

Here, Wilhelm et al. performed an RNAi screen in C. elegans to identify genes mediating post-reproductive longevity. They found that the inhibition of vesicle nucleation in the post-reproductive animal prevents age-associated neuronal degeneration, which leads to increased health and life span.
Keywords: antagonistic pleiotropy, C. elegans, BEC-1, autophagy, aging, neurodegeneration
Abstract
Autophagy is a ubiquitous catabolic process that causes cellular bulk degradation of cytoplasmic components and is generally associated with positive effects on health and longevity. Inactivation of autophagy has been linked with detrimental effects on cells and organisms. The antagonistic pleiotropy theory postulates that some fitness-promoting genes during youth are harmful during aging. On this basis, we examined genes mediating post-reproductive longevity using an RNAi screen. From this screen, we identified 30 novel regulators of post-reproductive longevity, including pha-4. Through downstream analysis of pha-4, we identified that the inactivation of genes governing the early stages of autophagy up until the stage of vesicle nucleation, such as bec-1, strongly extend both life span and health span. Furthermore, our data demonstrate that the improvements in health and longevity are mediated through the neurons, resulting in reduced neurodegeneration and sarcopenia. We propose that autophagy switches from advantageous to harmful in the context of an age-associated dysfunction.
Aging represents the functional deterioration of an organism, which compromises fitness and predisposes for prevalent diseases such as cancer and neurodegeneration. Many genes that modulate aging have been identified in Caenorhabditis elegans through mutagenesis or RNAi screens. These screens inactivate genes across the whole life span from early development (Klass 1983; Ni and Lee 2010). Genes identified through these studies often work through a small set of common pathways, including the target of rapamycin (TOR) pathway and the insulin-like signaling pathway (Ni and Lee 2010). However, genetic pathways specific to aging in older individuals remain largely undiscovered. The existence of alleles belonging to such pathways is predicted by the antagonistic pleiotropy (AP) theory of aging. This evolutionary-based theory states that strong natural selection early in life enriches alleles that mediate fitness while simultaneously accumulating their harmful effects after reproduction, when natural selection is ineffectual (Williams 1957). Thus, some genes that reduce fitness when inhibited in young worms should conversely extend life span when inhibited in older worms. Two screens in late L4 worms identified AP genes critical to development (Chen et al. 2007; Curran and Ruvkun 2007). However, the potential of these screens was limited with regard to detecting AP genes, as they initiated RNAi early in life at a point of maximal selection pressure (Fig. 1A).
Figure 1.
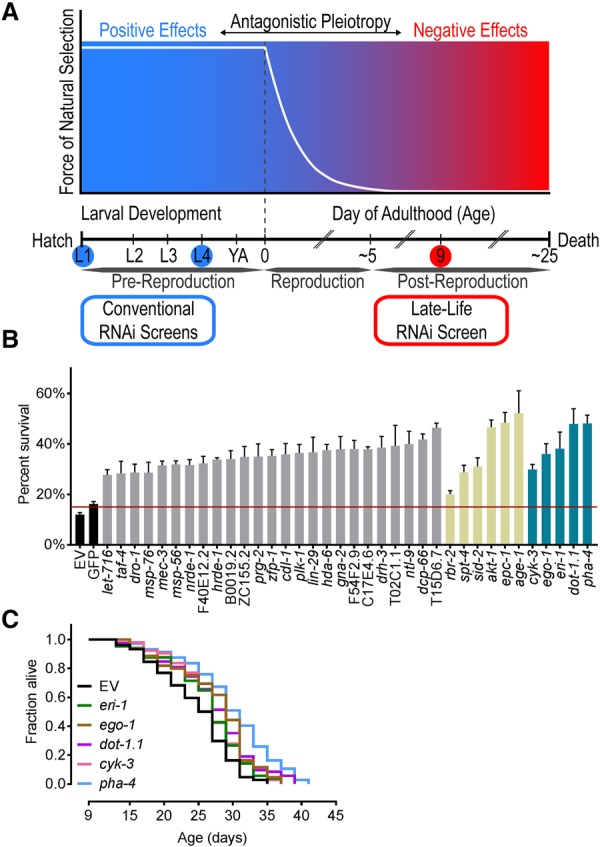
Screening for AP genes uncovers novel regulators of post-reproductive longevity. (A) Screening for AP genes. The force of natural selection (white line) declines over age. AP genes have positive fitness effects (blue region) early in life and negative effects (red region) late in life. Inhibiting genes post-reproductively can identify novel regulators of longevity. (Blue circles) Conventional RNAi screens’ initiation points; (red circle) initiation point of our RNAi screen. (B) Potential longevity genes identified from the AP RNAi screen. The percentage of rrf-3(pk1426) worms alive at day 30 is shown. Controls used were empty vector (EV), nontargeting GFP (black), novel longevity genes (gray), known longevity genes (khaki), and our top five candidate genes (blue). The red line indicates the threshold for consideration as a longevity candidate. Data represent an average of four replicates ± the SEM (Supplemental Table 2). (C) Day 9 RNAi against our top candidate genes—eri-1, dot-1.1, cyk-3, ego-1, and pha-4—extended mean treated life span (MTL). All life spans were rrf-3(pk1426). Days represent days after first egg lay. Life span statistics are in Supplemental Table 4.
Macroautophagy (referred to here as autophagy) is a conserved process that encloses cytoplasmic components and organelles in double-membrane structures called autophagesomes, which then fuse with the lysosome, where the contents are degraded and recycled (Mizushima et al. 1998). As such, autophagy is essential for proteostasis, in which long-lived, unfolded, misfolded, or damaged proteins are removed from circulation. Autophagy is required for normal development and health and is crucial for the extended life span by reduced germline, insulin, or TOR signaling (Rubinsztein et al. 2011). Autophagy is predominantly cytoprotective; however, when autophagy becomes dysregulated, it is associated with adverse effects such as cell death or failure to clear aggregated proteins, resulting in neurodegeneration (Komatsu et al. 2006). Autophagy has in fact been called a double-edged sword in that it can be detrimental in certain disease contexts, either being causal or exacerbating their pathologies (Shintani and Klionsky 2004). However, the role of autophagy in post-reproductive life span has not been investigated.
Here, we introduce a novel post-reproductive screening approach for the identification of unknown longevity genes functioning according to the AP model of aging (Fig. 1A). In our screen, we identified 30 novel regulators of longevity, including the FOXA transcription factor pha-4. Surprisingly, we found that PHA-4 inactivation increases life span through its role in autophagy via BEC-1. Importantly, the inhibition of pha-4 or bec-1 modulates life span conversely over aging, fulfilling the criteria for AP. Our data indicate that global autophagy becomes dysfunctional with age and is blocked at the later steps of autophagosomal degradation. We demonstrate that post-reproductive inhibition of the VPS-34/BEC-1/EPG-8 autophagic nucleation complex as well as its upstream regulators strongly extend C. elegans life span. Furthermore, we show that post-reproductive inhibition of bec-1 mediates longevity specifically through the neurons. Thus, in contrast to previous studies that suggest positive roles of autophagy during aging, our data indicate that suppression of early autophagy in aged worms results in improved neuronal integrity, contributing to enhanced global health and culminating in increased longevity.
Results
Screening for AP genes identifies regulators of post-reproductive longevity
To screen for potential AP genes that extend post-reproductive life span, we assembled an RNAi library targeting 800 gene regulatory factors involved in chromatin or transcriptional regulation (Supplemental). These targets were chosen because disruption of such factors is often detrimental to organismal fitness, a requirement for AP. As we inhibited these genes late in life, we were concerned that decreased feeding rates in aged worms (Huang et al. 2004) would preclude robust RNAi phenotypes. Consequently, the screen was carried out in rrf-3 mutant animals, which exhibit enhanced sensitivity to RNAi (Simmer et al. 2002). To generate the large numbers of post-reproductive and age-synchronized worms required for this screen, we developed a large-scale liquid culture system (Supplemental Fig. S1A–D). This system allowed for screening in fertile worms, excluding the potential cofounding effects of germline removal or 5-fluoro-2′-deoxyuridine (FUdR) treatment. We conducted gene inactivation in post-reproductive 9-d-old worms by sorting them into 24-well agar plates that contained the RNAi feeding library. Longevity candidates were identified by the presence of live worms at day 32, when most of the control worms were dead. We identified 36 longevity genes (5% of the library), 30 of which have never been linked previously with life span extension (Fig. 1B; Supplemental Table 2). Examining the listed phenotypes for these new longevity genes in WormBase (version WS258), we found that 19 are good AP candidates owing to phenotypes associated with lethality as well as with defects in development, reproduction, growth, and health (Supplemental Table 2). We validated the efficacy of our screen in identifying novel longevity regulators through conventional life span assays of our top five candidates (Fig. 1C). The observed extension of mean treated life span (MTL) for these candidates ranged from 12% (eri-1) to 33% (pha-4). We used the rrf-3 strain for follow-up experiments unless stated otherwise.
PHA-4 and BEC-1 exhibit AP and improve health span upon post-reproductive inhibition
We focused on PHA-4/FOXA as a prime AP candidate due to its crucial role in development (targeting >4000 genes) (Zhong et al. 2010) and because its early inactivation is known to reduce life span (Sheaffer et al. 2008). Activation of pha-4 induces autophagy and regulates longevity in diet-restricted and germline-less animals (Hansen et al. 2008; Lapierre et al. 2011). Additionally, the critical autophagy nucleation factor BEC-1 is a transcriptional target of PHA-4 (Zhong et al. 2010). In general, autophagy is associated predominantly with its positive effects on health and longevity (Rubinsztein et al. 2011). Given the strong association of pha-4 and autophagy, we determined whether autophagy could have a pleiotropic role in post-reproductive longevity.
As shown previously in young worms (Lapierre et al. 2011), we demonstrated that both key autophagy genes bec-1 and unc-51 remain regulated by PHA-4 in aged worms (Supplemental Fig. S2A). Furthermore, GFP::LGG-1 focus formation, which marks autophagosomal membranes, was strongly reduced upon pha-4 and bec-1 inactivation at day 9 (Supplemental Fig. S2B–G). By inactivating bec-1 from day 9, we tested whether PHA-4 mediates its longevity effect through BEC-1. Inhibition of bec-1 caused a 63% MTL extension, twice that of pha-4 (Fig. 2A; Supplemental Fig. S2H). As BEC-1 acts downstream from PHA-4 in the autophagic cascade (Lapierre et al. 2011), the differences in MTL extension were unanticipated. The reduced life span effect of PHA-4 inactivation could be due to its broad spectrum of target genes (Zhong et al. 2010), some of which might have detrimental effects when dysregulated. In support of this, simultaneous inactivation of pha-4 and bec-1 at day 9 caused significant reductions in bec-1 life span extension (Fig. 2B).
Figure 2.
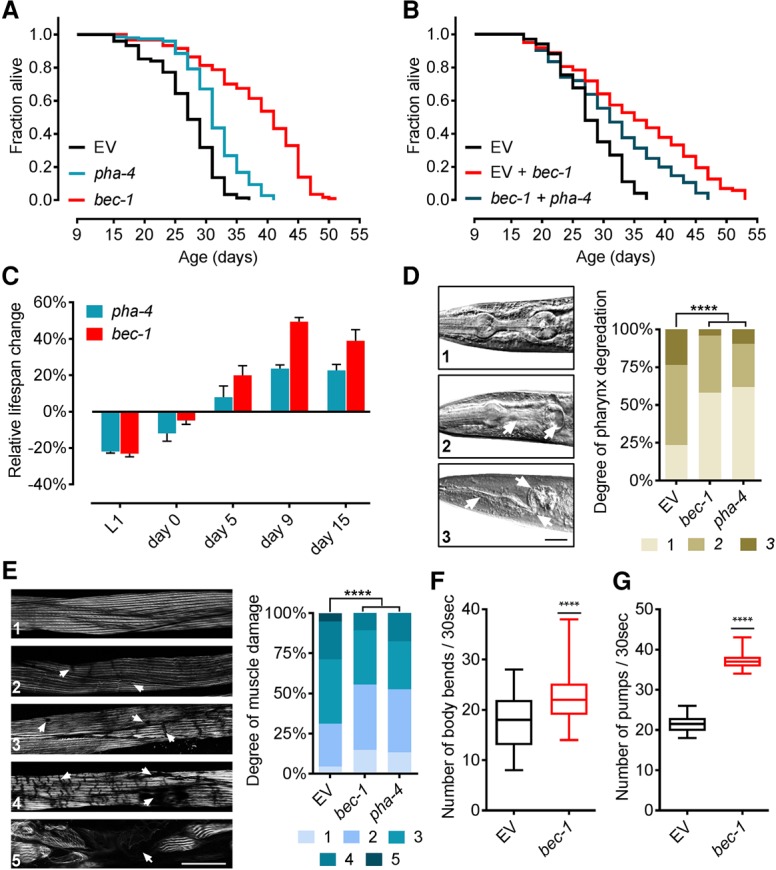
pha-4 and bec-1 extend life span in an AP manner and improve post-reproductive health span. (A) Day 9 RNAi against bec-1 extends MTL by twice that of pha-4. For knockdown validation, see Supplemental Figure S2, F and G. (B) Inactivation of pha-4 at day 9 in combination with bec-1 reduces MTL extension compared with bec-1 knockdown alone. Quantitative PCR (qPCR) validation is in Supplemental Table S12. (C) Inhibition of pha-4 and bec-1 at different times across life shows differing effects, depending on the RNAi initiation time point. Depicted is the relative percentage change in MTL ± SEM compared with control. qPCR validation is in Supplemental Table S12. (D) Day 9 RNAi-treated worms were scored at day 20 for pharynx degradation on a three-point scale. n = 90. White arrows indicate damage of the corpus, isthmus, and terminal bulb. Bar, 30 µm. Magnification, 63×. (****) P < 0.0001. (E) Day 9 RNAi-treated worms were scored at day 20 for phalloidin-stained muscle damage on a five-point scale. n = 120. White arrows indicat sites of muscle damage. Bar, 30 µm. Magnification, 63×. (****) P < 0.0001. (F) Day 20 quantification of body movement in rrf-3(pk1426) worms, as measured by body bend counts. Data are depicted as a box plot with minimum to maximum values. RNAi against bec-1 was initiated at day 9. n = 50. (****) P < 0.0001. (G) Day 20 quantification of pharynx pumping rate in rrf-3(pk1426) worms. Data are depicted as a box plot with minimum to maximum values. RNAi against bec-1 was initiated at day 9. n = 50. (****) P < 0.0001. All life spans were rrf-3(pk1426). Days represent days after first egg lay. Life span statistics are in Supplemental Tables S4 and S5.
The AP hypothesis predicts different longevity effects based on the gene inactivation time point. Consequently, we examined whether pha-4 and bec-1 knockdowns across life span exhibit matching AP effects. We observed a decrease in MTL in larvae and young adult worms, transitioning into a neutral and then positive effect during aging for both genes (Fig. 2C). We determined whether the observed life span extension of both genes translates into an improved health span. Inactivation of pha-4 or bec-1 at day 9 caused the preservation of a youthful pharynx and muscle structure (Fig. 2D,E). In addition, bec-1 knockdown at day 9 significantly improved mobility and pharynx pumping (Fig. 2F,G). These findings suggest that late-life inactivation of both genes extends life span and improves health span. These results collectively imply that pha-4 inactivation mediates life span extension through autophagy via BEC-1. As an integral autophagic component, we proceeded with BEC-1 to further investigate the role of autophagy in the observed life span extension.
Post-reproductive BEC-1 suppression mediates longevity through its canonical role in autophagy
BEC-1 exists in at least two distinct protein complexes, with either CED-9/Bcl-2 or VPS-34 and EPG-8 inhibiting caspase-mediated apoptosis or promoting autophagic nucleation, respectively (Takacs-Vellai et al. 2005; Yang and Zhang 2011). Inactivation of bec-1 triggers CED-4-dependent apoptotic cell death (Takacs-Vellai et al. 2005). Combinatorial suppression of bec-1 and ced-4 at day 9 did not alter bec-1-meditated MTL extension (Fig. 3A), suggesting an apoptosis-independent longevity mechanism. However, RNAi against either epg-8 or vps-34 at day 9 caused life span extensions similar to bec-1 knockdown (Fig. 3B). Inactivation of epg-8 or vps-34 from the first day of adulthood caused a significantly reduced life span (Supplemental Fig. S3A), implying that AP is a common feature of the VPS-34/BEC-1/EPG-8 autophagic nucleation complex.
Figure 3.
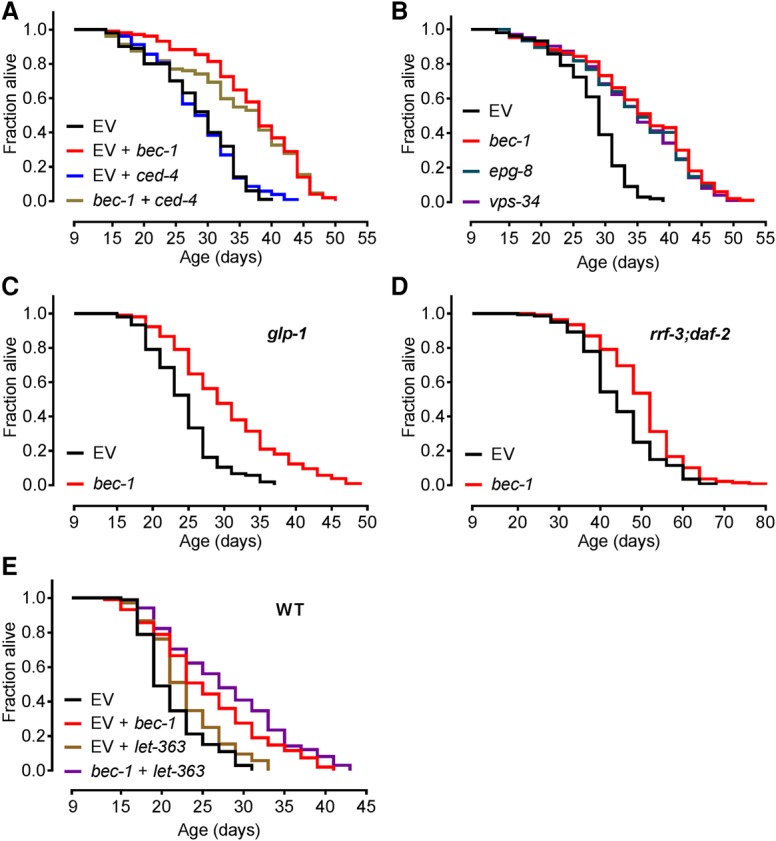
Inactivation of bec-1 extends life span through autophagy and is independent of classical longevity pathways. (A) bec-1 RNAi treatment shows no reduction in effect on MTL when combined with simultaneous ced-4 RNAi treatment at day 9 in rrf-3(pk1426) worms. qPCR validation is in Supplemental Table S12. (B) bec-1, vps-34, and epg-8 extend the MTL of rrf-3(pk1426) worms equally when inactivated at day 9. (C) Day 9 RNAi against bec-1 extends the MTL of glp-1(e2141) worms. (D) Day 9 RNAi against bec-1 extends the MTL of rrf-3(pk1426);daf-2(e1370) worms. (E) Combined inactivation of bec-1 and let-363(TOR) at day 9 in wild-type (N2) worms further extends their MTL compared with let-363 inactivation alone. qPCR validation is in Supplemental Table S12. Days represent days after first egg lay. Life span statistics are in Supplemental Tables 6 and 7.
We next investigated mitophagy as another principle form of autophagy involving BEC-1 (Palikaras et al. 2015) by inactivating either dct-1 or pink-1 at day 9. Neither condition affected MTL, indicating that mitophagy is unlikely to be involved (Supplemental Fig. S3B). Late-life reduction in autophagy via bec-1 may mediate its longevity effects through up-regulation of the ubiquitin–proteasome system (UPS), the other major protein degradation pathway. However, day 9 inactivation of bec-1 did not impact the chymotrypsin-like activity of the proteasome or the abundance of Lys48-linked polyubiquitin chains that target proteins for degradation (Supplemental Fig. S3C–E). Next, we examined whether day 9 inactivation of pha-4 or bec-1 causes unfolded protein or oxidative stress responses. Expression levels of genes involved in both stress resistance pathways were not significantly altered, which argues against their involvement in the observed longevity phenotype (Supplemental Fig. S3F,G).
Post-reproductive bec-1 inactivation further extends life span in autophagy-dependent longevity mutants
We tested whether bec-1 inhibition still exhibits an AP phenotype in worms with reduced germline, insulin, or TOR signaling. Longevity mediated via these pathways has been shown to depend on increased autophagy (Meléndez et al. 2003; Hars et al. 2007; Hansen et al. 2008; Sheaffer et al. 2008; Lapierre et al. 2011, 2013). In contrast to observations in young worms (Hansen et al. 2008; Lapierre et al. 2011), the germline-less glp-1 and insulin/IGF-1-like receptor daf-2 mutants showed a significant MTL extension upon day 9 bec-1 RNAi (Fig. 3C,D). Additionally, bec-1 inactivation in daf-2;daf-16 double mutants extended MTL (Supplemental Fig. S3H). Combinatorial inactivation of TOR/let-363 and bec-1 at day 9 also showed an additive effect on MTL in wild-type worms (Fig. 3E). Similar to reports in young animals (Mizunuma et al. 2014), day 9 RNAi against let-363 in rrf-3 mutants did not significantly extend MTL but also did not impact bec-1-mediated longevity (Supplemental Fig. S3I). Our data show a converse modulation of life span by bec-1 even in mutants whose longevity normally strictly depends on autophagy.
Inhibition of the autophagic cascade up until vesicle nucleation extends life span
After autophagy induction via the serine/threonine protein kinase UNC-51 and vesicle nucleation via the BEC-1 complex, autophagy is broken down into further steps that include vesicle expansion, lysosomal fusion, and degradation. We inactivated representative genes from each step at day 9 and examined life span effects (Fig. 4A; Supplemental Table 3; Supplemental Fig. S4A–E). Post-reproductive inhibition of autophagy genes caused a strong MTL extension up to and including the step of vesicle nucleation. After this, inhibition of the later autophagic flux did not similarly increase life span. Interestingly, day 9 inhibition of lgg-1, but not its homolog, lgg-2 (Alberti et al. 2010), significantly reduced life span (Supplemental Fig. S4C). Shortly after lgg-1 inactivation, we further detected a sick and sluggish phenotype in young as well as in old worms (data not shown). This negative phenotype was not observed upon post-reproductive inactivation of any other tested autophagy gene. Notably, this included genes that are critical for the functionality of LGG-1 in autophagy, such as ATG-4.1 (Wu et al. 2012), ATG-7 (Kim et al. 1999), and LGG-3 (Supplemental Fig. S4C; Mizushima et al. 1998). Also, lgg-1 day 9 RNAi still exerted negative effects when autophagy was independently repressed through simultaneous bec-1 inactivation (Supplemental Fig. S4F,G), which suggest critical nonautophagy functions of LGG-1. Combined, our results show that the inhibition of vesicle nucleation is integrally linked to post-reproductive longevity.
Figure 4.

Autophagy is dysfunctional in aged worms, and inhibition of its initiation increases life span. (A) Day 9 RNAi against genes involved in autophagosome regulation (I), induction (II), nucleation (III), expansion/maturation (IV), lysosomal fusion (V), and degradation (VI). Relative percentage change in MTL ± SEM compared with control is shown. Statistics are in Supplemental Table 3. Gene knockdown validation is shown in Supplemental Figure S4E. (B) Quantification of GFP::LGG-1 foci over time in hypodermal cells of rrf-3(pk1426);lgg-1p::GFP::lgg-1. Foci were quantified in young and old worms from images at 100× magnification. n = 50. Red lines show median and interquartile range. (****) P < 0.0001. (C) Representative Western blot of free GFP across life balanced to total GFP::LGG-1. The cleaved GFP bands correspond to the GFP::LGG-1 degradation products in the autolysosome. (D) Day 15 Quantification of GFP::LGG-1-positive foci in hypodermal cells of rrf-3(pk1426);lgg-1p::GFP::lgg-1 worms following day 9 RNAi knockdown. Foci were quantified from images with 100× magnification. n = 50. Red lines show median and interquartile range. (****) P < 0.0001. LGG-1 knockdown validation is shown in Supplemental Figure S4G. (E) Representative Western blot of free GFP (balanced to GFP::LGG-1) in young and old worms treated with chloroquine. The cleaved GFP bands correspond to the GFP::LGG-1 degradation products in the autolysosome. (F) Representative Western blot of day 9 RNAi against bec-1, vha-15, and lgg-1 and its effect on free GFP compared with GFP:LGG-1 measured at day 15. The cleaved GFP bands correspond to the GFP::LGG-1 degradation products in the autolysosome. (G) Day 9 inactivation of lysosomal degradation or acidification in rrf-3(pk1426) has no effect on MTL or the MTL increase mediated by bec-1 inhibition when used in combination. qPCR validation is shown in Supplemental Table S12. All Western blots used antibodies against GFP. Days represent days after first egg lay. Life span statistics are in Supplemental Tables S8 and S9.
Aged worms exhibit a dysfunctional late autophagic flux
Upon autophagosome formation, GFP::LGG-1 changes its diffuse cytoplasmatic localization to distinct foci that represent autophagic structures (Meléndez et al. 2003; Kang et al. 2007). Interestingly, we detected an age-dependent increase in these autophagic structures by quantifying GFP::LGG-1 foci in the hypodermis (Fig. 4B; Supplemental Fig. S5A,B). Next, we used Western blotting to monitor GFP::LGG-1 protein levels across aging. We observed a strong depletion in the GFP::LGG-1 protein pool with age (Supplemental Fig. S5C) that was not reflected, however, in a decreasing expression of lgg-1 mRNA (Supplemental Fig. S5D).
After fusion of the autophagosome with the lysosome, Atg8/LC3/LGG-1 reporter proteins are degraded by lysosomal hydrolases (Klionsky et al. 2016). Conversely, the coupled GFP fragment accumulates in the vacuole, as it is relatively resistant to the vacuolar enzymes (Klionsky et al. 2016). The efficiency of lysosomal degradation determines how long the free GFP moiety can be detected. Thus, the ratio of full-length protein to its cleaved reporter indicates the status of the flux (Klionsky et al. 2016). Compared with the full-length GFP::LGG-1 pool, free GFP increased from day 0 to 20 of adulthood (Fig. 4C).
Together, our data support three alternative autophagic flux states in old worms: an increased formation of autophagosomes due to enhanced autophagy induction; inefficient completion of autophagy, resulting in accumulation of autophagic structures; or a combination of both (Klionsky et al. 2016). To discriminate between these options, we monitored GFP::LGG-1 focus levels present in old worms upon day 9 inhibition of proteins involved at different stages of the autophagic flux. Inactivation of pha-4, unc-51, bec-1, and atg-7 at day 9 significantly reduced autophagosome numbers (Fig. 4D; Supplemental Fig. S6A–G). As epg-5 and cup-5 function in lysosomal fusion and degradation, respectively, their inactivation is associated with an accumulation of GFP::LGG-1 foci (Tian et al. 2010; Sun et al. 2011). In contrast, we did not observe a significant increase in GFP::LGG-1 foci upon epg-5 or cup-5 RNAi from day 9. This lack of accumulation suggests a blocked late autophagic flux, leading to inefficient autophagy completion. Next, we simulated an inefficient completion of autophagy through genetic or pharmacological perturbation of autophagic degradation by neutralizing lysosomal pH in young and old worms. In young animals, chloroquine treatment or the inactivation of the vacuolar ATPase subunit VHA-15 caused a significant increase in free GFP (Fig. 4E; Supplemental Fig. S7A). However, these treatments did not increase free GFP levels in aged worms (Fig. 4E,F). Hence, the level of free GFP in young worms with an artificially perturbed late autophagic flux mimics the intrinsic status in old worms.
To further substantiate our analysis of the autophagic flux across aging, we monitored the autophagic substrate SQST-1/p62. Blocking autophagy leads to the accumulation of the adaptor protein SQST-1, which recognizes and loads cargo into the autophagosome for degradation (Tian et al. 2010). We observed a marked increase in SQST-1 with age (Supplemental Fig. S7B), which is supported by a recent proteome analysis (Narayan et al. 2016). We did not detect a matched increase in sqst-1 mRNA expression levels (Supplemental Fig. S7C), which speaks for an accumulation on the protein level. RNAi against different autophagy genes at day 9 did not cause a marked increase in SQST-1 (Supplemental Fig. S7D). However, we observed a striking SQST-1 accumulation upon bec-1 inactivation at day 0 (Supplemental Fig. S7E). The progressive accumulation of SQST-1::GFP (Supplemental Fig. S7B) in spite of increasing numbers of autophagic structures (Fig. 4D) in aged worms corroborates a dysfunction of late-life autophagy. Transcriptional up-regulation of autophagy genes could indicate increased autophagic induction. Using a sequencing approach, we did not observe a systematic up-regulation of autophagy genes over age (Supplemental Fig. S5D), in line with proteomic observations (Walther et al. 2015). Notably, we detected an ∼20% increase in bec-1 expression across aging.
Our findings imply two possible mechanisms by which post-reproductive inhibition of autophagic vesicle nucleation could be beneficial: Inhibition either alleviates the pressure on the autophagic system to restore productive autophagy or prevents detrimental effects from the early termination of dysfunctional autophagy processes. Hence, we tested for restored productive autophagy by investigating whether bec-1-mediated life span extension requires lysosomal degradation. Simultaneous inactivation of bec-1 with either cup-5, vha-13, or vha-15 at day 9 did not affect the bec-1 longevity phenotype (Fig. 4G). While we could successfully inhibit lysosomal acidification in old worms, inactivation of vha-13 or vha-15 at day 9 had no effect on life span (Supplemental Figs. S4D, S7F,G). Conversely, the same inactivation strongly reduced the life span of young worms (Supplemental Fig. S7H). Thus, bec-1 inactivation at day 9 does not restore productive autophagy. Its AP effects most likely arise as harmful consequences of impaired late stage autophagy.
Aged wild-type worms show reduced neuronal RNAi and decreased longevity gains upon inhibition of autophagic nucleation
We also examined the effect of inhibition of autophagosome nucleation in wild-type worms. In agreement with published data (Meléndez et al. 2003; Sheaffer et al. 2008), RNAi against bec-1 or pha-4 results in a significant reduction of life span early in life (Supplemental Fig. S8A,B). Conversely, day 9 inhibition of autophagy genes in wild type resulted in life span-extending trends similar to those in the rrf-3 mutant but weaker (Supplemental Fig. S8C). bec-1, vps-34, unc-51, and atg-9 all extended life span by ∼30%, whereas pha-4 inactivation from day 9 did not significantly extend wild-type life span (Supplemental Fig. S8C). Combined, this suggests that the weaker effects on longevity in old worms compared with young worms could derive from differences in RNAi efficacy between wild-type and rrf-3 mutant worms. Loss of functional RRF-3 enhances global sensitivity to RNAi in young worms (Simmer et al. 2002). Upon day 9 RNAi against pha-4 and bec-1, we likewise observed a reduced knockdown efficiency in wild type compared with rrf-3 mutants at day 12, which equalized by day 15 (Supplemental Fig. S8D). Moreover, RNAi is particularly effective in the neurons of rrf-3 mutants compared with wild type, which are largely refractory to neuronal RNAi (Simmer et al. 2002). Using unc-119::gfp as a neuronal reporter, we confirmed that this enhanced neuronal RNAi sensitivity persists in aged rrf-3 worms (Supplemental Fig. S8E). Combined with the striking MTL differences between the two strains, we were prompted to investigate neuronal contribution to the observed longevity.
Inhibition of bec-1 extends health span and life span through neurons
We performed life span experiments in the sid-1(−);unc-119p::sid-1(+) mutant strain, which is hypersensitive to RNAi in neurons but refractory in all other tissues (Calixto et al. 2010). Intriguingly, neuron-specific inactivation of bec-1 and vps-34 at day 9 strongly extended MTL by up to 57% (Fig. 5A), whereas atg-7 did not alter life span, and lgg-1 significantly reduced MTL (Fig. 5A). We observed a marked reduction in life span upon neuron-specific bec-1 RNAi at day 0 (Supplemental Fig. S9A), which indicates a conserved neuronal AP effect for bec-1. These life span effects mirror those seen in our rrf-3 mutant experiments. Notably, inactivation of bec-1 at day 9, specifically in the germline niche, gut, muscle, or hypodermis, did not affect life span (Supplemental Fig. S9B). These results suggest that enhanced neuronal RNAi is likely the main contributor to extended longevity in rrf-3 mutants. Taken together, our observations imply that inhibition of autophagic nucleation extends life span primarily through neurons.
Figure 5.
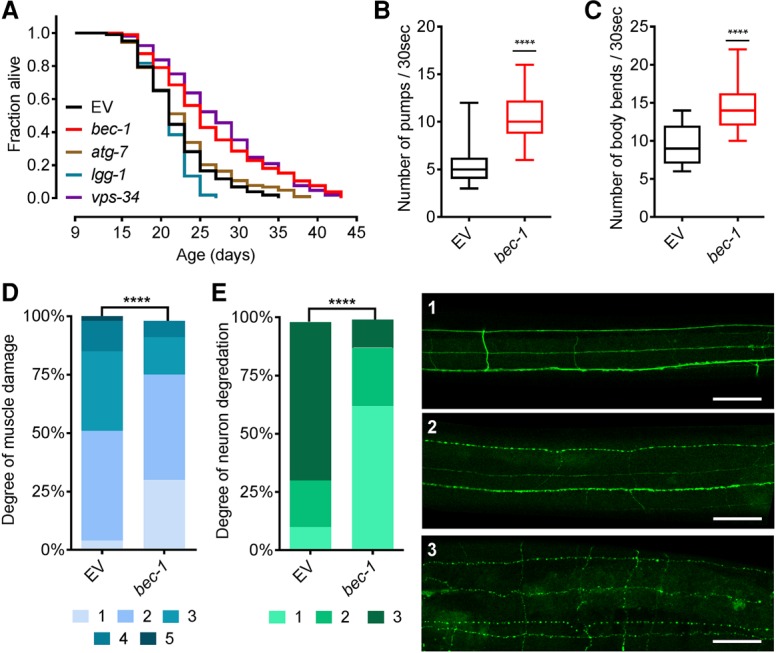
Neuronal inhibition of bec-1 mediates health and life span effects. (A) Day 9 RNAi against members of the autophagic flux extended MTL in sid-1(pk3321);unc-119p::sid-1 worms. (B) Day 20 quantification of pharynx pumping rate in sid-1(pk3321);unc-119p::sid-1 worms. Data are depicted as a box plot with minimum to maximum values. RNAi against bec-1 was initiated at day 9. n = 50. (****) P < 0.0001. (C) Quantification of worm mobility measured by body bend counts in sid-1(pk3321);unc-119p::sid-1 worms at day 20. Data are depicted as a box plot with minimun to maximum values. RNAi against bec-1 was initiated at day 9. n = 50. (****) P < 0.0001. (D) Day 20 quantification of phalloidin-stained muscle damage following day 9 RNAi against bec-1 in sid-1(pk3321);unc-119p::sid-1 worms. The scoring system is the same as used in Figure 2D. n = 100. (****) P < 0.0001. (E) Day 20 quantification of longitudinal neuron morphology and integrity in unc-119p::GFP;rrf-3(pk1426) worms treated with RNAi against bec-1 at day 9. Neurons were scored on a three-point scale based on the amount of visible bubbling and neuron integrity. Neurons were quantified from images with 100× magnification. n = 100. Bar, 40 µm. (****) P < 0.0001. Days represent days after first egg lay. Life span statistics are in Supplemental Table 11.
Next, we examined whether neuron-specific inactivation of the nucleation complex via bec-1 accounts for the improvement in global health. Day 9 inactivation of bec-1 in the neurons promoted a youthful muscle structure, improved mobility, and an increased pumping rate (Fig. 5B–D). However, the same inactivation solely in muscle tissue failed to generate similar improvements (Supplemental Fig. S9C–E). Additionally, we examined whether inhibition of the early flux through bec-1 would elicit changes in neuronal integrity. Intriguingly, bec-1 inhibition at day 9 resulted in more youthful neurons with a significant reduction in axonal blebbing and fragmentation (Fig. 5E; Supplemental Fig. S9F,G). Overall, our data suggest that neuroprotective effects of bec-1 inactivation mediate an improved muscle function and integrity.
Discussion
Novel screening approach for AP in post-reproductive C. elegans
Prevalent age-associated disorders such as cardiovascular diseases, cancer, and neurodegenerative diseases have a strong genetic contribution (Kulminski et al. 2016). With an increasingly aging population, disease prevention as opposed to disease treatment of the elderly is a promising approach for future medicine. However, such preventive therapies require an improved understanding of the genetics of aging. Based on the AP theory of aging, our study illustrates that genetic observations made in young organisms cannot easily be translated into an aged context. In contrast, here we show that key autophagy genes required for normal life span in young worms antagonistically shorten life in post-reproductive worms. In line with this finding, recent human genome-wide association studies showed that disease risk associated with a given allele is highly sensitive to chronological age (Kulminski et al. 2016; Rodríguez et al. 2017). However, high-throughput screens for genes affecting life span in C. elegans largely ignored the impact of chronological age, as they were carried out in young worms (Klass 1983; Ni and Lee 2010). To the best of our knowledge, we present here the first post-reproductive RNAi screen for genes that behave according to the AP theory of aging. This novel approach led to the identification of 30 previously unknown longevity genes that increase life span upon post-reproductive inactivation. These findings indicate that AP is a common genetic mechanism that modulates health and life span in an age-dependent manner.
Dysfunctional autophagy in aged worms
Our data provide evidence for active and possibly enhanced autophagic induction in old worms, resulting in accumulation of autophagic foci that are not cleared, likely due to a blockage in the late flux. This is in line with observations in hepatocytes of aging mice and rats (Terman 1995; Del Roso et al. 2003). We observed an age-related increase in free GFP deriving from the autophagy reporter protein GFP::LGG-1. Free GFP is generated through autolysosomal degradation of GFP::LGG-1/LC3 but accumulates only when the autophagic flux is increased or blocked. To discriminate between these two options, we assessed GFP::LGG-1 cleavage in the presence or absence of genetic and pharmacological inhibition of autolysosomal degradation. If the age-related accumulation of free GFP would emanate from a greater flux, autolysosomal inhibition should result in a further increase of the fluorescent moiety. In contrast, the disruption of autolysosomal acidity through the inhibition of vacuolar ATPase subunit VHA-15 or chloroquine treatment did not alter free GFP levels in aged worms (Fig. 4E,F). These results indicate increasingly dysfunctional rather than enhanced autolysosomal degradation with age. The accumulation of the autophagy receptor SQST-1 in old worms harkens back to studies that showed widespread protein accumulation in aged C. elegans (David et al. 2010; Walther et al. 2015) and implicates ineffective late-life autophagy. We also observed that genes crucially involved in autophagy degradation are dispensable late in life and have no effects on longevity (Supplemental Fig. S4D), which further argues for a dysfunctional autophagic process in aged worms. Moreover, the significant reduction of autophagic structures upon day 9 inactivation of the early autophagic flux (Fig. 4D; Supplemental Fig. S6A–G) indicates continuing vesicle nucleation with age.
Autophagy represents a quality control system that targets misfolded proteins and thereby prevents their toxic intracellular accumulation. Inhibition of the system in mice leads to the accumulation of toxic protein aggregates and cellular degeneration of cardiomyocytes and neurons (Komatsu et al. 2006; Nakai et al. 2007). As such, autophagy is a major factor that delays aging (Rubinsztein et al. 2011). During aging, organisms are increasingly challenged with large protein aggregates that are poor proteasomal substrates (David et al. 2010; Walther et al. 2015). Thus, one would expect that autophagic activity increases with age. However, many studies detected decreasing autophagic activity during aging (Rubinsztein et al. 2011). It is still unclear how worms could compensate for a dysfunctional autophagic recycling machinery in old age. Proteomic data across life span show an increasing abundance of proteasome components with age (Walther et al. 2015), suggesting that an up-regulation of the UPS could be one possibile way that aged animals maintain proteostasis. However, we did not detect further up-regulation of the UPS activity upon bec-1 inactivation from day 9 (Supplemental Fig. S3D,E), which could indicate that this alternative proteostasis pathway is already compensating for dysfunctional autophagy in old worms.
Autophagosome nucleation exhibits AP
Through downstream analysis of our screening hit, pha-4, we identified that the observed longevity effects are mediated through inhibition of the key autophagy gene bec-1. Next, we found that BEC-1 mediates its longevity effects through the VPS-34/BEC-1/EPG-8 autophagic nucleation complex rather than through its interaction with the anti-apoptotic protein CED-9/Bcl-2. Notably, BEC-1 and VPS-34 have also been shown to function together in endocytosis (Ruck et al. 2011). However, EPG-8 is not acting in this process (Yang and Zhang 2011), arguing against an involvement of endocytosis in the observed longevity. Hence, our data suggest that post-reproductive inactivation of bec-1 mediated its longevity through its role in autophagy. Surprisingly, day 9 inactivation of bec-1 also extends life span in long-lived worms with reduced germline, insulin, or TOR signaling whose longevity was linked previously to enhanced autophagy (Meléndez et al. 2003; Hars et al. 2007; Hansen et al. 2008; Sheaffer et al. 2008; Lapierre et al. 2011, 2013). Our findings do not contradict these reports, as they studied longevity effects of autophagy in young worms. Rather, our data suggest that, even in the presence of life span-extending mutations, autophagy eventually becomes dysfunctional at old age, altering the process from beneficial to harmful.
Autophagy genes involved in steps subsequent to vesicle nucleation show minor or no longevity effects upon day 9 inactivation even if their inhibition also reduces autophagosome numbers. This is illustrated by ATG-7, which acts immediately downstream from vesicle nucleation and is needed for autophagic vesicle elongation (Kim et al. 1999). Similar to results in young worms (Kang et al. 2007), we observed significantly reduced GFP::LGG-1 focus numbers upon day 9 inactivation of ATG-7 (Fig. 4D). However, day 9 RNAi against atg-7 had only minor effects on life span. Similarly, day 9 inactivation of LGG-1, which by itself is critical for vesicle expansion and completion (Meléndez et al. 2003), did not positively affect C. elegans life span. Interestingly, lgg-1 is the only autophagy gene tested that shows negative effects after day 9. This is potentially due to the roles of LC3/LGG-1 outside autophagy, such as in the endoplasmic reticulum (ER)-associated degradation (ERAD) tuning process (De Haan et al. 2010). As such, there seems to be a decoupling of autophagosome elongation and longevity. Our data indicate that autophagosome numbers are presumably not critical for life span extension but rather that it is important at which stage the flux is inhibited. This suggests that, in the context of age-related autophagic dysfunction, the vesicle nucleation event causes harmful life span-shortening effects.
Notably, post-reproductive inactivation of pha-4 resulted in weaker life span extension than observed for bec-1 RNAi and also reduced bec-1 longevity upon combinatorial inactivation from day 9 (Fig. 2A,B). Besides its role as an upstream transcriptional regulator of bec-1, the transcription factor PHA-4 also targets gene classes that are involved in metabolic processes and defense responses (Zhong et al. 2010). In addition to reducing autophagy, one would expect that post-reproductive inactivation of PHA-4 results in a dysregulation of its nonautophagy target genes. This likely explains the reduced longevity of pha-4 compared with bec-1 RNAi as well as its subtractive life span effect when corepressed with bec-1.
Inhibition of the autophagy nucleation complex extends life span through neurons
Dysfunctional autophagy has been linked to neuronal degeneration in cell culture as well as in animal models of neurodegenerative diseases (Nixon 2013). Similarly, accumulation of dysfunctional autophagic structures were observed in human Alzheimer's, Parkinson's, and Huntington's disease patients (Nixon 2013). Interestingly, the late autophagic flux rather than the process of autophagosome formation has been frequently reported to be impaired in these neurodegenerative diseases. However, the exact mechanistic link between impairment of the late autophagic flux and neurodegeneration is still a matter of debate and seems to largely depend on the disease context (Nixon 2013). Proposed mechanisms that link late flux dysfunction to neuronal damage include defective retrograde transport, harmful accumulation of autophagosomes and autolysosomes, ineffective organelle clearance, increased reactive oxygen species production, incomplete degradation as a source of cytotoxic products, and cell death through leakage of lysosomal enzymes (Nixon 2013). Interestingly, the inhibition of autophagosome formation has been shown to ameliorate neurodegeneration under disease conditions that show a pathologic accumulation of autophagic vesicles (Yang et al. 2007; Lee and Gao 2009). We demonstrate that neuron-specific inactivation of autophagic flux genes recapitulated the longevity effects seen through their global inactivation, implying that autophagy in the nervous system is as impaired as in the entire organism. Further research is still needed to establish the specific autophagy status in aged C. elegans neurons. Our study suggests that age-associated dysfunctional autophagy causes neurodegeneration and shortens organismal life span. Here we provide the first evidence that inhibition of autophagic vesicle nucleation ameliorates neuronal decline during aging. Furthermore, we show that neuron-specific inactivation of the vesicle nucleation complex through bec-1 is sufficient to extend health and life span in aging C. elegans.
An age-related decline in proteostasis has been associated with the accumulation of misfolded and aggregated proteins that are widely implicated in neurodegeneration (Nixon 2013). When autophagy is dysfunctional in old age, neurons may largely depend on other mechanisms to reduce the cytotoxicity of these aggregates, such as protein sequestration (Arrasate et al. 2004; Walther et al. 2015) or their removal via exophere formation (Melentijevic et al. 2017). Along these lines, ongoing vesicle nucleation that feeds into a dysfunctional autophagic flux may interfere with these neuroprotective pathways (Arrasate et al. 2004; Melentijevic et al. 2017) and thereby aggravate neuronal decline with advancing age. However, the potential involvement of these sequestration pathways in the longevity phenotype described here still remains to be investigated. Also, we cannot exclude unknown life span-shortening signaling events emanating from the autophagic vesicle nucleation complex in late life.
Our study shows that autophagy behaves in an antagonistic manner over life. Specifically, we show that autophagy switches its role from beneficial to detrimental within neurons. Consequently, neuronal inhibition of the autophagic nucleation complex reduces neurodegeneration, ameliorates sarcopenia, and strongly increases life span (Fig. 6). These core findings are of immediate relevance, as they will require a shift in our understanding of the genetic regulation of longevity and specifically autophagy regarding its role in health and aging.
Figure 6.
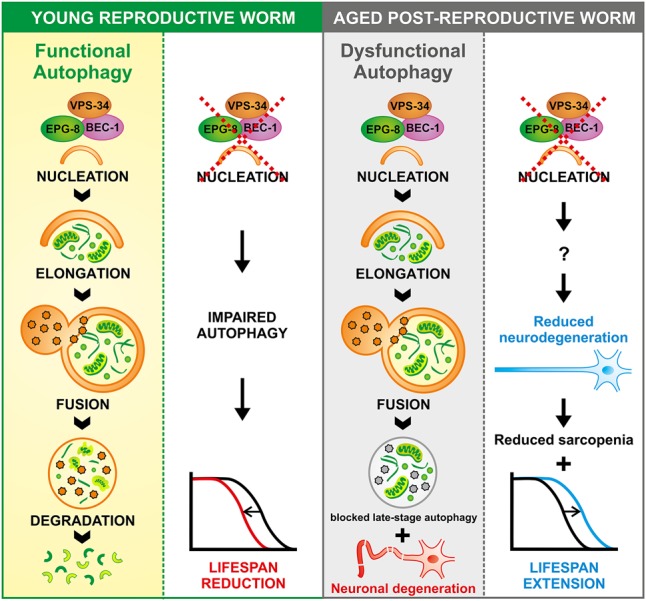
Model depicting age-associated antagonistic effects of autophagy on longevity. (Left panel) Young worms have functional autophagy, and inhibition of vesicle nucleation in these young worms shortens life span. (Right panel) Old worms still have effective autophagy induction but exhibit an age-related dysfunction in autophagic degradation. This is accompanied by age-associated neurodegeneration. Inhibition of autophagic nucleation in old worms through an unknown mechanism leads to improved neuronal integrity and an accompanying decrease in sarcopenia. This improvement in health acts either causally or in concert with an increase in post-reproductive life span.
Materials and methods
C. elegans strains
C. elegans strains were maintained at 20°C using standard procedures (Brenner 1974) unless indicated differently. A complete list of the strains used in this study is in the Supplemental Material.
RNAi screen
Eight-hundred dsRNA-expressing HT115 Escherichia coli bacteria specific to C. elegans gene regulatory factors was prepared from the Ahringer and Vidal libraries (Supplemental Table 1). All clones were sequence-validated using the M13 forward primer. The library was grown overnight in 2× YT medium in deep 96-well plates and seeded onto 24-well NGM agar plates with 100 µg/mL ampicillin and 1 mM β-D-isothiogalactopyranoside (IPTG) at 2× native density. A minimum of four replicate wells per RNAi clone was seeded. Nontargeting controls were comprised of four plates of both EV and gfp RNAi. gfp RNAi was a kind gift from Scott Kennedy. rrf-3 mutant worms were synchronized via liquid culture until day 9, cleaned, and sorted with the COPAS Biosorter with 20 worms per well. Live/dead scoring of all wells was performed on day 32, when most control animals were dead. Scoring was performed manually by flushing each well with M9 buffer and immediately counting moving and dead worms. Only worms that could be identified as live or dead were scored; missing worms were not scored. Contaminated wells or offspring-containing wells were not scored; combined, these conditions represented ∼10% of all wells.
Life span assays
Worms were synchronized using liquid culture sedimentation. dsRNA-expressing bacteria were grown overnight, seeded on NGM agar plates with 100 µg/mL ampicillin and 1 mM IPTG, and incubated overnight at room temperature. Day 0 was determined by the appearance of first internal eggs. A total of 105–140 animals was placed on three to four replicate plates, with 35 worms per 6-cm plate. Worms were picked to new plates and scored every 2 d. For double-RNAi treatment, bacteria were diluted in a 1:1 ratio before being plated at 1× native density. All assays were performed at 20°C. For the temperature-sensitive glp-1 mutant, worms were maintained at the restrictive temperature of 25°C until day 0, when they were transferred to 20°C. In the life span assays indicated in Supplemental Table S4 as “plate only,” worms were maintained and synchronized only on plates. Worms were scored as alive until there was no movement after repeated prodding with an eyelash. Worms were censored when they crawled off the plate, were bagged, burst, were dropped on transfer, were contaminated, and burrowed. All RNAi clones used for life span assays were from the Ahringer libary and were sequence-validated using the M13 forward primer.
LGG-1::GFP microscopy
Worms were grown as described and transferred to the respective RNAi treatments at day 9. On the day of analysis, 100 worms from each knockdown condition were paralyzed using 0.5% NaAz and mounted on a 2% agar pad on a glass microscope slide. GFP signals were acquired using a STED superresolution microscope (Leica) at 100× magnifications. Images were taken of the hypodermis, which was identified by locating the plane between the muscle and cuticle. At least 60 total regions were imaged from 50 different worms per replicate with two biological replicates. All images used for comparative quantifications were taken on the same day with the same settings and by the same user.
Pharynx imaging and analysis
Worms were grown as described and transferred to the respective RNAi treatments at day 9. On day 20, 50 age-synchronized worms from each knockdown condition were paralyzed using 0.5% NaAz and mounted on a 2% agar pad on a glass microscope slide. The images of the pharynges were acquired on a SP5 microscope (Leica) using the differential intense contrast (DIC) filter and a 63×/1.4 NA oil immersion objective. At least 30 whole worms per condition were imaged in each replicate with three technical replicates. Images were scored blind for pharynx degradation based on a three-point scale of damage related to changes in the structure of the corpus, isthmus, or terminal bulb. Worms with no obvious damage to any of the three parts were scored as 1, worms with damage to only one or two parts were scored as 2, and worms with damage to all three parts were scored as 3. For comparative analysis, all worms were imaged at the same time with the same settings and by the same user. This experiment was carried out in two independent biological replicates.
Muscle cell imaging and analysis
Worms were grown as described and transferred to the respective RNAi treatments at day 9. On day 20, 30 age-synchronized worms from each knockdown condition were incubated in fixation buffer (160 mM KCl, 100 mM Tris HCl at pH 7.4, 40 mM NaCl, 20 mM Na2EGTA, 1 mM EDTA, 10 mM spermidine HCl, 30 mM Pipes at pH 7.4, 1% Triton X-100, 50% methanol) for 1 h at room temperature with rotation. Worms were washed twice with PBS and incubated in a 1:200 dilution of Phalloidin–Atto 565 (Sigma) in PBS–0.5% Triton X-100 for 4 h at room temperature with rotation. Worms were then mounted on a 2% agar pad on a glass microscope slide. The 561-nm signals were acquired using a STED CW superresolution microscope (Leica) and a 63×/1.4 NA oil immersion objective. At least 40 images comprising two to four cells from 20 different worms were imaged per replicate with three technical replicates. Images were scored blind based on a five-point scale of muscle fiber degradation. Cells showing no obvious degradation were scored as 1; cells with kinks or striations were scored as 2; cells with small lesions combined with striations or other damage were scored as 3; cells with muscle fiber breaks, gross striations, and lesions were scored as 4; and cells in which the muscle fibers were no longer intact were scored as 5. For comparative analysis, all worms were imaged at the same time with the same settings and by the same user. This experiment was carried out in two independent biological replicates.
Pharynx pumping assay
Worms were grown in liquid culture and transferred to the respective RNAi treatments at day 9. On day 20, 50 age-synchronized worms from each knockdown condition were transferred individually to an agar plate seeded with a bacterial lawn. The number of contractions in the terminal bulb of the pharynx was scored during a 30-sec period immediately upon transfer using a stereomicroscope. This experiment was carried out in two independent biological replicates.
Movement scoring/thrashing assay
Worms were grown as described and transferred to the respective RNAi treatments at day 9. On day 20, 50 age-synchronized worms from each knockdown condition were transferred individually in a 20-µL drop M9 buffer on a Petri dish. After a 30-sec recovery period, the numbers of body bends were scored during a 30-sec period using a stereomicroscope. A body bend was defined as a change in the reciprocating motion of bending at the mid-body. This experiment was carried out in two independent biological replicates.
Imaging and analysis of the axonal network
Worms were grown as described and transferred to the respective RNAi treatments at day 9. On day 20, >100 age-synchronized worms from each knockdown condition were paralyzed using 0.5% NaAz and mounted on a 2% agar pad on a glass microscope slide. GFP signal was acquired using a STED superresolution microscope (Leica) at magnifications of 40×/1.2 NA oil immersion objective. Head regions of at least 50 different worms were imaged per knockdown condition and replicate. Axonal degeneration was scored on a three-point scale based on the amount of visible bubbling and neuron integrity. An intact axon without visible bubbling, kinks, or gaps was type 1; a slightly damaged axon with a medium amount of bubbling, occasional kinks, or very few to no gaps was type 2; and a largely disrupted axon with a high amount of bubbling and frequent gaps was type 3. All images used for comparative quantifications were taken on the same day with the same settings and by the same user. This experiment was carried out in two independent biological replicates.
Western blotting
Worms were grown as described and transferred to the respective RNAi treatments at day 9. For each time point or knockdown condition, samples from 200 synchronized animals were harvested, washed three times with M9 buffer and once with ddH2O, and suspended in 2× standard Laemmli buffer via 10 min of boiling. The samples were subjected to standard SDS-PAGE and Western blotting. The antibodies used were monoclonal mouse anti-GFP (1:10,000; Roche), mouse monoclonal (6G6) anti-RFP (1:1000; Chromotek), mouse monoclonal anti-α-Tubulin (1:10,000; Sigma), and rabbit monoclonal anti-ubiquitin (Lys48-specific Apu2 clone; 1:1000; Millipore). Antibodies were diluted in PBST (5% milk powder, 0.1% Tween20), which was also used as a blocking agent. For the time-course analysis of cleaved GFP, worms were maintained in liquid culture as described, and a portion of the culture was harvested at each time point following a 40% Percoll wash. This wash was repeated in the event that the cleaned worms contained <95% living worms. Each Western blot is representative of similar results obtained in two independent biological replicates.
Chloroquine treatment
To pharmacologically inhibit the lysosome, animals were treated with chloroquine diphosphate salt (Sigma-Aldrich). Briefly, worms were grown as described in liquid culture until day 0 or day 14 and subsequently incubated with 20 mM chloroquine or DMSO vehicle control for 24 h. The drug treatment was performed in M9 liquid medium supplemented with EV HT115 bacteria at a density of 3 × 109 cells per milliliter at 20°C while shaking. The worms were harvested at day 15 following a 40% Percoll wash. This wash was repeated in the event that the cleaned worms contained <95% living worms.
Statistics
All statistical analysis was performed with Graph Prism 6. P-values for life span curves were calculated using the log-rank (Mantel-Cox) test. MTL was calculated as the mean remaining life span of the worms from the day of first treatment. P-values for quantification of GFP::LGG-1 foci were calculated using the nonparametric Mann-Whitney U-test. P-values for muscle health, pharynx integrity, and neuronal integrity were calculated using a χ2 test with two tails and 95% confidence interval. P-values for quantitative PCRs (qPCRs), body bend counts, pharynx pumping, and chymotrypsin assay were calculated using a two-tailed t-test. Significance was scored as follows for all experiments: P < 0.0001 (****), P < 0.001 (***), P < 0.01 (**), and P < 0.05 (*).
Supplementary Material
Acknowledgments
We thank the Institute of Molecular Biology Core Facilities for their support, especially the Media Laborarory and the Genomics, the Microscopy, and the Bioinformatics Core Facilities. Particular thanks to Kolja Becker for designing the worm count program for life span assays. We thank Alicia Meléndez (Queens College, City University of New York) for providing us with the BEC-1::RFP reporter strain, and Scott Kennedy (Harvard Medical School) for providing bacteria expressing dsRNA constructs targeting GFP. Some strains were provided by the Caenorhabditis Genetics Center, which is funded by National Institutes of Health Office of Research Infrastructure Programs (P40 OD010440). We thank BioGraphix (http://www.biographix.cz) for the graphical abstract in Figure 6. This work was supported by the Boehringer Ingelheim Foundation and a Marie Curie Reintegration grant (321683, call FP7-PEOPLE-2012-CIG). The Large Particle Sorter (Biosorter, Union Biometrica) used for our large-scale RNAi longevity screen was financed through the Deutsche Forschungsgemeinschaft Major Research Instrumentation Program (INST 247/768-1 FUGG).
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.301648.117.
References
- Alberti A, Michelet X, Djeddi A, Legouis R. 2010. The autophagosomal protein LGG-2 acts synergistically with LGG-1 in dauer formation and longevity in C. elegans. Autophagy 6: 622–633. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. 2004. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431: 805–810. [DOI] [PubMed] [Google Scholar]
- Brenner S. 1974. The genetics of Caenorhabditis elegans. Genetics 77: 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calixto A, Chelur D, Topalidou I, Chen X, Chalfie M. 2010. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods 7: 554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Pan KZ, Palter JE, Kapahi P. 2007. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell 6: 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran SP, Ruvkun G. 2007. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3: 0479–0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David DC, Ollikainen N, Trinidad JC, Cary MP, Burlingame AL, Kenyon C. 2010. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol 8: 47–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haan CAM, Molinari M, Reggiori F. 2010. Autophagy-independent LC3 function in vesicular traffic. Autophagy 6: 994–996. [DOI] [PubMed] [Google Scholar]
- Del Roso A, Vittorini S, Cavallini G, Donati A, Gori Z, Masini M, Pollera M, Bergamini E. 2003. Ageing-related changes in the in vivo function of rat liver macroautophagy and proteolysis. Exp Gerontol 38: 519–527. [DOI] [PubMed] [Google Scholar]
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. 2008. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 4: e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hars ES, Qi H, Ryazanov AG, Jin S, Cai L, Hu C, Liu LF. 2007. Autophagy regulates ageing in C. elegans. Autophagy 3: 93–95. [DOI] [PubMed] [Google Scholar]
- Huang C, Xiong C, Kornfeld K, Gordon JI. 2004. Measurements of age-related changes of physiological processes that predict lifespan of Caenorhabditis elegans. Proc Natl Acad Sci 101: 8084–8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang C, You NJ, Avery L. 2007. Dual roles of autophagy in the survival of Caenorhabditis elegans during starvation. Genes Dev 21: 2161–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Dalton VM, Eggerton KP, Scott SV, Klionsky DJ. 1999. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol Biol Cell 10: 1337–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klass MR. 1983. A method for the isolation of longevity mutants in the nematode Caenorhabditis elegans and initial results. Mech Ageing Dev 22: 279–286. [DOI] [PubMed] [Google Scholar]
- Klionsky D, Agholme L, Agnello M, Agostinis P, Aguirre-ghiso JA, Ahn HJ, Ait-mohamed O, Brown EJ, Brumell JH, Brunetti-pierri N, et al. 2016. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8: 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. 2006. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: 880–884. [DOI] [PubMed] [Google Scholar]
- Kulminski AM, He L, Culminskaya I, Loika Y, Kernogitski Y, Arbeev KG, Loiko E, Arbeeva L, Bagley O, Duan M, et al. 2016. Pleiotropic associations of allelic variants in a 2q22 region with risks of major human diseases and mortality. PLoS Genet 12: e1006314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Gelino S, Meléndez A, Hansen M. 2011. Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr Biol 21: 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu C-C, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, et al. 2013. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun 4: 2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-A, Gao F-B. 2009. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J Neurosci 29: 8506–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meléndez A, Tallóczy Z, Seaman M, Eskelinen E-L, Hall DH, Levine B. 2003. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391. [DOI] [PubMed] [Google Scholar]
- Melentijevic I, Toth ML, Meghan AL, Guasp RJ, Harinath G, Nguyen KC, Taub T, Parker AJ, Neri C, Gabel CV, et al. 2017. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542: 367–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizunuma M, Neumann-Haefelin E, Moroz N, Li Y, Blackwell TK. 2014. mTORC2–SGK-1 acts in two environmentally responsive pathways with opposing effects on longevity. Aging Cell 13: 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, Klionsky DJ, Ohsumi M, Ohsumi Y. 1998. A protein conjugation system essential for autophagy. Nature 395: 395–398. [DOI] [PubMed] [Google Scholar]
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. 2007. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13: 619–624. [DOI] [PubMed] [Google Scholar]
- Narayan V, Ly T, Pourkarimi E, Murillo AB, Gartner A, Lamond AI, Kenyon C. 2016. Deep proteome analysis identifies age-related processes in C. elegans. Cell Syst 3: 144–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, Lee SS. 2010. RNAi screens to identify components of gene networks that modulate aging in Caenorhabditis elegans. Briefings Funct Genomics Proteomics 9: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA. 2013. The role of autophagy in neurodegenerative disease. Nat Med 19: 983–997. [DOI] [PubMed] [Google Scholar]
- Palikaras K, Lionaki E, Tavernarakis N. 2015. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521: 525–528. [DOI] [PubMed] [Google Scholar]
- Rodríguez JA, Marigorta UM, Hughes DA, Spataro N, Bosch E, Navarro A. 2017. Antagonistic pleiotropy and mutation accumulation influence human senescence and disease. Nat Ecol Evol 1: 55. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Mariño G, Kroemer G. 2011. Autophagy and aging. Cell 146: 682–695. [DOI] [PubMed] [Google Scholar]
- Ruck A, Attonito J, Garces KT, Núnez L, Palmisano NJ, Rubel Z, Bai Z, Nguyen KCQ, Sun L, Grant BD, et al. 2011. The Atg6/Vps30/Beclin 1 ortholog BEC-1 mediates endocytic retrograde transport in addition to autophagy in C. elegans. Autophagy 7: 386–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheaffer KL, Updike DL, Mango SE, Friedman JR, Kaestner KH, Panowski SH, Wolff S, Aguilaniu H, Durieux J, Dillin A, et al. 2008. The Target of Rapamycin pathway antagonizes pha-4/FoxA to control development and aging. Curr Biol 18: 1355–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ. 2004. Autophagy in health and disease: a double-edged sword. Science 306: 990–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RHA. 2002. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12: 1317–1319. [DOI] [PubMed] [Google Scholar]
- Sun T, Wang X, Lu Q, Ren H, Zhang H. 2011. CUP-5, the C. elegans ortholog of the mammalian lysosomal channel protein MLN1/TRPML1, is required for proteolytic degradation in autolysosomes. Autophagy 7: 1308–1315. [DOI] [PubMed] [Google Scholar]
- Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Müller F. 2005. Inactivation of the autophagy Gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol 15: 1513–1517. [DOI] [PubMed] [Google Scholar]
- Terman A. 1995. The effect of age on formation and elimination of autophagic vacuoles in mouse hepatocytes. Gerontology 41: 319–326. [DOI] [PubMed] [Google Scholar]
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. 2010. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 141: 1042–1055. [DOI] [PubMed] [Google Scholar]
- Walther DM, Kasturi P, Zheng M, Pinkert S, Vecchi G, Ciryam P, Morimoto RI, Dobson CM, Vendruscolo M, Mann M, et al. 2015. Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161: 919–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams GC. 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: 398–411. [Google Scholar]
- Wu F, Li Y, Wang F, Noda NN, Zhang H. 2012. Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. J Biol Chem 287: 29457–29467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Zhang H. 2011. The coiled-coil domain protein EPG-8 plays an essential role in the autophagy pathway in C. elegans. Autophagy 7: 159–165. [DOI] [PubMed] [Google Scholar]
- Yang Y, Fukui K, Koike T, Zheng X. 2007. Induction of autophagy in neurite degeneration of mouse superior cervical ganglion neurons. Eur J Neurosci 26: 2979–2988. [DOI] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HYK, Preston E, et al. 2010. Genome-wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA-4/FOXA in development and environmental response. PLoS Genet 6: e1000848. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.