

Abstract
Obtaining accurate drug response data in large cohorts of cancer patients is very challenging; thus, most cancer pharmacogenomics discovery is conducted in preclinical studies, typically using cell lines and mouse models. However, these platforms suffer from serious limitations, including small sample sizes. Here, we have developed a novel computational method that allows us to impute drug response in very large clinical cancer genomics data sets, such as The Cancer Genome Atlas (TCGA). The approach works by creating statistical models relating gene expression to drug response in large panels of cancer cell lines and applying these models to tumor gene expression data in the clinical data sets (e.g., TCGA). This yields an imputed drug response for every drug in each patient. These imputed drug response data are then associated with somatic genetic variants measured in the clinical cohort, such as copy number changes or mutations in protein coding genes. These analyses recapitulated drug associations for known clinically actionable somatic genetic alterations and identified new predictive biomarkers for existing drugs.
Precision cancer medicine has yielded some spectacular successes. For example, the use of tyrosine kinase inhibitors in BCR-ABL1–positive chronic myeloid leukemia (CML) has transformed the treatment of a previously lethal disease. However, such successes have been isolated, for example, according to OncoKB (Chakravarty et al. 2016), as of April 2017 there are only 12 cancer genes with FDA-approved drug treatments (Relling and Evans 2015). Thus, there is an urgent need to develop new methods and expand this list.
High-throughput sequencing technologies are now being applied to every field of biology. Many studies have applied these technologies to cancer; one of the largest to date is The Cancer Genome Atlas (TCGA) (The Cancer Genome Atlas Research Network et al. 2013), which has characterized more than 10,000 primary tumors. TCGA and similar studies have elucidated many previously unknown aspects of tumor biology, particularly by uncovering driver mutations (Tamborero et al. 2013) and reclassifying and creating subtypes of cancers using molecular data (Hoadley et al. 2014). However, because of the difficulty in collecting drug response data in large patient cohorts, these data have not been used extensively for discovering new drug biomarkers or in supporting precision medicine. Drug screening against patients raises serious logistical and ethical issues. Altering chemotherapeutic regimes can result in patients no longer receiving optimal therapy. Hence, precisely measuring drug response using randomized trials in clinical cohorts is not typically possible on a very large scale. This limits the ability to identify predictors of drug response when many potential markers are screened (e.g., a genome-wide screen) (Geeleher et al. 2014b, 2016a; Gray and Mills 2015), and severely limits our ability to discover novel drug biomarkers directly in cancer patients.
Alternatively, collecting drug response information in preclinical disease models such as cancer cell lines is much more straightforward. Similar to TCGA, the molecular characteristics of cancer cell lines have been cataloged using high-throughput sequencing technologies. The largest studies have been the Genomics of Drug Sensitivity in Cancer (GDSC) (Garnett et al. 2012), the Cancer Cell Line Encyclopedia (CCLE) (Barretina et al. 2012), and the Cancer Therapeutics Response Portal (CTRP) (Seashore-Ludlow et al. 2015). These have assayed nearly 1000 cancer cell lines with multiple genomics technologies. Crucially, they also screened these cell lines with hundreds of anti-cancer agents, thus collecting drug response information. However, the sample sizes available in these types of preclinical studies still lag far behind clinical studies such as TCGA.
Here, we propose a conceptually novel methodology that allows us to use clinical cancer sequencing data sets (e.g., TCGA) for pharmacogenomics discovery, without having to collect drug response information in patients. Our new approach implements a machine learning–based approach, similar to the method described by Geeleher et al. (2014b), where gene expression–based predictive models of drug response were constructed from nearly 1000 cancer cell lines from GDSC; these models were then applied to gene expression values from over 10,000 TCGA tumor samples, yielding an imputed drug response value in each TCGA sample, for each of 138 drugs. This allowed us to use TCGA to directly study pharmacogenomics on an unprecedented scale and to uncover new genomic variants that predict chemotherapeutic response. This approach overcomes both the difficulty in collecting drug response in large clinical cohorts and of limited sample sizes in preclinical studies. We refer to this approach as an “imputed drug-wide association study” (IDWAS). Importantly, IDWAS is not limited to TCGA and can be applied to any cancer patient sequencing study where gene expression data has been collected, which will broaden the utility of such data sets.
Results
Gene expression–based models from cell lines to predict an in vivo phenotype
Despite growing evidence (Geeleher et al. 2014a,b; Falgreen et al. 2015; Azuaje 2016), it is not widely accepted that it is possible to predict in vivo drug response using gene expression–based predictive models derived from cell lines—where expression and drug response have been measured. This lingering uncertainty may be due to the very controversial history of this type of analysis (Coombes et al. 2007) and ongoing debates surrounding the reliability of drug screens in cell lines (Haibe-Kains et al. 2013; Stransky et al. 2015; Geeleher et al. 2016b). One initial objective is to produce further evidence that such prediction is indeed possible.
Thus, we hypothesized that if it is possible to predict in vivo drug response using cell line–derived gene expression–based models, it should be possible to accurately classify tumor tissue of origin using similar models. To test this, we fit a logistic ridge regression model (see Methods) on a subset of cell lines from the GDSC cohort. We restricted our analysis to tissues represented by at least 30 cell lines that could be unambiguously mapped to a cancer type annotated by TCGA. This yielded a cell line derived classifier for cancers of the breast, blood, lung, skin and central nervous system (CNS). We then applied this classifier to “remove unwanted variation” (RUV) (Risso et al. 2014) batch-corrected (see Methods) RNA-seq data from TCGA. By using this approach, we classified these clinical tumors with a far higher level of accuracy than expected by chance (3136 of 3761 samples correctly classified, P < 2.2 × 10−16) (Supplemental Fig. S1). This level of accuracy clearly demonstrates that the transcriptome in cell lines can be informative of an in vivo phenotype. Notably, our batch correction of the TCGA data improved the number of correctly classified samples from 3029 to 3136 (P = 1.5 × 10−3 from Fisher's exact test) (Supplemental Fig. S1).
Drug sensitivity can be accurately imputed in a TCGA breast cancer cohort
To demonstrate that our proposed IDWAS strategy (Fig. 1) is possible, we must first demonstrate that drug response can be accurately imputed in clinical samples. One of the biggest successes of precision cancer medicine are drugs targeting ERBB2 (also frequently called HER2)-overexpressing breast cancers (e.g., trastuzumab and lapatinib). One of these drugs, lapatinib, was screened against the GDSC cell lines. Given the large number of breast cancer samples in TCGA, we were interested in establishing whether applying gene expression–based models—predictive of lapatinib response—to TCGA could yield predictions that were consistent with over a decade of clinical observation. In TCGA, ERBB2 status was reported using immunohistochemistry, as is typical in the clinical setting. Hence, we applied the GDSC-derived models of lapatinib response to the RNA-seq data (see Methods) from TCGA breast cancer samples and compared imputed response between the ERBB2+ and ERBB2− groups. Remarkably, lapatinib was predicted to be more sensitive in the ERBB2+ group (P = 6.7 × 10−13), suggesting that drug response is being accurately imputed (Fig. 2A). These data also contained a ERBB2 “equivocal” group, which could not be accurately classified by immunohistochemistry; strikingly, these samples were predicted to be at an intermediate level of lapatinib response to the ERBB2+ and ERBB2− groups. Unsurprisingly, patients annotated as having received a ERBB2-targeted therapy (lapatinib or trastuzumab) were predicted to be more sensitive to lapatinib, compared with the other TCGA breast cancer samples (P = 3.3 × 10−7) (Supplemental Table S1). These results led to the obvious question of whether these predictions were drug specific, which would be necessary if this approach was to be viable for the discovery of novel associations. Thus, we compared imputed drug response for all 138 drugs screened in the GDSC cell line cohort between the ERBB2+ and ERBB2− groups in TCGA. Indeed, the strongest association was achieved by lapatinib (Fig 2B; Supplemental Table S2), suggesting that this approach could be viable for the discovery of novel clinically relevant associations.
Figure 1.

Discovering new drug biomarkers directly in clinical cancer genomics data sets (e.g., TCGA) using IDWAS. Data flow diagram describing the proposed novel methodology (IDWAS) for discovering novel pharmacogenomic variants. Boxes represent data, and circles represent processes. Predictive models of drug response are fit in preclinical data (in this case GDSC cancer cell lines) and applied to tumor gene expression in a clinical cancer data set (in this case TCGA). These imputed drug response data are then associated with measured somatic variants in the clinical data set in order to discover novel biomarkers of drug response.
Figure 2.
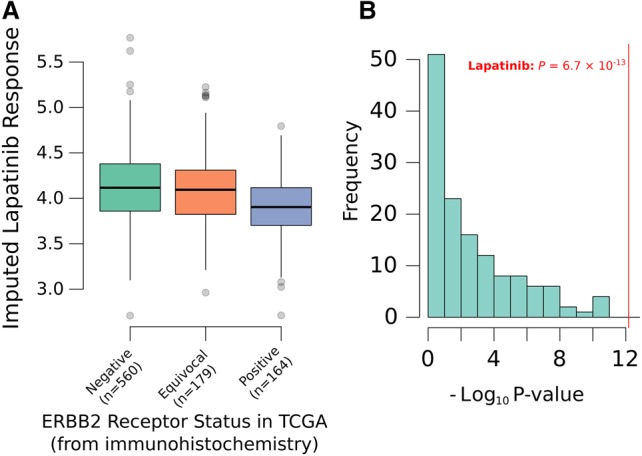
Imputed lapatinib response in TCGA breast cancer patients. (A) A boxplot showing the value for imputed lapatinib response in all TCGA breast cancer patients, where ERBB2 status was measured by immunohistochemistry. The imputed drug response data are consistent with how this drug behaves clinically (P = 6.7 × 10−13). Note that lower values on the y-axis imply greater drug sensitivity. (B) A histogram of P-values achieved for all 138 drugs when imputed drug response is compared between ERBB2+ and ERBB2− breast cancer patients in TCGA. Lapatinib, the only one of these drugs known to be differentially effective between these cohorts in the clinic, achieves the strongest association, suggesting that drug response is being accurately imputed.
IDWAS can be used to identify genomic aberrations that cause drug response
In the clinic, ERBB2 status is typically established by immunohistochemistry or by fluorescent in situ hybridization (FISH) (Vergara-Lluri et al. 2012). However, overexpression of ERBB2 typically occurs because of copy number amplification (CNA) of a large region of Chromosome 17 known as the ERBB2 amplicon (Kallioniemi et al. 1992). Thus, we were interested in whether this region could be identified as a biomarker using IDWAS in TCGA, which would suggest that this approach may be suitable for finding somatic genomic alterations associated with drug response. Thus, we tested the association of CNA of all genes with imputed lapatinib response. Indeed, genes in the ERBB2 amplicon were most strongly associated with imputed lapatinib response (P = 4.3 × 10−11 for ERBB2). Furthermore, imputed response increases steadily with copy number of ERBB2 (Fig. 3A). As expected, many genes in the ERBB2 amplicon were strongly associated with imputed lapatinib response (Supplemental Table S3), making it difficult to identify the causative gene by interrogating a ranked list of P-values. The numbers of samples in which a CNA is called also varies from gene to gene, meaning that the power to find an association differs between genes in the locus (Supplemental Table S3). However, plotting the effect size (for the association of amplification and imputed drug response) in the region revealed a smooth increase and decrease in the magnitude of the association around ERBB2, the known causative gene (Fig 3B). Thus, the precise causative gene could be identified by interrogating these effect sizes. This is important because it would not have been possible to identify this gene in such an unsupervised way from the cell line data on which the models were fit, due to the smaller sample size (Fig. 3C,D). This suggests not only that genomic predictors of drug response can be identified using this approach but also that there is added value in imputing drug response in this much larger clinical cohort, over only searching for these associations in the smaller set of cell line data. Strikingly, when we performed this same association analysis between gene amplification and also conditioned on ERBB2 amplification, EGFR—the known secondary target of lapatinib—was identified as the most strongly associated gene (Supplemental Table S4). This suggests that IDWAS has the ability to find multiple biomarkers for a drug. Thus, IDWAS may provide suitable power to derive polygenic models of drug response, which are likely necessary to achieve clinically relevant predictions for most drugs (Geeleher et al. 2015).
Figure 3.

Association between copy number amplification and lapatinib response for genes in ERBB2 amplicon. (A) Boxplot of association of ERBB2 amplification and imputed lapatinib sensitivity. (B) Each gene was tested for its association between copy number amplification and imputed lapatinib response in the TCGA breast cancer cohort (n = 1089). The resulting effect size is plotted for each gene in the ERBB2 amplicon (on Chr17q12). The biggest effect size is for ERBB2, the known causative gene, suggesting that IDWAS can be used to identify genomic predictors of drug response. Lower values on the y-axis imply greater lapatinib response in copy number–amplified samples. (C) Scatterplot of the measured lapatinib response in GDSC (P = 2.7 × 10−3 for the association of ERBB2 amplification and lapatinib IC50 in the GDSC breast cancer cell lines; n = 13). (D) Similar to B, but for the ERBB2 amplicon in the GDSC cell lines. ERBB2 is not easily identified as the causative gene; indeed, eight genes have a P-value as low as ERBB2, and nine genes have an effect size that is as low or lower.
IDWAS can be used to identify novel predictors of drug response
Next, we examined the association between all CNA and imputed response to all 138 drugs (Supplemental Table S5) in the TCGA breast cancer cohort. In addition to ERBB2 and lapatinib, we identified a number of top associations with a strong biological rationale, for example, imputed response to Nutlin-3a and amplification of mir-21. Nutlin-3a targets TP53, and there are multiple studies showing that mir-21, which is a known oncogene (Asangani et al. 2008), also targets TP53 (Ma et al. 2013), thus giving a clear mechanism for this observed association. Compound CGP.082996 is associated with the MYC locus; this compound is a CDK4 inhibitor (Hanaford et al. 2016), and CDK4 is a target of MYC (Hermeking et al. 2000). The second strongest association with any CNA is between a locus on Chromosome 8 and resistance to vinorelbine. Vinorelbine is used in the treatment of refractory breast cancer (Degardin et al. 1994), and this locus is amplified in ∼15% of breast cancers (Supplemental Fig. S2). Interrogating the effect sizes within the locus (Fig. 4A) suggests that ERLIN2 is the causative gene. The magnitude of the association also increased with increasing CNA (Fig. 4B) While amplification of this gene has not previously been linked to vinorelbine resistance, it has recently been shown that ERLIN2 plays a role in stabilizing microtubules (Zhang et al. 2015) and that vinorelbine functions by destabilizing microtubules (Klotz et al. 2012). ERLIN2 CNA in vivo is also associated with change in ERLIN2 expression (Supplemental Fig. S3) and with breast cancer patient survival (P = 8.9 × 10−3) (Supplemental Fig. S4). Given the potential clinical relevance of this novel association and a clear mechanistic basis, we overexpressed ERLIN2 in a CAMA-1 breast cancer cell line (Supplemental Fig. S5; see Methods) and, indeed, observed a strong association between ERLIN2 overexpression and vinorelbine resistance (P = 1 × 10−4 from ANOVA) (Fig. 4C). Of note, ERLIN2 amplification and vinorelbine resistance were only marginally associated in the GDSC breast cancer cell lines (P = 0.07) (Supplemental Fig. S6), and similar to ERBB2, it would not have been possible to identify ERLIN2 as the causative gene in the locus using a conventional analysis of the cell line data alone (Supplemental Fig. S7). This suggests that IDWAS can also be used to discover new pharmacogenomics biomarkers.
Figure 4.
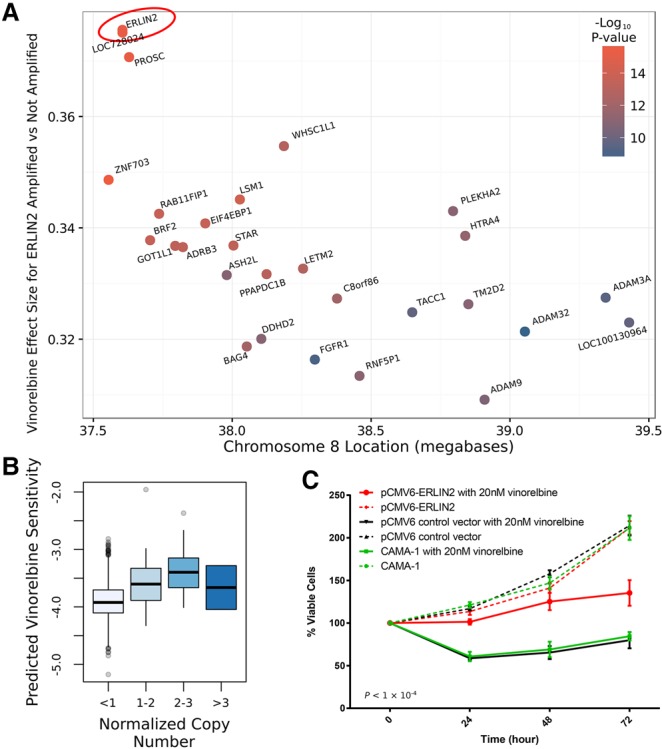
Copy number amplification of ERLIN2 causes vinorelbine resistance. (A) Each gene was tested for its association between CNA and vinorelbine response in the TCGA breast cancer cohort (n = 1089). The resulting effect size is plotted for each gene in Chr8p11.2. The biggest effect is for ERLIN2, suggesting that amplification of this gene may be causing resistance to vinorelbine. Higher values on the y-axis imply greater vinorelbine resistance in copy number–amplified samples. (B) Boxplot of association of ERLIN2 amplification and imputed vinorelbine response in TCGA. Note that the last box (n > 3) contains only two patients. (C) Percentage of viable CAMA-1 breast cancer cells treated with vinorelbine (solid lines) or untreated (broken lines). Viability is compared for ERLIN2-overexpressing cells (red), cells transfected with an empty plasmid (black), or untransfected cells (green). As predicted by IDWAS in the TCGA breast cancer patients, ERLIN2 overexpression causes vinorelbine resistance.
Elucidating the factors influencing the reliability of gene–drug associations
The associations we observed between imputed lapatinib response and ERBB2 status in breast cancer represents a particularly strong example for reasons that are important to highlight: First, measured response to this drug demonstrates a high level of predictability in 10-fold cross-validation in GDSC (rs = 0.48; P < 2.2 × 10−16). By using this metric, the level of “predictability” of drug response varies markedly between drugs (Supplemental Table S6). However, response to most drugs (125 of 138) can be predicted with statistical significance in cross-validation (P < 0.05 from Spearman's correlation test; Spearman's correlation is often underpowered in this context due to lack of variability in drug response for highly targeted drugs) (Geeleher et al. 2016b). For some drugs, for example, sorafenib, response could not be predicted with statistical significance in cross-validation; therefore, such models are almost certainly not applicable to in vivo data. Importantly, a good prediction in cross-validation does not guarantee accurate predictions in vivo, and in vivo prediction strongly depends on how well the preclinical data reflect in vivo biology, which can be difficult to judge. The next important factor is that ERBB2 amplification is well represented in TCGA. There are 164 ERBB2+ breast cancer samples in TCGA, providing sufficient statistical power. Also, ERBB2 amplification has a substantial effect on gene expression, with breast cancer samples harboring this mutation associated with the fourth, fifth, and sixth principal components derived from the corresponding gene expression data (P = 5.4 × 10−27, P = 7.3 × 10−10, and P = 1.73 × 10−8, respectively) (Supplemental Fig. S8). This will vary on a gene-by-gene basis, but many somatic mutations are strongly associated with changes in gene expression. However, were a somatic aberration to have no effect on gene expression, a gene expression–based model would not be useful for identifying drugs to target that aberration. Finally, while IDWAS can be used to estimate differential effectiveness between different groups of patients, this does not guarantee that the level of effectiveness achieved in either group will be sufficient for clinical utility. Directly comparing imputed drug response values for different drugs within one or more patients is also not meaningful; predictions should only be compared for a drug across patients, not between different drugs within patient(s). Currently, we also recommend fitting models on solid tumor cell lines when imputing in a solid tumor patient cohort, hematological cell lines for a hematological cohort, and all cell lines for a mixed cohort, for example, all of TCGA (Geeleher et al. 2014a). Future work applying this approach should strongly consider all of these potential pitfalls.
Expanding to nonsynonymous somatic mutations and a pan-cancer analysis of TCGA
Given that cancer research is typically disease specific, it is likely that the majority of researchers will be most interested in applying this approach to a specific type of cancer, as we have demonstrated for breast cancer. However, one appealing prospect of IDWAS is the possibility of applying such imputation in a pan-cancer analysis, for example, across all of TCGA (more than 10,000 samples from more than 30 cancer types). This would mean leveraging an unprecedented quantity of data, thus potentially identifying drug associations with rare alterations. For example, if one could reasonably expect to find an association for a somatic mutation that is present in about 50 TCGA patients, we could be powered to detect potential biomarkers that occur at a frequency of <0.3% across all tumor types. However, naively applying the models across all of TCGA does not yield meaningful predictions. This could be caused by factors such as very large systematic differences between the transcriptome of cancers from different tissues and possible technical effects due to samples being collected at different sites and processed by different groups (e.g., Supplemental Fig. S9). Several studies have also suggested that the comparison of drug response data between cell lines from different tissues does not typically produce results consistent with clinical observation (Jaeger et al. 2015; Yao et al. 2016). Given that these are cell line–derived predictive models, we would expect that the same problem exists in this imputed TCGA drug data.
However, we found that simply including cancer type as a covariate in gene–drug association analysis markedly improved this situation. Indeed, when controlling for cancer type, the association of ERBB2 and imputed lapatinib response could be recovered with about the same level of significance across all TCGA, as when assessed in breast cancer samples alone (P = 4.7 × 10−10), and this result was drug specific (Supplemental Fig. S10). Given this, we were interested whether any of the small numbers of additional known gene–drug associations could be recovered when interrogating the entire TCGA data set in this way. Thus, we leveraged the exome sequence data, available for many TCGA samples, to assess whether mutated genes were associated with expected drugs. We summarized these data at gene level and considered any gene with a protein coding change to be “mutated”; consequently, we were more likely to find genes whose inactivation is predictive of drug response, rather than gain-of-function mutations, which are rarer. Indeed, the results were consistent with expectation. The top two associations, when controlling for cancer type, were for Nutlin-3a and TP53 (P = 2.6 × 10−77) (Fig. 5A; Meijer et al. 2013) and for PD-0332991 (a.k.a. Palbociclib) and RB1 (P = 5.3 × 10−40) (Rocca et al. 2014). As expected, mutations in both of these genes were strongly associated with resistance to these drugs, and these associations are highly gene specific (Fig. 5A,B). Although the number of drugs in this data set with known actionable targets is small, several other expected associations were also recovered using this approach (Supplemental Table S7); for example, erlotinib and KRAS (P = 7 × 10−4) (Eberhard et al. 2005), PD-0325901 and BRAF (P = 1.1 × 10−8) (Henderson et al. 2010), KIT and tyrosine kinase inhibitors imatinib (P = 3.9 × 10−9; strongest association for this drug) (Blanke et al. 2008), nilotinib (P = 4.2 × 10−6; strongest association for this drug), and sunitinib (P = 0.03) (Demetri et al. 2006).
Figure 5.

Top gene–drug associations recovered across all of TCGA. Histograms showing the association between all genes with a somatic protein-coding change in at least 50 samples (n = 1739 genes) in TCGA and imputed drug response. (A–C) Associations when we control for cancer types; (D–F) associations when we control for GLDS, rather than cancer type. The canonical clinical target is highlighted in red; in all cases, the correct target is identified with a very high level of significance and specificity.
In addition to controlling for cancer type, we propose another potentially more effective method for using imputed drug response data across a pan-cancer data set. This is a type of correction that we have previously proposed for effectively discovering clinically relevant pharmacogenomic associations directly in cell line data. The method relies on modeling the general response of each sample (in this case, TCGA patients) to many drugs and then including this estimate as a covariate in subsequent statistical association analyses. We referred to this as controlling for “general levels of drug sensitivity” (GLDS; see Methods) (Geeleher et al. 2016a). By use of this approach, most of the top results remain largely consistent with those observed when controlling for cancer type alone (Fig. 5D,E). There are some other notable changes, for example, the associations of KRAS and imputed erlotinib response has improved, as has the specificity of this association (Fig. 5F). This is because KRAS mutation, a known clinical predictor for this drug, was heavily confounded by cancer type, thus benefitting from the GLDS approach, which obviates the need to include cancer type as a covariate. Other clinically relevant associations recovered using GLDS (Supplemental Table S8), but not when controlling for cancer type (with the correct directionality in all cases) include PLX4720 (a RAF inhibitor) and BRAF (P = 3.1 × 10−38) (Bollag et al. 2012), SB590885 and BRAF (P = 7.4 × 10−4) (Barollo et al. 2014), PD-0325901 and BRAF (P = 4.5 × 10−5), and gefitinib and EGFR (P = 8.6 × 10−3) (Lynch et al. 2004).
The numbers of significant associations identified by this IDWAS analysis in TCGA were larger than those that have been reported in previous pharmacogenomics screens, which is not surprising given over an order of magnitude increase in sample size. The analysis controlling for cancer type and GLDS identified 142 and 263 significant gene–drug associations, respectively (FDR < 0.05; also corrected for the number of drug models). This was of a total of 123,469 associations tested for 71 drugs exhibiting a Spearman's correlation of more than 0.3 in cross-validation and 1739 genes that were mutated in at least 50 TCGA samples. Some already validated associations were identified with striking specificity (e.g., Fig. 5), supporting the biological relevance of the results. However, given the very small number of currently known pharmacogenomic variants, the vast majority of the associations identified were novel. This highlights the increase in power that can be achieved using IDWAS compared with conventional approaches. While this list will likely contain false positives, these results provide a valuable starting point for functional validation—in addition to the validation work we have included in this study.
Using TCGA to study pharmacogenomics and drug response
One final consequence of our work is that TCGA can now be used to study mechanisms of drug response, similar to drug screening data sets such as GDSC and CCLE. We have provided imputed drug response estimates for all TCGA samples, corrected for both cancer type (Supplemental Table S9) and GLDS (Supplemental Table S10). Leveraging TCGA as a pharmacogenomics data set could prove to be a useful resource in several avenues of cancer research. Indeed, following the example of studies like TCGA and GDSC, we have made all analyses, code, and results publicly accessible (see Software Availability). This is particularly important in providing a clear understanding of the merits and limitations of this complex methodology, as well as facilitating other researchers who wish to improve upon or apply these ideas. We also provide an R package, “idwas,” aimed at facilitating the application of this approach to data sets other than TCGA.
Discussion
Recent reviews have concluded that computational models built on cell line–derived data are applicable to the prediction of clinical response in cancer patient cohorts (Azuaje 2016). It is also now clear that for drug response and disease prognosis, gene expression data provide the best predictive potential of any kind of high-throughput genomics data (Costello et al. 2014; Yuan et al. 2014; Zhao et al. 2015) but are difficult to apply in the clinic because of the instability of RNA. By leveraging these observations, we have developed IDWAS, an analytical method that overcomes several obstacles currently facing cancer pharmacogenomics research. IDWAS involves creating predictive models of drug response in a preclinical setting, where drug response data are easily obtained, and then using gene expression–based models to impute drug response in clinical samples—where drug response information is difficult to obtain. These imputed drug response data can then be used for discovery of novel pharmacogenomics variants. The reasons why this offers advantages over studying drug response in the preclinical setting alone include (1) improved power due to increased samples sizes, especially when applied to a large data set like TCGA; (2) the possibility of studying pharmacogenomics in cancers where the number of cell lines (or any preclinical model) is limited; and (3) ability to leverage classes of data that are not available in the preclinical setting. Overall, our results have shown that IDWAS can be used to both recapitulate known associations and to find new biomarkers of potential clinical relevance. The approach means data sets like TCGA can now be used to directly study pharmacogenomics, and we have identified new potential drug biomarkers that can be used as the basis for functional validation studies. Furthermore, there are many more clinical cancer genomics data sets upon which this method can be applied, and the number of these is growing constantly.
Importantly, the methods we have applied for imputing clinical drug response represent the minimum changes over some of our previously published methods (Geeleher et al. 2014b). The methods we previously developed were also carefully developed based on the results of previous benchmarking studies. Here, we have explicitly avoided fitting multiple models to the data and selecting that which performed best, a practice that leads to irreproducible results (Head et al. 2015). Thus, there is likely scope to improve on our proposed implementation of IDWAS, in particular, the drug imputation models: for example, accounting for the skewed drug response data that results from highly targeted agents (Geeleher et al. 2014b, 2016b). Further development of imputation models could be well suited to community challenges, where competing models are tested on hold-out data sets (Skocik et al. 2016).
The data we present here were generated using the initial release of the GDSC cell line data set, which is suitable to develop and present this methodology. However, there are other cell line data sets (e.g., CCLE, CTRP) that contain additional drugs, which could be interrogated using this methodology. Furthermore, an IDWAS need not be restricted to building models in cell lines. We are investigating fitting similar models on data gathered from mouse patient-derived xenografts (Gao et al. 2015). Another exciting new preclinical model is tumor organoids, although large drug-screening data sets are yet to be released (Walsh et al. 2016). Given suitable preclinical data, this means that IDWAS can be extended to drugs where, for example, cell lines are not a suitable model (e.g., an angiogenesis inhibitor) or where the microenvironment plays a strong role.
One additional potential application of IDWAS is in predicting drugs that may be effective against new molecular subtypes of cancer that have been defined by studies like TCGA; we have not extensively discussed this application here because there are not yet many validated drug treatment options for these newly proposed cancer subtypes. We have also reported limitations of IDWAS, which are important to consider when applying these types of analyses in the future. Important considerations include the level of predictability of the drug being studied and how well the preclinical model is likely to reflect in vivo biology—in terms of the behavior of both the disease and the drug being studied. Clinical data sets such as TCGA often have ascertainment biases, such as favoring large or late-stage tumors. The quality of the drug screening and preclinical and clinical genomics data are also clearly key, as no method will overcome poor data; however, our results support the utility and quality of both the GDSC and TCGA data sets. Nonetheless, the IDWAS approach should be considered hypothesis generating, and it should not be assumed that new associations necessarily represent true positives. While we have shown that some known clinically actionable gene–drug associations were recovered with strong specificity, new IDWAS findings still require prospective experimental and clinical work.
This study will likely spawn new avenues of research, such as optimizing the statistical models used for imputing drug response and the application of these methods to the many other clinical cancer sequencing efforts. Overall, IDWAS is a promising novel methodology that can be used to broaden the utility of existing cancer genomics data sets, generate new hypotheses, and speed up pharmacogenomics discovery.
Methods
Batch correcting the TCGA gene expression data set
TCGA gene expression data (The Cancer Genome Atlas Research Network et al. 2013) were obtained from firebrowse.org. We obtained the Illumina HiSeq RNA-seq v2 data (2015/08/21 release), which had been summarized at gene level using the RSEM software. These TCGA gene expression data were generated at many different sequencing centers and thus were processed in many different batches. Hence, to make meaningful comparisons between these samples, batch correction was performed. Batch IDs are included in TCGA, but in some cases, these are perfectly confounded by cancer type (i.e., all samples from a single tissue have been run in a single batch), thus correcting for batch ID would render some of the data useless. Hence, we have implemented a method based on the “remove unwanted variation” (RUV) principles described by Risso et al. (2014). This approach models unwanted variation (e.g., batch effects) using a set of genes referred to as “negative controls,” whose expression is not expected to vary across samples. In our case, we have identified a set of 250 negative control genes empirically by the set of constitutively expressed genes that exhibit the lowest variability across all TCGA samples. These genes would be expected to be affected primarily by technical variability, which we wish to remove, rather than by both technical and biological variability, as would be the case for genes showing a higher level of variability. As suggested by Risso et al. (2014), we calculated the first 10 principal components of these negative control genes to capture the unwanted technical variability. To determine a batch-corrected expression matrix, we then use the residuals of a generalized linear model (GLM) with Poisson link function, where these 10 principle components have been (iteratively) regressed against the expression of every gene. One additional step we added for these data was to calculate the 10 principal components on an expression matrix that had been standardized (i.e., set mean and variance of each gene to zero and one, respectively) by cancer type. This was necessary so as not to remove variability associated with cancer type, which is substantial and clearly biologically relevant. Code to reproduce this analysis is available in our supplemental R code (see Software Availability).
Predictive models of cancer type and imputing drug response
The models used for predicting drug response and cancer type are ridge regression models based on those that we have previously described (Geeleher et al. 2014b). Here, the only change that was necessary was to slightly alter the approach to allow models fit on microarray data (in cell lines) to be applied to RNA-seq data (in TCGA). In order to assess which of several plausible approaches produced the best predictions, we applied a number of different approaches to a set of breast cancer TCGA samples for which both microarray and RNA-seq data were available. We compared the microarray-derived prediction using the approach described in Geeleher et al. (2014b) to the prediction from the proposed RNA-seq–based approach, using the microarray-derived predictions as a gold standard. Only a minor modification of the method described in Geeleher et al. (2014b) was required to produce highly comparable results. Specifically, instead of standardizing the mean and variance of each gene using an empirical Bayesian approach (designed for microarray data), we standardized the mean and variance of each gene to zero and one, respectively. This modification was included in an updated version of an R package, pRRophetic (Geeleher et al. 2014a), which we have released with this paper. A predictive model of cancer type was constructed using logistic ridge regression on the GDSC cell lines, and predictive models used to impute drug response were constructed using linear ridge regression on the GDSC cell lines (Garnett et al. 2012).
Finding associations between imputed drug response and copy number/nonsynonymous somatic mutations
The associations between imputed drug response in TCGA and CNA or somatic mutations were calculated using linear models using R. CNAs were calculated from TCGA copy number data (Zack et al. 2013) obtained from firebrowse.org. These data were generated using Affymetrix SNP 6.0 arrays (2015/08/21 release). We summarized the data at gene level and gave a gene a missing value if the entire gene was not contained unambiguously within a single copy number region. These analyses were performed using the GenomicRanges (Lawrence et al. 2013) package in R. Genes with a normalized copy number greater than one were considered amplified. We obtained somatic mutation calls from firebrowse.org (2016/01/28 release). We summarized these data at gene level and considered a gene to be mutated in a patient if it contained any mutation that affects the protein amino acid sequence. When the gene–drug association analysis was applied across all of TCGA, we controlled for cancer type by including this as a covariate (encoded as a factor) in the linear models. When we controlled for GLDS, this was calculated as previously described (Geeleher et al. 2016a) and included as a covariate in the linear models.
Figures and data analysis
Most of the computational analyses were performed using the Bionimbus Protected Data Cloud (Heath et al. 2014). All basic statistical analyses (linear regression models, correlation tests, Wilcoxon rank-sum tests) were performed using the base functions in R version 3.2.2 (R Core Team 2016). False-discovery rates were estimated using the Benjamini and Hochberg method. Figures were created using the base graphics functions or the ggplot2 package in R and cBioPortal (Gao et al. 2013). Furthermore, given the complexity of these methods, transparency and reproducibility of the analysis are essential. Thus, we have documented all analysis using R Markdown, which has allowed us to construct a set of HTML documents using the knitr package (https://www.rforge.net/doc/packages/knitr/knitr-package.html). For easy reproduction of results, we have also included a script to automate the downloading of the same data that we used in our analysis.
ERLIN2 overexpression experiments
Functional validation of the association of ERLIN2 amplification with vinorelbine resistance was conducted in a human CAMA-1 breast cell line, obtained from the American Type Culture Collection (ATCC, Manassas). CAMA-1 cells were cultured in MEM culture medium supplemented with 20% FBS and kept in a 37°C humidified incubator with 5% CO2. Gene overexpression was performed by independently transfecting ERLIN2 constructed plasmid pCMV6-AC-GFP-ERLIN2 (catalog no. RG221700) and control vector pCMV6-AC-GFP (catalog no. PS100010) purchased from OriGene Technologies. Transfection of the plasmid was achieved using Lipofectamine 3000 (Invitrogen). Stably transfected cells were selected by resistance to neomycin (G418) at 200 µg/mL (Research Products International) for 4 wk after a 48-h transfection. Neomycin-resistant cells were passaged at several different dilutions and seeded on 96-well cell culture plate (Corning) containing 200 µg/mL of neomycin. Cells growing from GFP-positive single colonies (cloned and stable transfected) were isolated and expanded. RNA and DNA were collected to confirm up-regulation of ERLIN2 levels by quantitative RT-PCR (qRT-PCR) using TaqMan gene expression assay (catalog no. Hs 00200360_m1, Applied Biosystems) and TaqMan copy number assay (catalog no. Hs02147573_cn, Applied Biosystems). Cell viability was measured after 20 nM vinorelbine treatment using the CellTiter-Glo luminescent cell viability assay (Promega). Two-way ANOVA was performed to compare cell viabilities obtained post vinorelbine treatment in transfected cells and control cells.
Software availability
The R scripts to reproduce our analysis are included in our Supplemental Material. A file “index.html” explains how to run the scripts, what each of the scripts does, and what order they should be run. Additionally, all scripts and tools (including updated R packages) required to reproduce our results and analysis are available on Open Science Framework. We have released an updated version of our R package pRRophetic, available from https://osf.io/yatu3/ (DOI: 10.17605/OSF.IO/YATU3). A new R package, idwas, which can be used to apply our method to new data sets, is available at https://osf.io/5xvsg/ (DOI: 10.17605/OSF.IO/5XVSG). The scripts to reproduce this analysis are also available from https://osf.io/pwm4z/ (DOI: 10.17605/OSF.IO/PWM4Z).
Supplementary Material
Acknowledgments
This work was supported by a research grant from the Avon Foundation for Women. R.S.H. also received support from NIH/NIGMS grant K08GM089941, NIH/NCI grant R21 CA139278, NIH/NIGMS grant U01GM61393, and a Circle of Service Foundation Early Career Investigator award. P.G. received support from the Chicago Biomedical Consortium grant PDR-020.
Author contributions: P.G. and R.S.H. conceived the study. P.G. performed the analysis and drafted the paper. Z.Z., R.F.G., G.M., S.B., and A.N. assisted with analysis. F.W. and P.G. performed the wet-laboratory experiments. R.L.G. and Z.Z. assisted with data acquisition, analytical pipelines, and implementation. All authors edited and approved the final manuscript. R.S.H. supervised the study.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.221077.117.
References
- Asangani IA, Rasheed SAK, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H. 2008. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27: 2128–2136. [DOI] [PubMed] [Google Scholar]
- Azuaje F. 2016. Computational models for predicting drug responses in cancer research. Brief Bioinform pii: bbw065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barollo S, Bertazza L, Baldini E, Ulisse S, Cavedon E, Boscaro M, Pezzani R, Mian C. 2014. The combination of RAF265, SB590885, ZSTK474 on thyroid cancer cell lines deeply impact on proliferation and MAPK and PI3K/Akt signaling pathways. Invest New Drugs 32: 626–635. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. 2012. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483: 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanke CD, Demetri GD, von Mehren M, Heinrich MC, Eisenberg B, Fletcher JA, Corless CL, Fletcher CDM, Roberts PJ, Heinz D, et al. 2008. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol 26: 620–625. [DOI] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, Hirth P. 2012. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 11: 873–886. [DOI] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, Stuart JM. 2013. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Phillips SM, Gao JJ, Kundra R, Zhang H, Rudolph JE, Yaeger RD, Soumerai T, Nissan MH, Chandarlapaty S, et al. 2016. OncoKB: annotation of the oncogenic effect and treatment implications of somatic mutations in cancer. J Clin Oncol 34: 11583. [Google Scholar]
- Coombes KR, Wang J, Baggerly KA. 2007. Microarrays: retracing steps. Nat Med 13: 1276–1277. [DOI] [PubMed] [Google Scholar]
- Costello JC, Heiser LM, Georgii E, Gönen M, Menden MP, Wang NJ, Bansal M, Ammad-ud-din M, Hintsanen P, Khan SA, et al. 2014. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol 32: 1202–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degardin M, Bonneterre J, Hecquet B, Pion JM, Adenis A, Horner D, Demaille A. 1994. Vinorelbine (navelbine) as a salvage treatment for advanced breast cancer. Ann Oncol 5: 423–426. [DOI] [PubMed] [Google Scholar]
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, et al. 2006. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368: 1329–1338. [DOI] [PubMed] [Google Scholar]
- Eberhard DA, Johnson BE, Amler LC, Goddard AD, Heldens SL, Herbst RS, Ince WL, Jänne PA, Januario T, Johnson DH, et al. 2005. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J Clin Oncol 23: 5900–5909. [DOI] [PubMed] [Google Scholar]
- Falgreen S, Dybkær K, Young KH, Xu-Monette ZY, El-Galaly TC, Laursen MB, Bødker JS, Kjeldsen MK, Schmitz A, Nyegaard M, et al. 2015. Predicting response to multidrug regimens in cancer patients using cell line experiments and regularised regression models. BMC Cancer 15: 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. 2013. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al. 2015. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med 21: 1318–1325. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. 2012. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483: 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Cox N, Stephanie Huang R. 2014a. PRRophetic: an R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS One 9: e107468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Cox NJ, Huang R. 2014b. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol 15: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Loboda A, Lenkala D, Wang F, LaCroix B, Karovic S, Wang J, Nebozhyn M, Chisamore M, Hardwick J, et al. 2015. Predicting response to histone deacetylase inhibitors using high-throughput genomics. J Natl Cancer Inst 107: djv247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Cox NJ, Huang RS. 2016a. Cancer biomarker discovery is improved by accounting for variability in general levels of drug sensitivity in pre-clinical models. Genome Biol 17: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geeleher P, Gamazon ER, Seoighe C, Cox NJ, Huang RS. 2016b. Consistency in large pharmacogenomic studies. Nature 540: E1–E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JW, Mills GB. 2015. Large-scale drug screens support precision medicine. Cancer Discov 5: 1130–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJWL, Quackenbush J. 2013. Inconsistency in large pharmacogenomic studies. Nature 504: 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaford AR, Archer TC, Price A, Kahlert UD, Maciaczyk J, Nikkhah G, Kim JW, Ehrenberger T, Clemons PA, Dančík V, et al. 2016. DiSCoVERing innovative therapies for rare tumors: combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin Cancer Res 22: 3903–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head ML, Holman L, Lanfear R, Kahn AT, Jennions MD. 2015. The extent and consequences of p-hacking in science. PLoS Biol 13: e1002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath AP, Greenway M, Powell R, Spring J, Suarez R, Hanley D, Bandlamudi C, McNerney ME, White KP, Grossman RL. 2014. Bionimbus: a cloud for managing, analyzing and sharing large genomics datasets. J Am Med Inform Assoc 21: 969–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson YC, Chen Y, Frederick MJ, Lai SY, Clayman GL. 2010. MEK inhibitor PD0325901 significantly reduces the growth of papillary thyroid carcinoma cells in vitro and in vivo. Mol Cancer Ther 9: 1968–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermeking H, Rago C, Schuhmacher M, Li Q, Barrett JF, Obaya AJ, O'Connell BC, Mateyak MK, Tam W, Kohlhuber F, et al. 2000. Identification of CDK4 as a target of c-MYC. Proc Natl Acad Sci 97: 2229–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Wolf DM, Cherniack AD, Tamborero D, Ng S, Leiserson MDM, Niu B, McLellan MD, Uzunangelov V, et al. 2014. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 158: 929–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger S, Duran-Frigola M, Aloy P, Weinstein J, Garnett M, Edelman E, Heidorn S, Greenman C, Dastur A, Lau K, et al. 2015. Drug sensitivity in cancer cell lines is not tissue-specific. Mol Cancer 14: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallioniemi OP, Kallioniemi A, Kurisu W, Thor A, Chen LC, Smith HS, Waldman FM, Pinkel D, Gray JW. 1992. ERBB2 amplification in breast cancer analyzed by fluorescence in situ hybridization. Proc Natl Acad Sci 89: 5321–5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz DM, Nelson SA, Kroboth K, Newton IP, Radulescu S, Ridgway RA, Sansom OJ, Appleton PL, Nathke IS. 2012. The microtubule poison vinorelbine kills cells independently of mitotic arrest and targets cells lacking the APC tumour suppressor more effectively. J Cell Sci 125: 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ. 2013. Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat SM, Supko JG, Haluska FG, et al. 2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med 350: 2129–2139. [DOI] [PubMed] [Google Scholar]
- Ma X, Choudhury SN, Hua X, Dai Z, Li Y. 2013. Interaction of the oncogenic miR-21 microRNA and the p53 tumor suppressor pathway. Carcinogenesis 34: 1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer A, Kruyt FAE, van der Zee AGJ, Hollema H, Le P, ten Hoor KA, Groothuis GMM, Quax WJ, de Vries EGE, de Jong S. 2013. Nutlin-3 preferentially sensitises wild-type p53-expressing cancer cells to DR5-selective TRAIL over rhTRAIL. Br J Cancer 109: 2685–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Relling MV, Evans WE. 2015. Pharmacogenomics in the clinic. Nature 526: 343–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, Dudoit S. 2014. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32: 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca A, Farolfi A, Bravaccini S, Schirone A, Amadori D. 2014. Palbociclib (PD 0332991): targeting the cell cycle machinery in breast cancer. Expert Opin Pharmacother 15: 407–420. [DOI] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. 2015. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov 5: 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skocik M, Collins J, Callahan-Flintoft C, Bowman H, Wyble B. 2016. I tried a bunch of things: the dangers of unexpected overfitting in classification. bioRxiv 10.1101/078816. [DOI] [PubMed]
- Stransky N, Ghandi M, Kryukov GV, Garraway LA, Lehár J, Liu M, Sonkin D, Kauffmann A, Venkatesan K, Edelman EJ, et al. 2015. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, Perez-Llamas C, Deu-Pons J, Kandoth C, Reimand J, Lawrence MS, Getz G, Bader GD, Ding L, et al. 2013. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci Rep 3: 149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergara-Lluri ME, Moatamed NA, Hong E, Apple SK. 2012. High concordance between HercepTest immunohistochemistry and ERBB2 fluorescence in situ hybridization before and after implementation of American Society of Clinical Oncology/College of American Pathology 2007 guidelines. Mod Pathol 25: 1326–1332. [DOI] [PubMed] [Google Scholar]
- Walsh AJ, Cook RS, Sanders ME, Arteaga CL, Skala MC. 2016. Drug response in organoids generated from frozen primary tumor tissues. Sci Rep 6: 18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao F, Tonekaboni SAM, Safikhani Z, Smirnov P, El-Hachem N, Freeman M, Manem VSK, Haibe-Kains B. 2016. Tissue specificity of in vitro drug sensitivity. bioRxiv 10.1101/085357. [DOI] [PMC free article] [PubMed]
- Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, Byers LA, Xu Y, Hess KR, Diao L, et al. 2014. Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol 32: 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang C-Z, Wala J, Mermel CH, et al. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cai J, Zheng Z, Polin L, Lin Z, Dandekar A, Li L, Sun F, Finley RL, Fang D, et al. 2015. A novel ER–microtubule-binding protein, ERLIN2, stabilizes Cyclin B1 and regulates cell cycle progression. Cell Discov 1: 15024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Shi X, Xie Y, Huang J, Shia B, Ma S. 2015. Combining multidimensional genomic measurements for predicting cancer prognosis: observations from TCGA. Brief Bioinform 16: 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.