemic dysfunction, ALS affects lower MNs in the spinal cord in addition to upper MNs in the motor cortex, and symptoms of dysfunction in both regions are necessary to make a diagnosis (Fig. 1; Rowland and Shneider 2001). Second, ALS primarily affects adults, with an average age at onset of 60 yr (Svetoni et al. 2016), although, in some more aggressive forms of disease, patients are afflicted in early adulthood or adolescence (Swinnen and Robberecht 2014). Third, ALS is nearly always fatal, usually after an average disease duration of 3–5 yr (Rowland and Shneider 2001), while patients with less severe forms of SMA have normal life spans (Zerres et al. 1997). In adults, a significant fraction of ALS patients exhibits an array of cognitive, behavioral, and language defects that can result in diagnosis of FTD, thus placing them along an ALS–FTD spectrum (Ling et al. 2013). Finally, ALS is often defined by two dichotomies that attempt to simplify its complex etiology: One (the familial/sporadic division) separates patients on the basis of belonging to a lineage where ALS is recurrent (∼10%) or not (∼90%), while the other delineates patients based on the presence (∼97%) or absence (the remainder) of aggregates of the RBP TDP-43 (Ling et al. 2013).
TDP-43
Aggregations are hallmarks of many but not all neurodegenerative diseases, and aggregated TDP-43 inclusions represent the single greatest unifying factor throughout ALS molecular pathology. TDP-43 proteinopathy is also predominantly associated with frontotemporal lobar dementia (FTLD) subtype Ub+(FTLD-U)/TDP+ (FTLD–TDP), a pathological form of the heterogeneous dementia marked by the loss of cortical (and other) neurons. There is considerable intrafamily and clinical overlap between ALS and FTD, particularly FTLD-TDP, and the two diseases are thought to be different clinical manifestations of a common pathological mechanism (Al-Chalabi et al. 2012). The presence of aggregated TDP-43 in both familial and sporadic ALS with or without accompanying TDP-43 mutations (as described below) represents a central paradox in the effort to characterize the molecular events that are necessary and/or sufficient to cause disease (Blokhuis et al. 2013).
TDP-43 is a ubiquitously expressed critical protein. It is highly conserved across species (Ayala et al. 2005), and genetic knockout leads to embryonic lethality in mice (Kraemer et al. 2010). TDP-43 negatively regulates its own mRNA expression by binding its 3′ untranslated region (UTR), an interaction that requires a C-terminal Gly-rich region (Ayala et al. 2011; Polymenidou et al. 2011). Tight regulation of TDP-43 levels is thus important to the cell; one consequence of elevated TDP-43 expression is increased skipping of exon 9 in the mRNA encoding the cystic fibrosis transmembrane regulator (Buratti et al. 2001), among other similar roles in splicing inhibition. Overexpression of TDP-43 can be toxic in a wide array of cell, yeast, and animal models (Ash et al. 2010; Li et al. 2010; Wils et al. 2010; Armakola et al. 2011), thus making it even harder to disentangle the effects of mutant versions of the protein in any context that alters overall levels of the protein. As this highlights, changes in TDP-43 abundance are not detrimental to MNs alone, again prompting speculation that MNs are more intrinsically sensitive to alterations in RBP concentration (Sephton and Yu 2015).
The identification of TDP-43 as the major component of the hallmark neuronal cytoplasmic inclusions in ALS enabled several important discoveries regarding the nature of pathological TDP-43. These include the protein's nuclear clearance, post-translational cleavage, and hyperphosphorylation (Arai et al. 2006; Neumann et al. 2006). Whereas pure loss of SMN is sufficient to at least explain the outcome of SMA and loss of nuclear TDP-43 has been argued to underlie disease (Vanden Broeck et al. 2014), the aggregation and modifications that TDP-43 undergoes pose a novel gain of function (GOF) that may be toxic, a subject of considerable inquiry. Identification of TDP-43 also led to the discovery of causative mutations in the gene that encodes it, TARDBP, thus strongly supporting a causal role in disease (Fig. 2; Kabashi et al. 2008; Sreedharan et al. 2008). Nonetheless, the uncanny pathological similarities between TARDBP mutants (themselves highly diverse) and patients with mutations in other ALS genes or with sporadic forms of disease make it difficult to determine whether TDP-43 proteinopathy represents the cause or effect of the underlying pathology of these syndromes (Banks et al. 2008).
Figure 2.
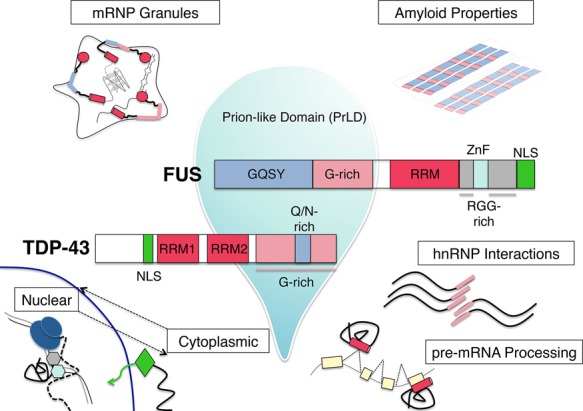
Functional domains of the RBPs TDP-43 and FUS and their cellular roles. Domain architecture of the disease-related proteins TDP-43 and FUS, with emphasis on the prion-like domains (PrLDs) that contribute to liquid–liquid phase separation, represented by a liquid droplet. Various cellular roles of these proteins, including nuclear–cytoplasmic shuttling, pre-mRNA processing, hnRNP interactions, mRNP granule formation, and amyloid-like fibrillization, are diagrammed, with the domains implicated in these processes highlighted.
Currently, there are ∼55 identified TARDBP mutations associated with ALS. These are primarily dominant missense mutations found mostly in familial ALS but also in some sporadic cases (accounting for ∼2% of all ALS) and rare FTD patients (Buratti 2015). The vast majority of these mutations clusters in the C terminus of the protein, which is sometimes referred to as an hnRNP-interacting domain (Ayala et al. 2005; Buratti et al. 2005). The C terminus is also found in an ∼25-kDa proteolytic fragment along with a portion of the second of two RNA recognition motifs (RRMs). Only two rare mutations have been found in the RRMs, both in RRM1 (Buratti 2015). While the mutated amino acids are close to residues important for RNA binding (Lukavsky et al. 2013), they are not suspected to influence RNA recognition (Sun and Chakrabartty 2017). The heavily mutated C-terminal region of the protein is rich in glycine (Gly-rich) and glutamine/asparagine (Q/N) residues (Fig. 2). The entire C-terminal region of ∼160 amino acids is considered low complexity in that it is highly enriched in only four amino acids (glycine, glutamine, serine, and asparagine), which are similar to the residues enriched in yeast prion domains (March et al. 2016). Some mutations are predicted to increase propensity for phosphorylation (Kabashi et al. 2008) and spontaneous aggregation (Johnson et al. 2009), while others are proposed to influence amyloid-like states that are available to TDP-43 via a short stretch of mostly Q/N (Fuentealba et al. 2010; Budini et al. 2012; Mompean et al. 2016). These observations suggest that while changes in splicing and gene expression may be seen in disease, they are not due to differences in RNA recognition. Instead, mutations may indirectly affect the profiles of RNAs that are bound through increased aggregation and/or impaired nuclear import (Buratti 2015) or through disruption of protein interactions, although some common mutations in the hnRNP interaction region have been shown not to influence binding to one interacting protein, hnRNP A2 (Fig. 2; D'Ambrogio et al. 2009).
Proving that ALS/FTD is associated with TDP-43 LOF would require a clear understanding of what TDP-43's major functions are and how the protein behaves in the cell. TDP-43 binds long stretches of UG-rich RNA through its two RRMs (Lukavsky et al. 2013) in homodimeric form (Kuo et al. 2009). These sequences are often enriched near TDP-43-regulated exons (Buratti et al. 2001) but are also found in deep intronic positions downstream from skipped exons (Polymenidou et al. 2011; Tollervey et al. 2011a). The UG-binding motif has been confirmed in vivo by CLIP in both cell culture and human post-mortem brains (Tollervey et al. 2011a; Xiao et al. 2011). Interestingly, despite the axiomatic cytoplasmic aggregation in the FTLD–TDP patients studied, CLIP from both nuclear and cytoplasmic fractions revealed that control and patient brains did not demonstrate much cytoplasmic RNA binding, but instead both were enriched for intronic regions and noncoding transcripts such as NEAT1 and MALAT1. Furthermore, the finding that bound RNAs are enriched for multiple dispersed TDP-43-binding motifs suggests possible cooperative binding of multiple molecules to UG repeats that are spread out over 100-nucleotide distances (Tollervey et al. 2011a).
Loss of TDP-43 function due to nuclear clearance can lead to particular splicing signatures of disease (Polymenidou et al. 2011; Xiao et al. 2011; Ling et al. 2015) or to defective transport of distally targeted mRNAs (Ishiguro et al. 2016). Intriguingly, it has been postulated that an important function of TDP-43 is the repression of cryptic exons (Fig. 1), and increased inclusion of such exons was found in post-mortem ALS/FTD individuals (Ling et al. 2015) to the potential detriment of cell survival (Tan et al. 2016). While this result presents an important observable metric of TDP-43 LOF in disease, such cryptic exons are not exclusively regulated by TDP-43 and are bound by a number of other RBPs whose concentrations may vary in patients (Humphrey et al. 2017).
The patterns of cleavage that TDP-43 undergoes are an interesting feature of disease. However, its contribution has been called into question due to the minor prevalence in the spinal cord (Igaz et al. 2008). One proposed explanation for the toxicity of the C-terminal fragments (Zhang et al. 2009) is that their lack of a nuclear localization signal (NLS) prevents them from entering the nucleus, thus leading to their buildup and aggregation in the cytoplasm; indeed, an NLS-tagged 25-kDa fragment is less toxic and aggregation-prone than the fragment without an NLS (Kitamura et al. 2016). Furthermore, the ability to bind RNA may underlie the organization of TDP-43 oligomers, and loss of RNA-binding capability in truncated versions might promote aggregation (Kitamura et al. 2017). Once formed in the cytosol, these aggregates can then trap full-length TDP-43, disrupting its normal nucleo–cytoplasmic shuttling and leading to nuclear depletion (Winton et al. 2008). However, the existence of these fragments in the brains of patients with both mutant and wild-type TDP-43 alike suggests that cleavage is not specifically driven by mutations (Li et al. 2015).
FUS
The discovery of TDP-43 pathology and mutations established RBP dysfunction as a major theme in ALS/FTD. The knowledge that RBPs can harbor ALS mutations led to the concerted sequencing efforts that identified mutations in another RBP: TLS/FUS (or FUS) (Fig. 2; Kwiatkowski et al. 2009; Vance et al. 2009). FUS was originally identified as the site of the most common translocation event leading to liposarcoma (hence, the name fused in liposarcoma/translocated in liposarcoma) (Crozat et al. 1993; Rabbitts et al. 1993). FUS is a member of the TET/FET family of proteins, which includes two other similar proteins, EWS and TAF-15; is ubiquitously expressed; and has many proposed cellular functions, ranging from transcription to RNA processing to DNA damage repair (Tan and Manley 2009; Wang et al. 2013). The protein's ability to shuttle between the nucleus and cytoplasm might have important consequences for synaptic mRNA transport (Fujii et al. 2005; Masuda et al. 2015). Structurally, FUS consists of an N-terminal domain comprised of a QGSY region and the first of three RGG repeat regions, and an RRM and zinc finger motif account for its ability to bind nucleic acids. An NLS, which harbors a majority of characterized ALS mutations (see below), is located at the extreme C terminus (Fig. 2).
Like TDP-43, FUS has been characterized as both an RNA- and DNA-binding protein. With respect to RNA, while an affinity for UG-rich RNA sequences has been observed (Lerga et al. 2001; Lagier-Tourenne et al. 2012), it has also been argued that FUS does not have a specific binding recognition motif but rather binds RNAs promiscuously, with greater binding occupancy on highly expressed RNAs (Wang et al. 2015). The lack of a consensus FUS-binding motif was echoed by CLIP experiments, which identified a predisposition for binding to stem–loop structures (Hoell et al. 2011). Like TDP-43, FUS has been found to bind to long introns, with potentially important regulatory implications (Lagier-Tourenne et al. 2012; Rogelj et al. 2012). The brain is a site of increased transcriptional activity of genes with long introns, as expression of longer genes is overrepresented in neural tissue (Gabel et al. 2015). A cotranscriptional requirement of FUS and TDP-43 for processing long introns may therefore underlie a critical function of these proteins that is specific to the brain (Zylka et al. 2015).
FUS has also been implicated in DNA binding. Early studies suggested an ability to bind ssDNA, perhaps reflecting a function in DNA repair and/or transcription (Tan and Manley 2009). DNA FUS-responsive elements were later found enriched within promoter regions of genes shown to respond transcriptionally to FUS levels (Tan et al. 2012). One such gene, MECP2, biologically interesting due to its relationship to the neuroregressive disease Rett syndrome, was found to be misspliced in cells transfected with mutant, but not wild-type, FUS (Coady and Manley 2015). Interestingly, the splice isoform of MECP2 promoted by mutant FUS was found to have increased RNA stability yet express lower protein levels. The mRNA was found to be present with mutant FUS in cytoplasmic aggregates by both biochemical analysis and FISH, suggesting that the transcript is sequestered. These results suggest that mutant FUS aggregation can affect the stability and translation of mRNA targets through direct sequestration of RNAs as well as pre-mRNA splicing through sequestration of splicing factors (Coady and Manley 2015).
FUS aggregates are detected in ALS, although less frequently than TDP-43. Of the ∼3% of ALS cases that do not have TDP-43 aggregation, FUS is analogously aggregated in ∼50% of these (Mackenzie et al. 2007). However, unlike with TDP-43, FUS pathology never presents without FUS mutations, >50 of which have been described (Nolan et al. 2016). Whereas disease-causing TDP-43 mutations are clustered in the C-terminal G-rich and Q/N regions, FUS mutations occur throughout the protein, although the most common and well-described are in the C-terminal NLS (Shang and Huang 2016; Svetoni et al. 2016). Such mutations have been proposed to influence the cytoplasmic aggregation and nuclear clearance of the protein. These pathological features also occur in the ∼10% of FTD patients with an atypical FTLD-U pathological subtype whose inclusions are TDP-43-negative (Neumann et al. 2009). These patients do not carry mutations in FUS, reminiscent of ALS–TDP and FTLD–TDP cases where wild-type TDP-43 is mislocalized and aggregated (Mackenzie et al. 2010).
As with TDP-43, there is debate surrounding the relative contributions of GOF and LOF mechanisms with respect to how both FUS mutations and aggregates impact disease. One characteristic of FUS-ALS is that the gene harbors several mutations that lead to especially aggressive juvenile-onset forms of disease, such as the P525L mutation (Sharma et al. 2016), a deletion in exon 14 (Belzil et al. 2012), and the recently described Y526C mutation (Corcia et al. 2017). An important question is whether these juvenile mutations affect the same functions and biochemical properties of FUS as mutations that lead to more classical ALS, such as R521C, but to a more severe degree. In support of this, it was found that mice expressing P525L mutant FUS displayed greater aggregation and higher cytoplasmic to nuclear FUS ratio than mice expressing the less severe R521C FUS (Sharma et al. 2016). FUS cytoplasmic aggregation has been shown to influence the profile of the RNAs with which it is associated, as mutant FUS binds less to intronic nuclear RNA than wild type does and makes many more associations with 3′ UTRs (Hoell et al. 2011). In a recently described mouse model, FUS lacking an NLS exhibited mislocalization to the cytoplasm that led to changes in gene expression and splicing that largely mirrored those caused by FUS ablation. However, mislocalization also led to up-regulation of genes involved in mRNA translation and to a subset of unique splicing changes that may reflect disruptions to other RBPs as a consequence of the cytoplasmic FUS aggregation (Scekic-Zahirovic et al. 2016). It was also shown that both depletion and cytoplasmic mislocalization of FUS led to perinatal lethality due to respiratory insufficiency; however, mislocalization alone further resulted in perinatal MN death (Scekic-Zahirovic et al. 2016). In another mouse model, widespread postnatal elimination of FUS from the central nervous system as well as MN-specific elimination resulted in mice with normal postnatal survival without MN loss or denervation compared with control animals (Sharma et al. 2016). These results argue in favor of a mechanism in which FUS is pathogenic through a gain of mutant and/or cytoplasmic function.
RBPs and membraneless organelles
While LOF contributions to pathogenesis cannot be ruled out, the expanding evidence for GOF has prompted more biochemically focused research into the structure and interactions of mutant and wild-type FUS. This is due in part to the sequencing of large cohorts of patients and controls that has led to identification of (presumed) ALS-causing mutations in so-called intrinsically disordered regions (IDRs) (Ticozzi et al. 2009; Corrado et al. 2010). Due to the rarity of these mutations, in-depth post-mortem analyses and identification of FUS pathology have been lacking. Additionally, the presence of these rare variants in sporadic patients makes the case for causality weaker than if they were seen as driving a recurrent phenotype in families. Despite these reasons to retain skepticism, mutations in the FUS prion-like and Gly-rich regions offer a new opportunity for unification with the rapidly expanding topic of liquid–liquid phase separation (LLPS). The idea that mutant versions of TDP-43 and FUS lead to alterations of the biophysical properties of their IDRs has been gaining traction as part of a larger effort to characterize intracellular phase transitions undergone by membraneless organelles such as stress granules (SGs) and P bodies (Brangwynne et al. 2015).
The formation of liquid droplets that partition as a separate phase from the surrounding medium are of interest because they are mediated by low-complexity domains (LCDs), also known as IDRs. These include the QGSY-rich and RGG-rich domains of FUS and the Gly-rich and Q/N-rich domains of TDP-43, which have been shown to be important for SG formation (Fig. 2; Liu-Yesucevitz et al. 2010; Bentmann et al. 2012). The natural conformational heterogeneity of these domains permits them to make diverse combinations of charge–charge, dipole, and π-stacking interactions along the polypeptide backbone over short and long ranges. This predisposition to make self-interactions rather than solvent interactions has the potential to drive compaction and thus phase separation. Consequently, as concentrations of critical protein components rise, so does formation of these proteinaceous droplets, enabling spatial separation of important biochemical processes such as the partitioning of nucleic acids (Nott et al. 2016), which can also help drive formation of these species (Brangwynne et al. 2015; Shorter 2016).
Several types of membraneless granules may be relevant to RBPs and ALS/FTD. For example, TDP-43 (Liu-Yesucevitz et al. 2010; McDonald et al. 2011) and FUS (Baron et al. 2013) have both been found in SGs. These structures form in response to cellular stress as a means to temporarily halt translation of mRNAs not needed to respond to the situation at hand and thus can be viewed as temporary RNA storage sites. P bodies, on the other hand, while sharing some properties with SGs, are sites of mRNA degradation (Decker and Parker 2012; Li et al. 2013). RNA transport granules are another important class of membrane-less organelles with specific neuronal relevance in localized mRNA expression (Kiebler and Bassell 2006). Their ability to fuse into droplet states has become of great interest for disease because the properties driving this protective aggregation, which can involve phase transitions of mutant FUS and TDP-43, can also lead to amyloid-like states (Fang et al. 2014; Murakami et al. 2015). This can be imagined as a transition from liquid droplets to hydrogels to irreversible amyloid fibers, giving rise to a gradient in rigidity (Fig. 2).
The ability of SGs to form rapidly and reversibly is an important aspect of their role in maintaining RNA homeostasis. Important work on the molecular events that initiate SG formation in yeast implicated a prion-like Q/N-rich region of the protein Lsm4p in conferring the ability to aggregate reversibly (Decker et al. 2007). Despite the emphasis on the unstructured nature of IDRs, it has been proposed that transient, partially populated folded conformations, facilitated by particular amino acid stretches, may be disrupted by mutations, for instance, in TDP-43 (Conicella et al. 2016). TDP-43 fibrilization is thought to seed aggregates of itself (Furukawa et al. 2011) and cross-seed aggregations of other proteins (Fang et al. 2014). Such fibrillization by LCD-containing proteins is not necessary for phase separation and formation of granules but has been shown to increase in these conditions (Molliex et al. 2015). The ability to study the properties driving hydrogel formation and phase separation was augmented by the discovery that these molecules can be selectively precipitated using compounds such as biotinylated isoxazole (Han et al. 2012; Kato et al. 2012). Through application to extracts from different cells and tissues, it was shown that RNA-based granules contain many RBPs such as translation factors, ribosomal proteins, helicases, and 3′ UTR-binding factors but that there are core RBPs, such as SRSF5, PTBP1, Pur-α, and hnRNPs, that are essential to these granules, as treatment with RNases revealed that the LCDs themselves, and not the RNAs, provide the basis for aggregation (Han et al. 2012). However, the coexistence of RBDs and LCDs within the same proteins suggests that coalescence via association with the same RNAs serves to drive local concentrations to the point of phase transitions (Molliex et al. 2015; Zhang et al. 2015). Intriguingly, one of these essential granule proteins that exhibited greater enrichment after RNase treatment is Matrin-3 (MATR3) (Han et al. 2012), which is infrequently mutated in familial ALS (Fig. 1; Johnson et al. 2014). Matrin-3 is a DNA- and RNA-binding protein with a role in promoting mRNA stability (Salton et al. 2011) and has also been suggested to associate with TDP-43 (Ling et al. 2010), although the interaction was shown to be RNA-dependent (Johnson et al. 2014). While the precise nature of Matrin-3 in cellular granules with other disease-related proteins in unclear, such findings reinforce the convergence of different ALS genes in the same physical processes.
The focus on LCDs in RNA granules has provided a number of additional insights, which are not discussed in this review. These studies helped foster the view that disease is somehow triggered by injury, persistent stress, or activity-dependent demands from which cells with mutant TDP-43 or FUS are slow or unable to recover (Wolozin and Apicco 2015). Differential exposure to environmental toxins, damage, and stressors can therefore be seen as important modifiers of disease and may explain the incomplete penetrance of some ALS-causing variants (Harms et al. 2015). This line of research has also underscored the fact that aggregation is an intrinsic property of LCD-containing proteins but also a liability when taken to the extremes of concentration (Guo and Shorter 2015). Along these lines, several studies have investigated how mutant forms of FUS (Patel et al. 2015) and TDP-43 (Liu-Yesucevitz et al. 2014) as well as more rarely mutated proteins such as hnRNP A1/2 (Kim et al. 2013; Shorter and Taylor 2013) and EWS (Couthouis et al. 2012) perturb the dynamics of aggregate formation, dissipation, and strength. While great advances in our understanding of the different states accessible to RBPs under different conditions of crowding have been made using cell-free systems (Han et al. 2012; Kato et al. 2012) and structural biology (Mompean et al. 2015; Conicella et al. 2016), it will be important to see how these insights translate into in vivo systems and therapeutic targets.
Mutations affecting RBPs in trans
Microsatellite expansions in protein-coding regions
RNA is not merely a passive carrier of genetic information acted on by proteins but an important actor as well. RBPs have evolved to deal with the complexities associated with RNA, particularly its propensity to fold, sometimes nonproductively (Herschlag 1995), and its high demand for precise localization relevant to gene expression and catalysis. Just as it seems a combination of mutant and relocalized RBPs can cause aggregation and effect changes in RNA and gene expression, excess repetitive RNAs or those with highly stable structures (Mooers et al. 2005; Bernat and Disney 2015; Ciesiolka et al. 2017) can sequester RBPs in aggregates, leading to deficiencies in their normal functions (Fig. 1; Jiang et al. 2004; Morriss and Cooper 2017). The convergence of multiple mutations that cause diseases of the neuraxis on mechanisms of RNA and RBP homeostasis suggest that disruption of these pathways represents the most parsimonious candidate for a central mechanism in these diseases (La Spada and Taylor 2010). Furthermore, the fact that all known nucleotide expansion disorders take their tolls specifically via neurodegenerative and neuromuscular disorders (Budworth and McMurray 2013) reinforces the argument that the regulation of RNA metabolism is held to stricter standards in the nervous system.
The first disease-linked nucleotide microsatellite expansions were discovered >25 years ago. These were in fragile X syndrome of mental retardation (FXS) and X-linked spinal and bulbar muscular atrophy (SBMA) (La Spada and Taylor 2010). Both diseases reflect X-linked triplet repeats: poly-CGG in the FMR1 gene (Verkerk et al. 1991) and poly-CAG in the AR gene (La Spada et al. 1991) in FXS and SBMA, respectively (Fig. 1). These repeats were initially categorized as encoding polyamino acid tracts leading to proteins with a higher tendency to misfold and aggregate (Ashley et al. 1993; Sisodia 1998; Stenoien et al. 1999). This remains the prevailing notion in one of the most common inherited diseases and best-known polyglutamine (polyQ) CAG microsatellite disorders: Huntington's disease (HD) (Ross and Tabrizi 2011; Arrasate and Finkbeiner 2012). PolyQ aggregation is a recurring theme in neurodegeneratve diseases, implicated in possible phase separation of RNA granules (Zhang et al. 2015) and cross-seeding aggregates of proteins with Q/N regions (Furukawa et al. 2009; Fuentealba et al. 2010). However, whether aggregation of expanded polyQ proteins is entirely responsible for the pathogenesis of these disorders has been called into question (Klement et al. 1998; Saudou et al. 1998), and the observation that mutant Huntington aggregates are most prevalent in spared neuronal populations (Kuemmerle et al. 1999) has helped foster the view that aggregates may be protective in these and other diseases (Ross and Poirier 2005).
The polyQ paradigm has been challenged by recent work on the toxicity of the repeat-containing RNA itself (Nalavade et al. 2013; Marti 2016). For example, it has been proposed that the CAG repeat RNA forms stable hairpin structures with an endogenous neighboring CCG tract and sequesters the RBP Muscleblind-like 1 (MBNL1) in intranuclear aggregates (de Mezer et al. 2011), leading to altered splicing of MBNL1 targets (Mykowska et al. 2011). The hairpin structures are additionally thought to induce cleavage by the RNA silencing complex endonuclease DICER, generating short RNAs that lead to silencing of downstream targets (Fig. 1; Krol et al. 2007). In further support of RNA toxicity, a disorder closely resembling HD, HD-like 2, is caused by a CTG expansion in the junctophilin-3 gene (Holmes et al. 2001) that has been shown to give rise to RNA foci that also sequester MBNL1 (Rudnicki et al. 2007), reminiscent of other diseases (see also below). However, it has also been shown in transgenic mice that polyQ pathology arises from antisense CAG transcripts (Wilburn et al. 2011). Thus, the combined toxicities of RNA and polyQ may have a synergistic effect in disrupting RBP function through different modes of sequestration and by placing stress on the cell's autophagic mechanisms (Cortes and La Spada 2014).
Several observations have contributed to the view that expanded polypeptides cannot be the only mechanism behind triplet repeat diseases. For example, with regard to FXS, the CGG repeat in FMR1 is outside the ORF, in the 5′ UTR (Ashley et al. 1993), suggesting that the expansion might lead to decreased gene expression (Bell et al. 1991). However, recent advances in understanding repeat-associated non-AUG (RAN) translation have reopened the debate about whether expansions in noncoding regions can be translated to generate short repetitive polypeptides (Cleary and Ranum 2014; Sellier et al. 2017). The ubiquity of this mechanism across a multitude of triplet, quadruplet, and higher-order nucleotide expansions suggests a dual role of RNA as the toxic molecule itself and the intermediary to one, with important inputs from specific RBPs factoring into both.
Microsatellite expansions and RBP sequestration
Perhaps the most well-established RNA GOF mechanism is the one described for myotonic dystrophy type 1 (DM1) and DM2 (Udd and Krahe 2012). DM1 and DM2 are dominantly inherited disorders affecting multiple organs, characterized by muscle weakness, myotonia, cataracts, cardiac arrhythmias, and neuropsychological symptoms (Thornton et al. 2017), and are the most common forms of adult muscular dystrophy (Caillet-Boudin et al. 2014). The distinction between DM1 and DM2 refers to their causative genes. The mutation leading to DM1 is a CTG expansion in the 3′ noncoding region of the gene encoding the dystrophia myotonica protein kinase (DMPK) (Brook et al. 1992), while DM2 refers to a phenotypically similar disorder due to a CCTG expansion in an intervening sequence of the gene encoding CCHC-type zinc finger nucleic acid-binding protein (CNBP) (Fig. 1; Liquori et al. 2001). The two encoded proteins share no functional overlap, arguing against the idea that DM pathogenesis can be explained by loss of protein function (La Spada and Taylor 2010). Instead, RNA transcribed from these genes forms intranuclear foci that overlap with the proteins MBNL1, MBNL2, MBNL3 (Jiang et al. 2004), and CUG-binding protein 1 (CUGBP1) (Timchenko et al. 2001). In vitro, purified MBNL1 binds to extended hairpin structures consisting of G–C base pairs and U–U mismatches (Michalowski et al. 1999) with high affinity that is similar for both short (CUG4) and long (CUG90) repeat RNAs and higher for short CCUG repeat RNAs (Warf and Berglund 2007). However, it has been argued that CUGBP1 does not bind the dsRNA hairpins that predominate (Michalowski et al. 1999), and SELEX (systematic evolution of ligands by exponential enrichment) experiments identified enrichment of the trinucleotide UGU, not CUG (Marquis et al. 2006), raising some doubts about the in vivo sequestration of CUGBP1. That the observed foci and biochemical evidence of binding reflect functional sequestration of these proteins has been demonstrated in patients and cell models (Kanadia et al. 2003). Examples of missplicing of genes targeted by MBNL and CUGBP include events that directly correlate to disease phenotypes (Philips et al. 1998; Kuyumcu-Martinez and Cooper 2006). Furthermore, knockout of mouse Mbnl1 results in a phenotype that recapitulates several, but not all, aspects of disease (Kanadia et al. 2003), reflecting the fact that human disease is the result of compound splicing factor disruption (Lee et al. 2013).
The potential for RAN translation in DM1/2 has also been investigated. In DM1, this has been demonstrated in cell culture when repeat constructs lacking an ATG start codon are overexpressed, presumably requiring the hairpin-forming propensity of the CUG repeats (Zu et al. 2011). While low levels of polyQ aggregates were observed in tissues from DM1 transgenic mice and human DM1 patient myoblasts and skeletal muscle, whether these species contribute to disease has not been further pursued. No such potential for homopolymeric products exists in DM2, and although DM1 is sometimes more clinically severe than DM2 (Zu et al. 2011), the fact that both repeat expansions cause such similar phenotypes suggests that the mechanism is primarily RNA-driven.
An important issue in repeat expansion disorders is whether the number of expanded repeats correlates with any observable metrics of disease, such as age of onset, disease duration, or molecular features such as splicing dysregulation. In DM, expansion length correlates roughly with disease severity (Brook et al. 1992), although there is no apparent relationship between expansion length and the severity of splicing defects (Nakamori et al. 2013). However, confidence in such relationships is complicated by somatic instability of repeat length (Savic Pavicevic et al. 2013). Establishing the significance of such correlations will depend on the development of simpler, more accurate methods for measuring repeat length (Yum et al. 2017). Along with these endeavors, ongoing DM research has a broad curative focus, from developing DM gene silencing therapies (Thornton et al. 2017) to understanding the structure and role of extended hairpins and loops formed by the repeat sequences (Dere et al. 2004; Yuan et al. 2007; deLorimier et al. 2017) and how their modular structure may be therapeutically targeted (Garcia-Lopez et al. 2011; Childs-Disney et al. 2012, 2014).
RNA-centric mechanisms in C9ORF72 ALS–FTD
The advances made in DM have provided an important paradigm for subsequent repeat expansion discoveries. Most notable is the GGGGCC hexanucleotide expansion in the previously unstudied gene C9ORF72 (C9), which is by far the most common known cause of ALS and FTD and the best proof for the continuous nature of the two disorders (DeJesus-Hernandez et al. 2011; Renton et al. 2011). A tremendous amount of effort has gone into defining several nonmutually exclusive disease mechanisms suggested by this mutation (Haeusler et al. 2016; Taylor et al. 2016), yet the relative contributions of each mutation are unclear in vivo. C9 ALS/FTD is similar to most other forms of ALS in that it is marked by TDP-43 pathology but also has a distinct class of Ub+/p62+ cytoplasmic inclusions containing dipeptide repeat (DPR) proteins made by RAN translation (Kearse and Todd 2014; Schipper et al. 2016). Why C9 causes some people to get pure ALS or FTD or a mixture of both is a mystery. Further complicating matters, C9 has also been associated with Parkinson's disease (PD) (Wilke et al. 2016) and schizophrenia (Galimberti et al. 2014), and neuronal loss and gliosis have been described quite broadly throughout the brain and spinal cord in C9 ALS (Cooper-Knock et al. 2014).
DPR pathology has been a major focus of C9 ALS–FTD research. For instance, it presents an intriguing link to the low-complexity sequences and hydrogel properties involved in RNA granule formation. One possibility is that DPRs may actually disrupt cellular functions by interacting with proteins involved in membraneless organelles and cytoskeletal intermediate filaments (Lin et al. 2016). DPRs may also provide a useful diagnostic biomarker (Lehmer et al. 2017), and their demonstrated ability to spread between cells (Westergard et al. 2016) offers a simple explanation for the observation that ALS tends to spread from a region of onset to other parts of the nervous system (Brettschneider et al. 2015). However, an understanding of how expanded RNA repeats make DPRs is lacking (Freibaum and Taylor 2017). Furthermore, antibodies against poly-DPRs derived from multiple frames of the GGGGCC expansion recognize short epitopes that could be present in unrelated proteins such that detection on filter-bound/membrane assays may be misleading. Meanwhile, more informative visualization of foci in post-mortem human tissue is fraught with preservation, fixation, and background technicalities. While toxicity of DPRs has been shown in model organisms and cell culture (Wen et al. 2014; Lopez-Gonzalez et al. 2016), this could reflect nonphysiological effects resulting from overexpression. Understanding how DPRs are made (Green et al. 2016) and how abundant they truly are in degenerating regions (Gomez-Deza et al. 2015) will be important in order to gauge how critical they are to the pathology of the ALS/FTD spectrum.
Despite these caveats, DPR pathology and its mysterious origins may be linked to the structures formed by the RNA repeats and the proteins that bind them. The C9 repeats are notable for their ability to form highly stable G-quadruplex (G-Q) structures, which, interestingly, have been implicated in translational control (Bolduc et al. 2016; Song et al. 2016) and suggested to serve as internal ribosomal entry sites (Morris et al. 2010). Expanded, long arrays of G-Q-forming sequences may give rise to complex mixtures of G-Q structures and linear G-tract RNA, which have been reported to bind RBPs such as hnRNP H (Conlon et al. 2016), nucleolin (Haeusler et al. 2014), and other known G-Q binders (Brazda et al. 2014) as well as many other proteins (Fig. 1; Haeusler et al. 2016). The ability of G-Qs to self-interact (Petraccone 2013) combined with the potential for self-interaction among LCDs of proteins that experience high local concentration as a result of RNA binding, such as the Gly-rich domains of TDP-43 and other hnRNPs, may drive formation of compact RBP/RNA aggregates.
Numerous proteins have been suggested to bind to the C9 repeat RNA. However, functional sequestration of only one, hnRNP H, has been documented in post-mortem C9 patient brains, where it was detected in intranuclear aggregates, and a large fraction was found to be insoluble in extracts. Importantly, this correlated with missplicing of many known hnRNP H target exons (Conlon et al. 2016). hnRNP H levels affect splicing of hundreds of targets (Huelga et al. 2012), including many encoding proteins involved in splicing—among them TDP-43 and FUS (Uren et al. 2016). Thus, hnRNP H deficiency may result in splicing errors that extend beyond exons with which hnRNP H interacts physically. hnRNP H regulates splicing of several transcripts that encode proteins important to the proper functioning of neuromuscular junctions, which are vulnerable to denervation in ALS (Ohno et al. 2017). One of these transcripts, ACHE, which encodes acetylcholinesterase, contains an important hnRNP H-regulated exon (Nazim et al. 2017) that is misspliced in C9 ALS in a manner consistent with hnRNP H deficiency (EG Conlon and JL Manley, unpubl.). However, whether missplicing can account for the complex traits associated with C9 ALS/FTD is not known. Beyond splicing, hnRNP H deficiency may affect polyadenylation choices in C9 brains (Bagga et al. 1998; Prudencio et al. 2015) as well as increase the overall global level of RNA G-Qs. This latter possibility reflects a recently proposed role of hnRNP H in keeping RNA G-Qs unwound (Guo and Bartel 2016), and hnRNP H deficiency could thus reinforce C9-derived G-Q-based aggregates and promote folding of other genomic G-Qs.
Nucleotide expansions occur in multiple neurodegenerative diseases
G-Qs have been linked to additional expansion-related neurodegenerative diseases; for example, FXS (Fry and Loeb 1994; Simone et al. 2015). However, the significance of these G-Qs is unclear, as the CGG repeats in FMR1 are predicted to form a relatively unstable G-Q structure due to the weakness of two tetrad stack G-Qs compared with three or four (Mullen et al. 2012). CGG expansion in FMR1 causes two distinct disorders depending on the length of the expansion: FXS in the case of long (>230-repeat) expansions and fragile X tremor ataxia syndrome (FXTAS) in so-called premutation carriers (60–230 repeats) (Jin and Warren 2000). In the former case, the long 5′ UTR expansion forms a CpG island, resulting in transcriptional silencing and therefore reduced levels of FMRP. Supporting the significance of the G-Qs, FMRP is capable of binding transcripts containing them (Darnell et al. 2001; Vasilyev et al. 2015), which can facilitate their translocation, in some cases, to synapses (Zhang et al. 2014; Stefanovic et al. 2015). In FXTAS patients, FMR1 is not completely silenced, and the expansion is actively transcribed into a repeat-containing RNA that has been reported to bind multiple RBPs, such as hnRNP A2/B1, MBNL1 (Iwahashi et al. 2006), Sam68 (Sellier et al. 2010), Drosha, DGCR8 (Sellier et al. 2013), and more (Galloway and Nelson 2009). Evidence for the functional significance of this binding to human disease is somewhat limited; however, overexpression of hnRNP A2/B1 and CUGBP1 (Sofola et al. 2007) or Pur-α (Jin et al. 2007) in Drosophila models of FXTAS can suppress the neurodegenerative phenotypes. Only one protein, Sam68, has been shown to be functionally sequestered in humans, demonstrated by altered splicing of Sam68-responsive transcripts in the brains of FXTAS patients (Sellier et al. 2010).
Whether the bifurcated nature of FXS and FXTAS resulting from different lengths of the same expansion applies to other disorders, such as C9 ALS/FTD, is not known. The ability to directly measure expansion lengths, especially G-C rich expansions, with confidence based on sequencing data (Dolzhenko et al. 2016) is an important technological challenge. When solved, such information will hopefully prove useful in determining whether expansion length correlates with clinical presentation and/or molecular events such as splicing defects.
Repeat expansions are highly represented among spinocerebellar ataxias (SCAs). SCAs are a heterogeneous class of disorders marked by cerebellar atrophy, incoordination of gait, and loss of balance, among other noncerebellar symptoms (Sun et al. 2016). Many of these are caused by trinucleotide repeats, particularly CAG, which occurs in SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA12, and SCA17 (La Spada and Taylor 2010). The gene responsible for SCA2, ATXN2, contains a CAG polyQ expansion in its 5′ coding region that is between 36 and 52 repeats in disease, making it one of the shortest known CAG expansions (Pulst et al. 1996). Interestingly, intermediate lengths of CAG (27–33 repeats) in ATXN2 have been shown to increase the risk of ALS (Elden et al. 2010; Sproviero et al. 2017). The function of Ataxin-2 is still unclear, although it has been shown to have roles in mRNA stability (Yokoshi et al. 2014), P bodies, and stress granules (Nonhoff et al. 2007) and can associate with TDP-43 in an RNA-dependent manner (Elden et al. 2010). Decreased ATXN2 expression can suppress TDP-43 toxicity in yeast, flies (Elden et al. 2010), and mice (Becker et al. 2017), suggesting that ASOs targeting ATXN2 transcripts may be a viable therapeutic strategy in human ALS cases with TDP-43 pathology (Becker et al. 2017).
Higher-order repeats are seen in several genes, such as an ATTCT expansion in the ATXN10 gene responsible for SCA10 (Handa et al. 2005), a TGGAA expansion in SCA31 (Sato et al. 2009), and a GGCCTG expansion in NOP56, responsible for SCA36 (Kobayashi et al. 2011). It is notable that the UGGGCC repeats of the SCA36 expansion are similar to the GGGGCC repeats from C9 and thus would also presumably form G-Q structures and associate with a similar set of RBPs, although this has not been investigated. SCA36 presents as a “pure” SCA marked by ataxia and cerebellar degeneration as well as progressive loss of upper and lower MNs, which separates it from other SCAs that are predominantly cerebellar and suggests that SCA36 is a disorder at the intersection of SCA and MN disease (Ikeda et al. 2012). Despite loss of MNs from the anterior horn of the spinal cord, TDP-43 inclusions were not detected in surviving neurons of affected individuals, further differentiating this disorder from ALS (Ikeda et al. 2012).
Comparison of C9 ALS/FTD and SCA36 at the molecular level might yield interesting insights into what causes these two disorders to have a mixture of clinical overlaps and differences. At the most basic level, the two repeat expansions have different underlying repeat sequences. The difference of one G → U per repeat might have several consequences related to the proteins that bind the expanded RNA and their binding affinities, resulting in different degrees of sequestration. Furthermore, repetitive (G)4(N)2 tracts are predicted to give rise to more stable G-Q structures than (G)3(N)3 tracts due to the greater number of stacked planar quartets and shorter loops, which may influence the permanence of RNA foci and their aggregation properties. The expansions are also in different genes, and reduced C9 or NOP56 protein levels may influence the ALS/FTD and SCA36 phenotypes, respectively. Last, while the potential RAN translation products of the NOP56 expansion have not been studied (Arias et al. 2014), one frame each of the sense and antisense transcripts can hypothetically give rise to a Gly–Pro DPRs, which are commonly found in C9 carriers, and one frame of the antisense can give rise to Arg–Pro DPRs; both are thought to be toxic (Mizielinska et al. 2014). Such connections, while speculative, may inform the hunt for novel repeat expansions that are not as clearly and extensively linked to disease as C9 and NOP56. It is the hope that predictive biomarkers, such as patterns of RBP sequestration and splicing as well as DPRs, might provide evidence for unknown (potentially G-Q-forming) expansions.
Concluding remarks: RBP dosage as the central mechanism in neurodegenerative diseases
The examples discussed in this review reflect the increasingly compelling view that neurodegeneration can arise from alterations in RBP dosage. If we are to draw any aggregate conclusions from the model of SMA, the pleiotropic nature of ALS/FTD, and the contested mechanisms of TDP-43, FUS, and C9ORF72 mutations, it is that neurons and perhaps especially MNs simply cannot tolerate diminished RBP function. It is hard to imagine a way to reconcile TDP-43 aggregation in sporadic ALS and FTD as well as FUS aggregation in sporadic FTD without acknowledging their centrality to a common disease mechanism. We also note that a significant class of ALS without TDP-43 pathology is caused by mutations in superoxide dismutase 1 (SOD1), and the mechanism of disease is not believed to involve RBPs or their aggregation (Ling et al. 2013). Our reductionist treatment of these complex heterogeneous diseases is not intended to minimize the contributions of other mechanisms but rather to consolidate the ideas pertaining to the broad topic of RBP aggregation. Perhaps in thinking of this disease in such terms, we will become poised to draw new parallels between the aggregates that we discussed here and the SOD1 aggregates that characterize SOD1 mutation-affected patients (Bruijn et al. 1998).
It is our hope that as RBPs emerge as causative factors in more neurodegenerative and neurological disorders, such as TIA-1 in Alzheimer's (Vanderweyde et al. 2012), the potential for therapeutic intervention will be positively impacted by the collaborative efforts of neurologists, pathologists, and RNA biologists. From the vantage point of basic research, we may find that long-sought answers to basic questions about RBP dosage in alternative splicing, polyadenylation, and mRNA transport will come from the study of these unfortunate ailments. Through attempts to answer the question of how gene expression specifies cellular (neuronal) identity, we may uncover mechanisms that yield important benefits to those afflicted by these devastating diseases.
Acknowledgments
Work from the authors’ laboratory was supported by National Institutes of Health grant R35 GM118136 to J.L.M.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.304055.117.
References
- Al-Chalabi A, Jones A, Troakes C, King A, Al-Sarraj S, van den Berg LH. 2012. The genetics and neuropathology of amyotrophic lateral sclerosis. Acta Neuropathol 124: 339–352. [DOI] [PubMed] [Google Scholar]
- Andavan GS, Lemmens-Gruber R. 2011. Voltage-gated sodium channels: mutations, channelopathies and targets. Curr Med Chem 18: 377–397. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. 2006. TDP-43 is a component of ubiquitin-positive τ-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun 351: 602–611. [DOI] [PubMed] [Google Scholar]
- Arias M, Quintans B, Garcia-Murias M, Sobrido MJ. 2014. Spinocerebellar ataxia type 36. In GeneReviews (ed. Pagon RA, et al. ), https://www.ncbi.nlm.nih.gov/books/NBK231880 University of Washington, Seattle, WA. [Google Scholar]
- Armakola M, Hart MP, Gitler AD. 2011. TDP-43 toxicity in yeast. Methods 53: 238–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrasate M, Finkbeiner S. 2012. Protein aggregates in Huntington's disease. Exp Neurol 238: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ash PE, Zhang YJ, Roberts CM, Saldi T, Hutter H, Buratti E, Petrucelli L, Link CD. 2010. Neurotoxic effects of TDP-43 overexpression in C. elegans. Hum Mol Genet 19: 3206–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley CT, Sutcliffe JS, Kunst CB, Leiner HA, Eichler EE, Nelson DL, Warren ST. 1993. Human and murine FMR-1: alternative splicing and translational initiation downstream of the CGG-repeat. Nat Genet 4: 244–251. [DOI] [PubMed] [Google Scholar]
- Ayala YM, Pantano S, D'Ambrogio A, Buratti E, Brindisi A, Marchetti C, Romano M, Baralle FE. 2005. Human, Drosophila, and C. elegans TDP43: nucleic acid binding properties and splicing regulatory function. J Mol Biol 348: 575–588. [DOI] [PubMed] [Google Scholar]
- Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, Tollervey J, Ule J, Baralle M, Buratti E, et al. 2011. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J 30: 277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo FA, Carvalho LR, Grinberg LT, Farfel JM, Ferretti RE, Leite RE, Jacob Filho W, Lent R, Herculano-Houzel S. 2009. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513: 532–541. [DOI] [PubMed] [Google Scholar]
- Bagga PS, Arhin GK, Wilusz J. 1998. DSEF-1 is a member of the hnRNP H family of RNA-binding proteins and stimulates pre-mRNA cleavage and polyadenylation in vitro. Nucleic Acids Res 26: 5343–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banks GT, Kuta A, Isaacs AM, Fisher EM. 2008. TDP-43 is a culprit in human neurodegeneration, and not just an innocent bystander. Mamm Genome 19: 299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron DM, Kaushansky LJ, Ward CL, Sama RR, Chian RJ, Boggio KJ, Quaresma AJ, Nickerson JA, Bosco DA. 2013. Amyotrophic lateral sclerosis-linked FUS/TLS alters stress granule assembly and dynamics. Mol Neurodegener 8: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker LA, Huang B, Bieri G, Ma R, Knowles DA, Jafar-Nejad P, Messing J, Kim HJ, Soriano A, Auburger G, et al. 2017. Therapeutic reduction of ataxin-2 extends lifespan and reduces pathology in TDP-43 mice. Nature 544: 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell MV, Hirst MC, Nakahori Y, MacKinnon RN, Roche A, Flint TJ, Jacobs PA, Tommerup N, Tranebjaerg L, Froster-Iskenius U, et al. 1991. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell 64: 861–866. [DOI] [PubMed] [Google Scholar]
- Belzil VV, Langlais JS, Daoud H, Dion PA, Brais B, Rouleau GA. 2012. Novel FUS deletion in a patient with juvenile amyotrophic lateral sclerosis. Arch Neurol 69: 653–656. [DOI] [PubMed] [Google Scholar]
- Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. 2012. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem 287: 23079–23094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernat V, Disney MD. 2015. RNA structures as mediators of neurological diseases and as drug targets. Neuron 87: 28–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop NA, Lu T, Yankner BA. 2010. Neural mechanisms of ageing and cognitive decline. Nature 464: 529–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokhuis AM, Groen EJ, Koppers M, van den Berg LH, Pasterkamp RJ. 2013. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol 125: 777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc F, Garant JM, Allard F, Perreault JP. 2016. Irregular G-quadruplexes found in the untranslated regions of human mRNAs influence translation. J Biol Chem 291: 21751–21760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brangwynne CP, Tompa P, Pappu RV. 2015. Polymer physics of intracellular phase transitions. Nat Phys 11: 899–904. [Google Scholar]
- Brazda V, Haronikova L, Liao JC, Fojta M. 2014. DNA and RNA quadruplex-binding proteins. Int J Mol Sci 15: 17493–17517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettschneider J, Del Tredici K, Lee VM, Trojanowski JQ. 2015. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat Rev Neurosci 16: 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. 1992. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 68: 799–808. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. 1998. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science 281: 1851–1854. [DOI] [PubMed] [Google Scholar]
- Buckanovich RJ, Posner JB, Darnell RB. 1993. Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 11: 657–672. [DOI] [PubMed] [Google Scholar]
- Budini M, Romano V, Avendano-Vazquez SE, Bembich S, Buratti E, Baralle FE. 2012. Role of selected mutations in the Q/N rich region of TDP-43 in EGFP-12xQ/N-induced aggregate formation. Brain Res 1462: 139–150. [DOI] [PubMed] [Google Scholar]
- Budworth H, McMurray CT. 2013. A brief history of triplet repeat diseases. Methods Mol Biol 1010: 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E. 2015. Functional significance of TDP-43 mutations in disease. Adv Genet 91: 1–53. [DOI] [PubMed] [Google Scholar]
- Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. 2001. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J 20: 1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. 2005. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem 280: 37572–37584. [DOI] [PubMed] [Google Scholar]
- Caillet-Boudin ML, Fernandez-Gomez FJ, Tran H, Dhaenens CM, Buee L, Sergeant N. 2014. Brain pathology in myotonic dystrophy: when tauopathy meets spliceopathy and RNAopathy. Front Mol Neurosci 6: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR. 2002. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30: 377–384. [DOI] [PubMed] [Google Scholar]
- Chan RC, Black DL. 1997. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol Cell Biol 17: 4667–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Manley JL. 2009. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol 10: 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. 2012. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem Biol 7: 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs-Disney JL, Yildirim I, Park H, Lohman JR, Guan L, Tran T, Sarkar P, Schatz GC, Disney MD. 2014. Structure of the myotonic dystrophy type 2 RNA and designed small molecules that reduce toxicity. ACS Chem Biol 9: 538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou MY, Rooke N, Turck CW, Black DL. 1999. hnRNP H is a component of a splicing enhancer complex that activates a c-src alternative exon in neuronal cells. Mol Cell Biol 19: 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciesiolka A, Jazurek M, Drazkowska K, Krzyzosiak WJ. 2017. Structural characteristics of simple RNA repeats associated with disease and their deleterious protein Interactions. Front Cell Neurosci 11: 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JD, Ranum LP. 2014. Repeat associated non-ATG (RAN) translation: new starts in microsatellite expansion disorders. Curr Opin Genet Dev 26: 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coady TH, Manley JL. 2015. ALS mutations in TLS/FUS disrupt target gene expression. Genes Dev 29: 1696–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conicella AE, Zerze GH, Mittal J, Fawzi NL. 2016. ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 24: 1537–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL. 2016. The C9ORF72 GGGGCC expansion forms RNA G-quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS brains. Elife 5: e17820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR. 2017. RNA-binding proteins implicated in neurodegenerative diseases. Wiley Interdiscip Rev RNA 8: e1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper TA, Wan L, Dreyfuss G. 2009. RNA and disease. Cell 136: 777–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Knock J, Shaw PJ, Kirby J. 2014. The widening spectrum of C9ORF72-related disease; genotype/phenotype correlations and potential modifiers of clinical phenotype. Acta Neuropathol 127: 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcia P, Danel V, Lacour A, Beltran S, Andres C, Couratier P, Blasco H, Vourc'h P. 2017. A novel mutation of the C-terminal amino acid of FUS (Y526C) strengthens FUS gene as the most frequent genetic factor in aggressive juvenile ALS. Amyotroph Lateral Scler Frontotemporal Degener 18: 298–301. [DOI] [PubMed] [Google Scholar]
- Corrado L, Del Bo R, Castellotti B, Ratti A, Cereda C, Penco S, Soraru G, Carlomagno Y, Ghezzi S, Pensato V, et al. 2010. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J Med Genet 47: 190–194. [DOI] [PubMed] [Google Scholar]
- Cortes CJ, La Spada AR. 2014. The many faces of autophagy dysfunction in Huntington's disease: from mechanism to therapy. Drug Discov Today 19: 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couthouis J, Hart MP, Erion R, King OD, Diaz Z, Nakaya T, Ibrahim F, Kim HJ, Mojsilovic-Petrovic J, Panossian S, et al. 2012. Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis. Hum Mol Genet 21: 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coutinho-Mansfield GC, Xue Y, Zhang Y, Fu XD. 2007. PTB/nPTB switch: a post-transcriptional mechanism for programming neuronal differentiation. Genes Dev 21: 1573–1577. [DOI] [PubMed] [Google Scholar]
- Crozat A, Aman P, Mandahl N, Ron D. 1993. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363: 640–644. [DOI] [PubMed] [Google Scholar]
- D'Ambrogio A, Buratti E, Stuani C, Guarnaccia C, Romano M, Ayala YM, Baralle FE. 2009. Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res 37: 4116–4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB. 1996. Onconeural antigens and the paraneoplastic neurologic disorders: at the intersection of cancer, immunity, and the brain. Proc Natl Acad Sci 93: 4529–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB. 2001. Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499. [DOI] [PubMed] [Google Scholar]
- Decker CJ, Parker R. 2012. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol 4: a012286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker CJ, Teixeira D, Parker R. 2007. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol 179: 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Conti L, Baralle M, Buratti E. 2017. Neurodegeneration and RNA-binding proteins. Wiley Interdiscip Rev RNA 8: e1394. [DOI] [PubMed] [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, et al. 2011. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72: 245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deLorimier E, Hinman MN, Copperman J, Datta K, Guenza M, Berglund JA. 2017. Pseudouridine modification inhibits Muscleblind-like 1 (MBNL1) binding to CCUG repeats and minimally structured RNA through reduced RNA flexibility. J Biol Chem 292: 4350–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Mezer M, Wojciechowska M, Napierala M, Sobczak K, Krzyzosiak WJ. 2011. Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucleic Acids Res 39: 3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dere R, Napierala M, Ranum LP, Wells RD. 2004. Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2. J Biol Chem 279: 41715–41726. [DOI] [PubMed] [Google Scholar]
- Doktor TK, Hua Y, Andersen HS, Broner S, Liu YH, Wieckowska A, Dembic M, Bruun GH, Krainer AR, Andresen BS. 2017. RNA-sequencing of a mouse-model of spinal muscular atrophy reveals tissue-wide changes in splicing of U12-dependent introns. Nucleic Acids Res 45: 395–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Kingsbury Z, Humphray SJ, Schellevis RD, Brands WJ, Baker M, et al. 2016. Detection of long repeat expansions from PCR-free whole-genome sequence data. bioRxiv 10.1101/093831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombert B, Sivadasan R, Simon CM, Jablonka S, Sendtner M. 2014. Presynaptic localization of Smn and hnRNP R in axon terminals of embryonic and postnatal mouse motoneurons. PLoS One 9: e110846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G, Matunis MJ, Pinol-Roma S, Burd CG. 1993. hnRNP proteins and the biogenesis of mRNA. Annu Rev Biochem 62: 289–321. [DOI] [PubMed] [Google Scholar]
- Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X, Armakola M, Geser F, Greene R, Lu MM, et al. 2010. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature 466: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C, Zhang H, Su Y, Silani V, Singer RH, Rossoll W, Bassell GJ. 2011. The survival of motor neuron (SMN) protein interacts with the mRNA-binding protein HuD and regulates localization of poly(A) mRNA in primary motor neuron axons. J Neurosci 31: 3914–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, Wu CC, Liao JY, Chou SC, Lin V, et al. 2014. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun 5: 4824. [DOI] [PubMed] [Google Scholar]
- Freibaum BD, Taylor JP. 2017. The role of dipeptide repeats in C9ORF72-related ALS–FTD. Front Mol Neurosci 10: 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M, Loeb LA. 1994. The fragile X syndrome d(CGG)n nucleotide repeats form a stable tetrahelical structure. Proc Natl Acad Sci 91: 4950–4954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba RA, Udan M, Bell S, Wegorzewska I, Shao J, Diamond MI, Weihl CC, Baloh RH. 2010. Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J Biol Chem 285: 26304–26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii R, Okabe S, Urushido T, Inoue K, Yoshimura A, Tachibana T, Nishikawa T, Hicks GG, Takumi T. 2005. The RNA binding protein TLS is translocated to dendritic spines by mGluR5 activation and regulates spine morphology. Curr Biol 15: 587–593. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Matsumoto G, Kurosawa M, Nukina N. 2009. Cross-seeding fibrillation of Q/N-rich proteins offers new pathomechanism of polyglutamine diseases. J Neurosci 29: 5153–5162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Kaneko K, Watanabe S, Yamanaka K, Nukina N. 2011. A seeding reaction recapitulates intracellular formation of Sarkosyl-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J Biol Chem 286: 18664–18672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, Greenberg ME. 2015. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522: 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Reif A, Dell'osso B, Kittel-Schneider S, Leonhard C, Herr A, Palazzo C, Villa C, Fenoglio C, Serpente M, et al. 2014. C9ORF72 hexanucleotide repeat expansion is a rare cause of schizophrenia. Neurobiol Aging 35: 1214.e7–1214.e10. [DOI] [PubMed] [Google Scholar]
- Galloway JN, Nelson DL. 2009. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol 4: 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Lopez A, Llamusi B, Orzaez M, Perez-Paya E, Artero RD. 2011. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc Natl Acad Sci 108: 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Deza J, Lee YB, Troakes C, Nolan M, Al-Sarraj S, Gallo JM, Shaw CE. 2015. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun 3: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabowski PJ, Black DL. 2001. Alternative RNA splicing in the nervous system. Prog Neurobiol 65: 289–308. [DOI] [PubMed] [Google Scholar]
- Green KM, Linsalata AE, Todd PK. 2016. RAN translation-What makes it run? Brain Res 1647: 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Bartel DP. 2016. RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353: aaf5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo L, Shorter J. 2015. It's raining liquids: RNA tunes viscoelasticity and dynamics of membraneless organelles. Mol Cell 60: 189–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, et al. 2014. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Rothstein JD. 2016. The expanding biology of the C9orf72 nucleotide repeat expansion in neurodegenerative disease. Nat Rev Neurosci 17: 383–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, et al. 2012. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell 149: 768–779. [DOI] [PubMed] [Google Scholar]
- Handa V, Yeh HJ, McPhie P, Usdin K. 2005. The AUUCU repeats responsible for spinocerebellar ataxia type 10 form unusual RNA hairpins. J Biol Chem 280: 29340–29345. [DOI] [PubMed] [Google Scholar]
- Harada Y, Sutomo R, Sadewa AH, Akutsu T, Takeshima Y, Wada H, Matsuo M, Nishio H. 2002. Correlation between SMN2 copy number and clinical phenotype of spinal muscular atrophy: three SMN2 copies fail to rescue some patients from the disease severity. J Neurol 249: 1211–1219. [DOI] [PubMed] [Google Scholar]
- Harms MM, Miller TM, Baloh RH. 2015. TARDBP-related amyotrophic lateral sclerosis. In GeneReviews (ed. Pagon RA, et al. ), https://www.ncbi.nlm.nih.gov/books/NBK5942 University of Washington, Seattle, WA. [Google Scholar]
- Herculano-Houzel S. 2012. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. Proc Natl Acad Sci 109: 10661–10668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D. 1995. RNA chaperones and the RNA folding problem. J Biol Chem 270: 20871–20874. [DOI] [PubMed] [Google Scholar]
- Hoell JI, Larsson E, Runge S, Nusbaum JD, Duggimpudi S, Farazi TA, Hafner M, Borkhardt A, Sander C, Tuschl T. 2011. RNA targets of wild-type and mutant FET family proteins. Nat Struct Mol Biol 18: 1428–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes SE, O'Hearn E, Rosenblatt A, Callahan C, Hwang HS, Ingersoll-Ashworth RG, Fleisher A, Stevanin G, Brice A, Potter NT, et al. 2001. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat Genet 29: 377–378. [DOI] [PubMed] [Google Scholar]
- Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. 2007. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol 5: e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sahashi K, Rigo F, Hung G, Horev G, Bennett CF, Krainer AR. 2011. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature 478: 123–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelga SC, Vu AQ, Arnold JD, Liang TY, Liu PP, Yan BY, Donohue JP, Shiue L, Hoon S, Brenner S, et al. 2012. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep 1: 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J, Emmett W, Fratta P, Isaacs AM, Plagnol V. 2017. Quantitative analysis of cryptic splicing associated with TDP-43 depletion. BMC Med Genomics 10: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaz LM, Kwong LK, Xu Y, Truax AC, Uryu K, Neumann M, Clark CM, Elman LB, Miller BL, Grossman M, et al. 2008. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol 173: 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Ohta Y, Kobayashi H, Okamoto M, Takamatsu K, Ota T, Manabe Y, Okamoto K, Koizumi A, Abe K. 2012. Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology 79: 333–341. [DOI] [PubMed] [Google Scholar]
- Ishiguro A, Kimura N, Watanabe Y, Watanabe S, Ishihama A. 2016. TDP-43 binds and transports G-quadruplex-containing mRNAs into neurites for local translation. Genes Cells 21: 466–481. [DOI] [PubMed] [Google Scholar]
- Iwahashi CK, Yasui DH, An HJ, Greco CM, Tassone F, Nannen K, Babineau B, Lebrilla CB, Hagerman RJ, Hagerman PJ. 2006. Protein composition of the intranuclear inclusions of FXTAS. Brain 129: 256–271. [DOI] [PubMed] [Google Scholar]
- Jangi M, Fleet C, Cullen P, Gupta SV, Mekhoubad S, Chiao E, Allaire N, Bennett CF, Rigo F, Krainer AR, et al. 2017. SMN deficiency in severe models of spinal muscular atrophy causes widespread intron retention and DNA damage. Proc Natl Acad Sci 114: E2347–E2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ, Yang YY, Darnell RB. 2000. Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25: 359–371. [DOI] [PubMed] [Google Scholar]
- Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA. 2004. Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neurons. Hum Mol Genet 13: 3079–3088. [DOI] [PubMed] [Google Scholar]
- Jin P, Warren ST. 2000. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet 9: 901–908. [DOI] [PubMed] [Google Scholar]
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST. 2007. Purα binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 55: 556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. 2009. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem 284: 20329–20339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, et al. 2014. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci 17: 664–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Yoon BC, Holt CE. 2012. Axonal mRNA localization and local protein synthesis in nervous system assembly, maintenance and repair. Nat Rev Neurosci 13: 308–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. 2008. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet 40: 572–574. [DOI] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. 2003. A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980. [DOI] [PubMed] [Google Scholar]
- Kashima T, Manley JL. 2003. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34: 460–463. [DOI] [PubMed] [Google Scholar]
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. 2012. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell 149: 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse MG, Todd PK. 2014. Repeat-associated non-AUG translation and its impact in neurodegenerative disease. Neurotherapeutics 11: 721–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiebler MA, Bassell GJ. 2006. Neuronal RNA granules: movers and makers. Neuron 51: 685–690. [DOI] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, MacLea KS, Freibaum B, Li S, Molliex A, et al. 2013. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 495: 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Nakayama Y, Shibasaki A, Taki A, Yuno S, Takeda K, Yahara M, Tanabe N, Kinjo M. 2016. Interaction of RNA with a C-terminal fragment of the amyotrophic lateral sclerosis-associated TDP43 reduces cytotoxicity. Sci Rep 6: 19230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura A, Yuno S, Muto H, Kinjo M. 2017. Different aggregation states of a nuclear localization signal-tagged 25-kDa C-terminal fragment of TAR RNA/DNA-binding protein 43 kDa. Genes Cells 22: 521–534. [DOI] [PubMed] [Google Scholar]
- Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. 1998. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 95: 41–53. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. 2011. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 89: 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb SJ, Battle DJ, Dreyfuss G. 2007. Molecular functions of the SMN complex. J Child Neurol 22: 990–994. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, Schuck T, Wheeler JM, Robinson LC, Trojanowski JQ, Lee VM, Schellenberg GD. 2010. Loss of murine TDP-43 disrupts motor function and plays an essential role in embryogenesis. Acta Neuropathol 119: 409–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J, Fiszer A, Mykowska A, Sobczak K, de Mezer M, Krzyzosiak WJ. 2007. Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol Cell 25: 575–586. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. 1999. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol 46: 842–849. [PubMed] [Google Scholar]
- Kuo PH, Doudeva LG, Wang YT, Shen CK, Yuan HS. 2009. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res 37: 1799–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu-Martinez NM, Cooper TA. 2006. Misregulation of alternative splicing causes pathogenesis in myotonic dystrophy. Prog Mol Subcell Biol 44: 133–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. 2009. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 323: 1205–1208. [DOI] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Hutt KR, Vu AQ, Baughn M, Huelga SC, Clutario KM, Ling SC, Liang TY, Mazur C, et al. 2012. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat Neurosci 15: 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Taylor JP. 2010. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11: 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. 1991. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 352: 77–79. [DOI] [PubMed] [Google Scholar]
- Lee KY, Li M, Manchanda M, Batra R, Charizanis K, Mohan A, Warren SA, Chamberlain CM, Finn D, Hong H, et al. 2013. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol Med 5: 1887–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. 1997. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 16: 265–269. [DOI] [PubMed] [Google Scholar]
- Lehmer C, Oeckl P, Weishaupt JH, Volk AE, Diehl-Schmid J, Schroeter ML, Lauer M, Kornhuber J, Levin J, Fassbender K, et al. 2017. Poly-GP in cerebrospinal fluid links C9orf72-associated dipeptide repeat expression to the asymptomatic phase of ALS/FTD. EMBO Mol Med 9: 859–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerga A, Hallier M, Delva L, Orvain C, Gallais I, Marie J, Moreau-Gachelin F. 2001. Identification of an RNA binding specificity for the potential splicing factor TLS. J Biol Chem 276: 6807–6816. [DOI] [PubMed] [Google Scholar]
- Li Y, Ray P, Rao EJ, Shi C, Guo W, Chen X, Woodruff EA III, Fushimi K, Wu JY. 2010. A Drosophila model for TDP-43 proteinopathy. Proc Natl Acad Sci 107: 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YR, King OD, Shorter J, Gitler AD. 2013. Stress granules as crucibles of ALS pathogenesis. J Cell Biol 201: 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Yokoshi M, Okada H, Kawahara Y. 2015. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun 6: 6183. [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, et al. 2008. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 456: 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Yano M, Fak JJ, Mele A, Grabinski SE, Zhang C, Darnell RB. 2012. Ptbp2 represses adult-specific splicing to regulate the generation of neuronal precursors in the embryonic brain. Genes Dev 26: 1626–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Mori E, Kato M, Xiang S, Wu L, Kwon I, McKnight SL. 2016. Toxic PR poly-dipeptides encoded by the C9orf72 repeat expansion target LC domain polymers. Cell 167: 789–802 e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Albuquerque CP, Han JS, Lagier-Tourenne C, Tokunaga S, Zhou H, Cleveland DW. 2010. ALS-associated mutations in TDP-43 increase its stability and promote TDP-43 complexes with FUS/TLS. Proc Natl Acad Sci 107: 13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. 2013. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling JP, Pletnikova O, Troncoso JC, Wong PC. 2015. TDP-43 repression of nonconserved cryptic exons is compromised in ALS–FTD. Science 349: 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. 2001. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293: 864–867. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, et al. 2010. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One 5: e13250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, Kobrin K, Murphy GJ, Petrucelli L, Wolozin B. 2014. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci 34: 4167–4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Gonzalez R, Lu Y, Gendron TF, Karydas A, Tran H, Yang D, Petrucelli L, Miller BL, Almeida S, Gao FB. 2016. Poly(GR) in C9ORF72-related ALS/FTD compromises mitochondrial function and increases oxidative stress and DNA damage in iPSC-derived motor neurons. Neuron 92: 383–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotti F, Imlach WL, Saieva L, Beck ES, Hao le T, Li DK, Jiao W, Mentis GZ, Beattie CE, McCabe BD, et al. 2012. An SMN-dependent U12 splicing event essential for motor circuit function. Cell 151: 440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukavsky PJ, Daujotyte D, Tollervey JR, Ule J, Stuani C, Buratti E, Baralle FE, Damberger FF, Allain FH. 2013. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat Struct Mol Biol 20: 1443–1449. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, et al. 2007. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol 61: 427–434. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. 2010. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol 9: 995–1007. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Allaman I. 2015. A cellular perspective on brain energy metabolism and functional imaging. Neuron 86: 883–901. [DOI] [PubMed] [Google Scholar]
- March ZM, King OD, Shorter J. 2016. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res 1647: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis J, Paillard L, Audic Y, Cosson B, Danos O, Le Bec C, Osborne HB. 2006. CUG-BP1/CELF1 requires UGU-rich sequences for high-affinity binding. Biochem J 400: 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti E. 2016. RNA toxicity induced by expanded CAG repeats in Huntington's disease. Brain Pathol 26: 779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda A, Takeda J, Okuno T, Okamoto T, Ohkawara B, Ito M, Ishigaki S, Sobue G, Ohno K. 2015. Position-specific binding of FUS to nascent RNA regulates mRNA length. Genes Dev 29: 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, Rouleau GA, Vande Velde C. 2011. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet 20: 1400–1410. [DOI] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, O'Donovan MJ. 2011. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69: 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalowski S, Miller JW, Urbinati CR, Paliouras M, Swanson MS, Griffith J. 1999. Visualization of double-stranded RNAs from the myotonic dystrophy protein kinase gene and interactions with CUG-binding protein. Nucleic Acids Res 27: 3534–3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, et al. 2014. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345: 1192–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliex A, Temirov J, Lee J, Coughlin M, Kanagaraj AP, Kim HJ, Mittag T, Taylor JP. 2015. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 163: 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mompean M, Hervas R, Xu Y, Tran TH, Guarnaccia C, Buratti E, Baralle F, Tong L, Carrion-Vazquez M, McDermott AE, et al. 2015. Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP-43. J Phys Chem Lett 6: 2608–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mompean M, Baralle M, Buratti E, Laurents DV. 2016. An amyloid-like pathological conformation of TDP-43 is stabilized by hypercooperative hydrogen bonds. Front Mol Neurosci 9: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooers BH, Logue JS, Berglund JA. 2005. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc Natl Acad Sci 102: 16626–16631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris MJ, Negishi Y, Pazsint C, Schonhoft JD, Basu S. 2010. An RNA G-quadruplex is essential for cap-independent translation initiation in human VEGF IRES. J Am Chem Soc 132: 17831–17839. [DOI] [PubMed] [Google Scholar]
- Morriss GR, Cooper TA. 2017. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum Genet 10.1007/s00439-017-1807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen MA, Assmann SM, Bevilacqua PC. 2012. Toward a digital gene response: RNA G-quadruplexes with fewer quartets fold with higher cooperativity. J Am Chem Soc 134: 812–815. [DOI] [PubMed] [Google Scholar]
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, et al. 2015. ALS/FTD mutation-induced phase transition of FUS liquid droplets and reversible hydrogels into irreversible hydrogels impairs RNP granule function. Neuron 88: 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mykowska A, Sobczak K, Wojciechowska M, Kozlowski P, Krzyzosiak WJ. 2011. CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucleic Acids Res 39: 8938–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, et al. 2013. Splicing biomarkers of disease severity in myotonic dystrophy. Ann Neurol 74: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalavade R, Griesche N, Ryan DP, Hildebrand S, Krauss S. 2013. Mechanisms of RNA-induced toxicity in CAG repeat disorders. Cell Death Dis 4: e752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazim M, Masuda A, Rahman MA, Nasrin F, Takeda JI, Ohe K, Ohkawara B, Ito M, Ohno K. 2017. Competitive regulation of alternative splicing and alternative polyadenylation by hnRNP H and CstF64 determines acetylcholinesterase isoforms. Nucleic Acids Res 45: 1455–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. 2006. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133. [DOI] [PubMed] [Google Scholar]
- Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IR. 2009. A new subtype of frontotemporal lobar degeneration with FUS pathology. Brain 132: 2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan M, Talbot K, Ansorge O. 2016. Pathogenesis of FUS-associated ALS and FTD: insights from rodent models. Acta Neuropathol Commun 4: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonhoff U, Ralser M, Welzel F, Piccini I, Balzereit D, Yaspo ML, Lehrach H, Krobitsch S. 2007. Ataxin-2 interacts with the DEAD/H-box RNA helicase DDX6 and interferes with P-bodies and stress granules. Mol Biol Cell 18: 1385–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott TJ, Craggs TD, Baldwin AJ. 2016. Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat Chem 8: 569–575. [DOI] [PubMed] [Google Scholar]
- Nussbacher JK, Batra R, Lagier-Tourenne C, Yeo GW. 2015. RNA-binding proteins in neurodegeneration: seq and you shall receive. Trends Neurosci 38: 226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Rahman MA, Nazim M, Nasrin F, Lin Y, Takeda JI, Masuda A. 2017. Splicing regulation and dysregulation of cholinergic genes expressed at the neuromuscular junction. J Neurochem 142: 64–72. [DOI] [PubMed] [Google Scholar]
- Ottesen EW. 2017. ISS-N1 makes the first FDA-approved drug for spinal muscular atrophy. Transl Neurosci 8: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. 2015. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162: 1066–1077. [DOI] [PubMed] [Google Scholar]
- Pedrotti S, Bielli P, Paronetto MP, Ciccosanti F, Fimia GM, Stamm S, Manley JL, Sette C. 2010. The splicing regulator Sam68 binds to a novel exonic splicing silencer and functions in SMN2 alternative splicing in spinal muscular atrophy. EMBO J 29: 1235–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni L. 2007. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep 8: 340–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petraccone L. 2013. Higher-order quadruplex structures. Top Curr Chem 330: 23–46. [DOI] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, Cooper TA. 1998. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science 280: 737–741. [DOI] [PubMed] [Google Scholar]
- Piazzon N, Rage F, Schlotter F, Moine H, Branlant C, Massenet S. 2008. In vitro and in cellulo evidences for association of the survival of motor neuron complex with the fragile X mental retardation protein. J Biol Chem 283: 5598–5610. [DOI] [PubMed] [Google Scholar]
- Polymenidou M, Lagier-Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, et al. 2011. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat Neurosci 14: 459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior TW. 2010. Perspectives and diagnostic considerations in spinal muscular atrophy. Genet Med 12: 145–152. [DOI] [PubMed] [Google Scholar]
- Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P, et al. 2015. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci 18: 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, et al. 1996. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet 14: 269–276. [DOI] [PubMed] [Google Scholar]
- Rabbitts TH, Forster A, Larson R, Nathan P. 1993. Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4: 175–180. [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, et al. 2011. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS–FTD. Neuron 72: 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Litterman NK, Norabuena EM, Mull JL, Galazo MJ, Sun C, Ng SY, Makhortova NR, White A, Lynes MM, et al. 2017. Single-cell analysis of SMN reveals its broader role in neuromuscular disease. Cell Rep 18: 1484–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogelj B, Easton LE, Bogu GK, Stanton LW, Rot G, Curk T, Zupan B, Sugimoto Y, Modic M, Haberman N, et al. 2012. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep 2: 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. 2005. Opinion: what is the role of protein aggregation in neurodegeneration? Nat Rev Mol Cell Biol 6: 891–898. [DOI] [PubMed] [Google Scholar]
- Ross CA, Tabrizi SJ. 2011. Huntington's disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10: 83–98. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. 2001. Amyotrophic lateral sclerosis. N Engl J Med 344: 1688–1700. [DOI] [PubMed] [Google Scholar]
- Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL. 2007. Huntington's disease-like 2 is associated with CUG repeat-containing RNA foci. Ann Neurol 61: 272–282. [DOI] [PubMed] [Google Scholar]
- Ruggiu M, McGovern VL, Lotti F, Saieva L, Li DK, Kariya S, Monani UR, Burghes AH, Pellizzoni L. 2012. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol Cell Biol 32: 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal L, Briese M, Kneitz S, Glinka M, Sendtner M. 2014. Subcellular transcriptome alterations in a cell culture model of spinal muscular atrophy point to widespread defects in axonal growth and presynaptic differentiation. RNA 20: 1789–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton M, Elkon R, Borodina T, Davydov A, Yaspo ML, Halperin E, Shiloh Y. 2011. Matrin 3 binds and stabilizes mRNA. PLoS One 6: e23882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, et al. 2009. Spinocerebellar ataxia type 31 is associated with ‘inserted’ penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85: 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. 1998. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95: 55–66. [DOI] [PubMed] [Google Scholar]
- Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, et al. 2013. Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2: e01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic Pavicevic D, Miladinovic J, Brkusanin M, Svikovic S, Djurica S, Brajuskovic G, Romac S. 2013. Molecular genetics and genetic testing in myotonic dystrophy type 1. Biomed Res Int 2013: 391821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scekic-Zahirovic J, Sendscheid O, El Oussini H, Jambeau M, Sun Y, Mersmann S, Wagner M, Dieterle S, Sinniger J, Dirrig-Grosch S, et al. 2016. Toxic gain of function from mutant FUS protein is crucial to trigger cell autonomous motor neuron loss. EMBO J 35: 1077–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schipper LJ, Raaphorst J, Aronica E, Baas F, de Haan R, de Visser M, Troost D. 2016. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: a systematic neuropathological review. Neuropathol Appl Neurobiol 42: 547–560. [DOI] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. 2010. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A, et al. 2013. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep 3: 869–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Buijsen RA, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A, et al. 2017. Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to fragile X tremor ataxia syndrome. Neuron 93: 331–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sephton CF, Yu G. 2015. The function of RNA-binding proteins at the synapse: implications for neurodegeneration. Cell Mol Life Sci 72: 3621–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Chiang PM, Price DL, Wong PC. 2010. Altered distributions of Gemini of coiled bodies and mitochondria in motor neurons of TDP-43 transgenic mice. Proc Natl Acad Sci 107: 16325–16330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang Y, Huang EJ. 2016. Mechanisms of FUS mutations in familial amyotrophic lateral sclerosis. Brain Res 1647: 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A, Lyashchenko AK, Lu L, Nasrabady SE, Elmaleh M, Mendelsohn M, Nemes A, Tapia JC, Mentis GZ, Shneider NA. 2016. ALS-associated mutant FUS induces selective motor neuron degeneration through toxic gain of function. Nat Commun 7: 10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpee TO. 2014. Toward functional classification of neuronal types. Neuron 83: 1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter J. 2016. Membraneless organelles: phasing in and out. Nat Chem 8: 528–530. [DOI] [PubMed] [Google Scholar]
- Shorter J, Taylor JP. 2013. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis 1: e25200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simone R, Fratta P, Neidle S, Parkinson GN, Isaacs AM. 2015. G-quadruplexes: emerging roles in neurodegenerative diseases and the non-coding transcriptome. FEBS Lett 589: 1653–1668. [DOI] [PubMed] [Google Scholar]
- Simone C, Ramirez A, Bucchia M, Rinchetti P, Rideout H, Papadimitriou D, Re DB, Corti S. 2016. Is spinal muscular atrophy a disease of the motor neurons only: pathogenesis and therapeutic implications? Cell Mol Life Sci 73: 1003–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisodia SS. 1998. Nuclear inclusions in glutamine repeat disorders: are they pernicious, coincidental, or beneficial? Cell 95: 1–4. [DOI] [PubMed] [Google Scholar]
- So BR, Wan L, Zhang Z, Li P, Babiash E, Duan J, Younis I, Dreyfuss G. 2016. A U1 snRNP-specific assembly pathway reveals the SMN complex as a versatile hub for RNP exchange. Nat Struct Mol Biol 23: 225–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. 2007. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55: 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Perreault JP, Topisirovic I, Richard S. 2016. RNA G-quadruplexes and their potential regulatory roles in translation. Translation 4: e1244031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, Al-Sarraj S, Andersen PM, Bonini NM, Conforti FL, et al. 2017. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging 51: 178 e171–178 e179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. 2008. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319: 1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanovic S, Bassell GJ, Mihailescu MR. 2015. G quadruplex RNA structures in PSD-95 mRNA: potential regulators of miR-125a seed binding site accessibility. RNA 21: 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. 1999. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8: 731–741. [DOI] [PubMed] [Google Scholar]
- Sun Y, Chakrabartty A. 2017. Phase to phase with TDP-43. Biochemistry 56: 809–823. [DOI] [PubMed] [Google Scholar]
- Sun YM, Lu C, Wu ZY. 2016. Spinocerebellar ataxia: relationship between phenotype and genotype - a review. Clin Genet 90: 305–314. [DOI] [PubMed] [Google Scholar]
- Svetoni F, Frisone P, Paronetto MP. 2016. Role of FET proteins in neurodegenerative disorders. RNA Biol 13: 1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Robberecht W. 2014. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10: 661–670. [DOI] [PubMed] [Google Scholar]
- Tan AY, Manley JL. 2009. The TET family of proteins: functions and roles in disease. J Mol Cell Biol 1: 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AY, Riley TR, Coady T, Bussemaker HJ, Manley JL. 2012. TLS/FUS (translocated in liposarcoma/fused in sarcoma) regulates target gene transcription via single-stranded DNA response elements. Proc Natl Acad Sci 109: 6030–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Yalamanchili HK, Park J, De Maio A, Lu HC, Wan YW, White JJ, Bondar VV, Sayegh LS, Liu X, et al. 2016. Extensive cryptic splicing upon loss of RBM17 and TDP43 in neurodegeneration models. Hum Mol Genet 25: 5083–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JE, Thomas NH, Lewis CM, Abbs SJ, Rodrigues NR, Davies KE, Mathew CG. 1998. Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur J Hum Genet 6: 467–474. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr, Cleveland DW. 2016. Decoding ALS: from genes to mechanism. Nature 539: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CA, Wang E, Carrell EM. 2017. Myotonic dystrophy: approach to therapy. Curr Opin Genet Dev 44: 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticozzi N, Silani V, LeClerc AL, Keagle P, Gellera C, Ratti A, Taroni F, Kwiatkowski TJ Jr, McKenna-Yasek DM, Sapp PC, et al. 2009. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 73: 1180–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timchenko NA, Cai ZJ, Welm AL, Reddy S, Ashizawa T, Timchenko LT. 2001. RNA CUG repeats sequester CUGBP1 and alter protein levels and activity of CUGBP1. J Biol Chem 276: 7820–7826. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Curk T, Rogelj B, Briese M, Cereda M, Kayikci M, Konig J, Hortobagyi T, Nishimura AL, Zupunski V, et al. 2011a. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci 14: 452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollervey JR, Wang Z, Hortobagyi T, Witten JT, Zarnack K, Kayikci M, Clark TA, Schweitzer AC, Rot G, Curk T, et al. 2011b. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res 21: 1572–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuiji H, Iguchi Y, Furuya A, Kataoka A, Hatsuta H, Atsuta N, Tanaka F, Hashizume Y, Akatsu H, Murayama S, et al. 2013. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med 5: 221–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu WY, Simpson JE, Highley JR, Heath PR. 2017. Spinal muscular atrophy: factors that modulate motor neurone vulnerability. Neurobiol Dis 102: 11–20. [DOI] [PubMed] [Google Scholar]
- Turunen JJ, Niemela EH, Verma B, Frilander MJ. 2013. The significant other: splicing by the minor spliceosome. Wiley Interdiscip Rev RNA 4: 61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udd B, Krahe R. 2012. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11: 891–905. [DOI] [PubMed] [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB. 2003. CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215. [DOI] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, de Araujo PR, Vogel C, Qiao M, Burns SC, Smith AD, Penalva LO. 2016. High-throughput analyses of hnRNP H1 dissects its multi-functional aspect. RNA Biol 13: 400–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. 2009. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 323: 1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Robberecht W, Van Den Bosch L. 2017. Modelling amyotrophic lateral sclerosis: progress and possibilities. Dis Model Mech 10: 537–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck L, Callaerts P, Dermaut B. 2014. TDP-43-mediated neurodegeneration: towards a loss-of-function hypothesis? Trends Mol Med 20: 66–71. [DOI] [PubMed] [Google Scholar]
- Vanderweyde T, Yu H, Varnum M, Liu-Yesucevitz L, Citro A, Ikezu T, Duff K, Wolozin B. 2012. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J Neurosci 32: 8270–8283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderweyde T, Youmans K, Liu-Yesucevitz L, Wolozin B. 2013. Role of stress granules and RNA-binding proteins in neurodegeneration: a mini-review. Gerontology 59: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilyev N, Polonskaia A, Darnell JC, Darnell RB, Patel DJ, Serganov A. 2015. Crystal structure reveals specific recognition of a G-quadruplex RNA by a β-turn in the RGG motif of FMRP. Proc Natl Acad Sci 112: E5391–E5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, et al. 1991. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914. [DOI] [PubMed] [Google Scholar]
- Wang W, Kwon EJ, Tsai LH. 2012. MicroRNAs in learning, memory, and neurological diseases. Learn Mem 19: 359–368. [DOI] [PubMed] [Google Scholar]
- Wang WY, Pan L, Su SC, Quinn EJ, Sasaki M, Jimenez JC, Mackenzie IR, Huang EJ, Tsai LH. 2013. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat Neurosci 16: 1383–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwartz JC, Cech TR. 2015. Nucleic acid-binding specificity of human FUS protein. Nucleic Acids Res 43: 7535–7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warf MB, Berglund JA. 2007. MBNL binds similar RNA structures in the CUG repeats of myotonic dystrophy and its pre-mRNA substrate cardiac troponin T. RNA 13: 2238–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, et al. 2014. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84: 1213–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D. 2016. Cell-to-cell transmission of dipeptide repeat proteins linked to C9orf72-ALS/FTD. Cell Rep 17: 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL, et al. 2011. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron 70: 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilke C, Pomper JK, Biskup S, Puskas C, Berg D, Synofzik M. 2016. Atypical parkinsonism in C9orf72 expansions: a case report and systematic review of 45 cases from the literature. J Neurol 263: 558–574. [DOI] [PubMed] [Google Scholar]
- Wils H, Kleinberger G, Janssens J, Pereson S, Joris G, Cuijt I, Smits V, Ceuterick-de Groote C, Van Broeckhoven C, Kumar-Singh S. 2010. TDP-43 transgenic mice develop spastic paralysis and neuronal inclusions characteristic of ALS and frontotemporal lobar degeneration. Proc Natl Acad Sci 107: 3858–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. 2008. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283: 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B, Apicco D. 2015. RNA binding proteins and the genesis of neurodegenerative diseases. Adv Exp Med Biol 822: 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Krainer AR. 1999. AT-AC pre-mRNA splicing mechanisms and conservation of minor introns in voltage-gated ion channel genes. Mol Cell Biol 19: 3225–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu P, Zuo X, Deng H, Liu X, Liu L, Ji A. 2013. Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull 97: 69–80. [DOI] [PubMed] [Google Scholar]
- Xiao S, Sanelli T, Dib S, Sheps D, Findlater J, Bilbao J, Keith J, Zinman L, Rogaeva E, Robertson J. 2011. RNA targets of TDP-43 identified by UV-CLIP are deregulated in ALS. Mol Cell Neurosci 47: 167–180. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Chen S, Yu Y, Yan B, Haertlein TC, Carrasco MA, Tapia JC, Zhai B, Das R, Lalancette-Hebert M, et al. 2012. FUS-SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep 2: 799–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Yin GL, Darnell RB. 1998. The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci 95: 13254–13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo G, Holste D, Kreiman G, Burge CB. 2004. Variation in alternative splicing across human tissues. Genome Biol 5: R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoshi M, Li Q, Yamamoto M, Okada H, Suzuki Y, Kawahara Y. 2014. Direct binding of Ataxin-2 to distinct elements in 3′ UTRs promotes mRNA stability and protein expression. Mol Cell 55: 186–198. [DOI] [PubMed] [Google Scholar]
- Yuan Y, Compton SA, Sobczak K, Stenberg MG, Thornton CA, Griffith JD, Swanson MS. 2007. Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucleic Acids Res 35: 5474–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yum K, Wang ET, Kalsotra A. 2017. Myotonic dystrophy: disease repeat range, penetrance, age of onset, and relationship between repeat size and phenotypes. Curr Opin Genet Dev 44: 30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. 1997. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci 146: 67–72. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lee JY, Tian B. 2005. Biased alternative polyadenylation in human tissues. Genome Biol 6: R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Lotti F, Dittmar K, Younis I, Wan L, Kasim M, Dreyfuss G. 2008. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell 133: 585–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, et al. 2009. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci 106: 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gaetano CM, Williams KR, Bassell GJ, Mihailescu MR. 2014. FMRP interacts with G-quadruplex structures in the 3′-UTR of its dendritic target Shank1 mRNA. RNA Biol 11: 1364–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Elbaum-Garfinkle S, Langdon EM, Taylor N, Occhipinti P, Bridges AA, Brangwynne CP, Gladfelter AS. 2015. RNA controls polyQ protein phase transitions. Mol Cell 60: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J, et al. 2016. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature 529: 48–53. [DOI] [PubMed] [Google Scholar]
- Zhou H, Mangelsdorf M, Liu J, Zhu L, Wu JY. 2014. RNA-binding proteins in neurological diseases. Sci China Life Sci 57: 432–444. [DOI] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, et al. 2011. Non-ATG-initiated translation directed by microsatellite expansions. Proc Natl Acad Sci 108: 260–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zylka MJ, Simon JM, Philpot BD. 2015. Gene length matters in neurons. Neuron 86: 353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]