

Abstract
Polymer–protein conjugation has been extensively explored toward a better protein drug with improved pharmacokinetics. However, a major problem with polymer–protein conjugation is that the polymers drastically reduce the bioactivity of the modified protein. There is no perfect solution to prevent the bioactivity loss, no matter the polymer is conjugated in a non-site specific way, or a more complex site-specific procedure. Here the authors report for the first time that when zwitterionic carboxybetaine polymer (PCB) is conjugated to insulin through simple conventional coupling chemistry. The resulting PCB–insulin does not show a significant reduction of in vitro bioactivity. The obtained PCB–insulin shows two significant advantages as a novel pharmaceutical agent. First, its therapeutic performance is remarkable. For PCB–insulin, there is a 24% increase of in vivo pharmacological activity of lowering blood glucose compared with native insulin. Such uncommonly seen increase has rarely been reported and is expected to be due to both the improved pharmacokinetics and retained bioactivity of PCB–insulin. Second, the production is simple from manufacturing standpoints. Conjugation procedure involves only one-step coupling reaction without complex site-specific linkage technique. The synthesized PCB–insulin conjugates do not require chromatographic separation to purify and obtain particular isoforms.
The major purpose for polymer–protein conjugation is to improve pharmacokinetics (PK) and retain bioactivity in order to improve the pharmacological activity of the modified protein.[1] The most widely used conjugation strategy is PEGylation, by using polyethylene glycol (PEG) to conjugate proteins.[2] Despite improved PK, PEGylation is known for its sacrificing the conjugated protein activity.[3] Significant results indicate that conjugating PEG, and other commonly seen polymers as well, on proteins through conventional covalent coupling chemistry resulting in polymer chains nonselectively attached on the protein surface groups (e.g., amines), and potential attachment near to the protein’s active site significantly inhibit the protein from performing designated biological function (e.g., amine group on Gly A1 of insulin). There are alternative methods allowing for site-specific conjugation to proteins, e.g., involving reengineering the proteins, or laborsome separation, but those procedures are drastically complex.[4] When PEG is conjugated to proteins in a site selective manner, however, the resulting PEG–proteins are still subject to bioactivity loss. Eli Lily has developed a PEGylated insulin Lispro (LY2605541) where a 20 kDa PEG was conjugated at the amino group of Lys B28 of insulin Lispro. For LY2605541 high molar concentration is still required to achieve comparable glucose lowering effect to the unconjugated insulin as observed in the in vitro insulin receptor binding studies.[5] It appears that no matter which site of the protein the polymer is conjugated to, most resulting polymer–protein conjugates inevitably suffer from bioactivity loss.
Here we report that the conjugation of zwitterionic carboxybetaine polymer (PCB) on insulin through conventional coupling chemistry improves the PK of the protein, but does not compromise the insulin in vitro bioactivity. Remarkably, this conjugation strategy increases the in vivo glucose lowering activity for ≈24% compared with native insulin at equal molar dose when intravenously (i.v.) injected. Such high in vivo therapeutic activity has rarely been reported for any prior polymer–insulin conjugates. As an additional advantage, the synthesis of PCB–insulin does not require complex site-specific linkage technique and can be implemented through simple one-step coupling reaction. The resulting PCB–insulin conjugates do not require tedious chromatographic separations to obtain purified isoforms, enabling a simplified scaling-up, and manufacturing procedure.
Zwitterionic polymer materials have attracted increasing attention as novel nonfouling materials.[6] Particles with surface modified with zwitterionic polymers, such as PCB, have been found to effectively avoid the interaction with plasma proteins and the uptake by the reticuloendothelial system (RES),[6a,7] and show significantly prolonged blood circulation half-life (improved PK),[7,8] and reduced immunogenicity.[9] Here the general strategy in obtaining PCB–insulin conjugate is by reacting an N-hydroxysuccinimide (NHS) ester terminated PCB, with the available amine groups of human recombinant insulin (Figure 1A). To prepare PCB–insulin conjugate, we first synthesized NHS ester terminated PCB (PCB–NHS) following established procedures.[8] The MW for PCB–NHS is ≈5000 Da as previously reported: Mn = 4890 Da determined by 1H NMR, Mn = 5410 Da, polydispersity = 1.03 characterized by GPC.[8] Then PCB–NHS was reacted with the amine group of insulin at PCB–NHS: insulin molar ratio of 1.2:1 to synthesize PCB–insulin conjugate. The reaction was conducted at Na2CO3 solution (pH > 10) at which condition the acylation is at any of the three amines (GlyA1, PheB1, or LysB29), but prioritized at epsilon amine of LysB29.[10] In a similar way, PEG–insulin control was obtained by reacting a commercially available PEG–NHS (MW, 5000 Da) with insulin. We developed a purification method using a 6–8 kDa Molecular weight cut-off (MWCO) dialysis kit to remove unreacted insulin (≈5800 Da) and unconjugated PCB–NHS or PEG–NHS (about 5000 Da). The purified PCB–insulin and PEG–insulin were analyzed by Matrix-assisted laser desorption/ionization-Time of Flight Mass (MALDI-TOF MS), and their purity was confirmed by the disappearance of free insulin peak (m/z = 5813.44 Da) (Figure 1B). Based on MALDI-TOF results (Figure 1B), the major products for both PCB–insulin and PEG–insulin have one-polymer-chain modification per protein: only one peak was shown corresponding to insulin with one polymer chain conjugated. The obtained PCB–insulin and PEG–insulin were not further separated to obtain their respective purified isoforms (i.e., a polymer conjugated to LysB29 of insulin) before evaluating their PK and bioactivity.
Figure 1.
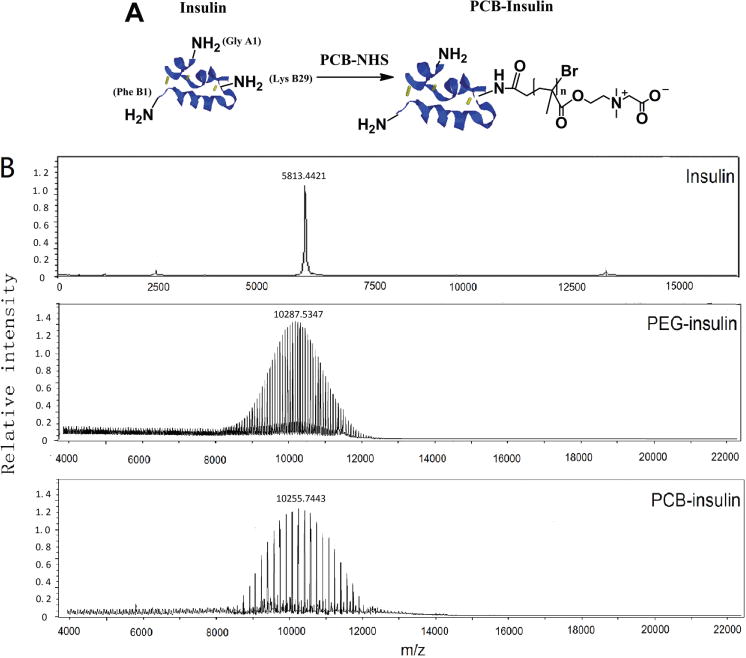
A) The schematic illustration of PCB conjugation with native recombinant human insulin. There are three amino acids with free amino groups (GlyA1, PheB1, and Lys B29) that are available for PCB conjugation. The disulfide bonds in insulin were labeled with yellow sticks. B) MALDI-TOF Mass spectra of native insulin, PEG–insulin, and PCB–insulin.
The obtained PCB–insulin was first evaluated for its improved blood circulating potential or PK. Hydrodynamic size of conjugated proteins is a major factor determining theirs in vivo blood circulation behavior.[5a,11] An increase in hydrodynamic size typically supersedes the renal ultrafiltration cut-off and reduces the glomerular filtration of the modified proteins. A prolonged blood circulation half-life has been extensively reported for PEGylated proteins.[12] We measured the hydrodynamic size of conjugated insulin with dynamic light scattering (DLS) method by using a Malvern Nano-ZS Zetasizer (Figure 2A). The hydrodynamic sizes of free insulin, PEG–insulin, and PCB–insulin in pH 8.4 (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES) buffer are 1.4 ± 0.2, 2.8 ± 0.7, and 2.2 ± 0.6 nm, respectively (average ± standard deviation, n = 3). Similar to PEG–insulin, there is a significant hydrodynamic size increase for PCB–insulin over native insulin, implying a potentially protracted blood circulating lifetime for PCB–insulin.
Figure 2.
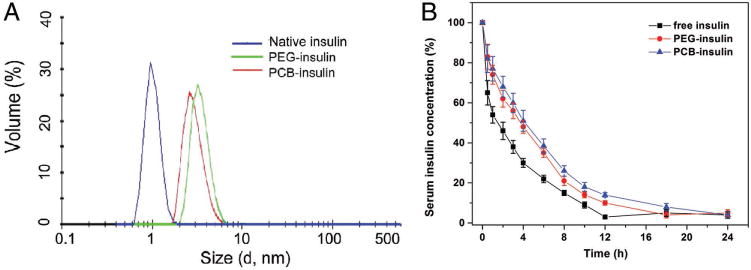
A) Hydrodynamic sizes for free insulin, PEG–insulin, and PCB–insulin, respectively, as measured by DLS. Samples were dissolved in 0.2 M pH8.4 HEPES buffer. B) The serum insulin concentration as a function of time after the tail vein injection of free insulin, PEG–insulin, and PCB–insulin, respectively. The data are presented as the percentage of the initial values of serum insulin concentration over time ± standard deviation (n = 6).
PK of PCB–insulin was examined in a diabetic mouse model. All animal experiments were performed according to the National Institute of Health guidelines for animal research under a protocol approved by the Institutional Animal Care and Use Committee at Wayne State University. C57BL/6 mice received a daily i.p. injection of 5 mg mL−1 streptozocin (STZ) at 50 mg kg−1 for 5 consecutive days. STZ-mice were placed on a nonfasted regular diet. 17 d after the first injection, weight and blood glucose were measured to confirm diabetic status. Only mice whose nonfasted blood glucose levels are above 300 mg dL−1 for two consecutive days were considered diabetic. Free insulin and purified polymer–insulin conjugates were injected into diabetic mice through the tail vein at an equal molar dose of 10 nmol kg−1. Blood samples were collected at various time intervals, and the concentration of serum insulin was quantified with human insulin Enzyme-linked immunosorbent assay (ELISA) assay (Figure 2B). The relative area under the curve (AUC) was calculated and the blood circulation half-life (t1/2) was obtained to fit a one-compartment model[8] (Table 1, Supporting Information). The AUCs for both PCB–insulin and PEG–insulin are larger than that of native insulin. The relative AUC for PCB–insulin is almost 1.43 times of that of native insulin. t1/2 for PCB–insulin (4.2 h) and PEG–insulin (3.6 h) are significantly longer than free insulin (2 h), indicating the prolonged blood circulation effect upon conjugation of both PCB and PEG.
The obtained PCB–insulin was secondly evaluated for potential bioactivity change. We initially studied the effect of polymer conjugation on the secondary structure of insulin, since proteins generally require stabilized structures to contain their particular bioactivity.[13] Circular dichroism (CD) spectroscopy has been widely involved in characterizing the secondary conformation of insulin and its derivatives,[12a,14] and was used here to evaluate free insulin, PCB–insulin, and PEG–insulin (spectra in Figure 3A; quantified results of secondary structures in Table S2, Supporting Information). We found that the percentage of α-helix structure for PEG–insulin is 46%, and is slightly less than that for free insulin which is 50%. The random coil structure proportion for PEG–insulin is 30% and is more than 24% for free insulin. A similar trend has been reported in the literature for PEG conjugated insulin,[12a] indicating a slightly destabilizing effect on the protein structure. For the CD spectrum of PCB–insulin (Figure 3A), there is an obvious decrease of the negative peak at 222 nm, indicating the increase of α-helix structure. Quantified results (Table S2, Supporting information) show that the α-helix structure proportion of PCB–insulin is 53% (slightly more than 50% of free insulin), and the random coil structure proportion is 23% (a little less than 24% of free insulin). This suggests that the conjugation of PCB has a moderate stabilizing effect on the secondary structure of insulin, and implies the potential retention of insulin bioactivity after PCB conjugation.
Figure 3.
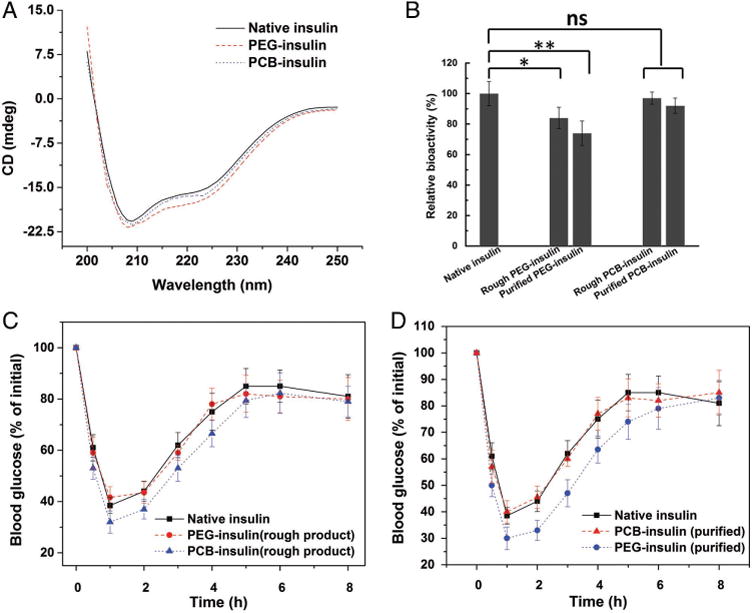
A) Circular dichroism (CD) spectra of native insulin, PEG–insulin, and PCB–insulin (0.2 mg mL−1 insulin or conjugates at 0.2 M pH8.2 PBS buffer), respectively. B) The bioactivity of native insulin, rough PEG–insulin, purified PEG–insulin, rough PCB–insulin, and purified PCB–insulin, respectively, as measured by phosphorylation of the insulin receptor in vitro. The relative activity was presented as the percentage relative to native insulin ± standard deviation (n = 6). *p < 0.05, **p < 0.01, n.s. = not significant. The relative blood glucose concentration as a function of time after the tail vein injection of free insulin, PEG–insulin, and PCB–insulin C) before and D) after purifying insulin conjugate products. The data are presented as the percentage of the initial values of blood glucose concentration ± standard deviation (n = 6).
In vitro activity of PCB–insulin was tested using a cell-based protein kinase B phosphorylation assay. Briefly, 25 000 CHO-M1 cells per well (American Type Culture Collection, ATCC) were seeded in a 96 well tissue culture treated plate and incubated for 24 h before being cultured in serum-free medium overnight. These cells were then treated with 50 µL of native insulin, rough products of PEG–insulin, and PCB–insulin without any purification (containing both conjugated and unconjugated insulin), and purified products of PEG–insulin and PCB–insulin (containing solely of conjugated insulin), respectively, for 45 min. The effective insulin concentration in each of the sample was maintained at 2 nmol mL−1. For rough conjugate products (unpurified), the effective insulin concentration can be easily controlled by the feeding molar content of insulin for the conjugation reaction. For purified conjugate products, we used both UV–vis absorbance (276 nm), and a Mercodia human insulin ELISA assay to quantify effective insulin concentration. Then treated cells were lysed, and a commercially available kit (Cisbio) was used to assay phosphorylated AKT at Ser473. Figure 3B indicates that there was no significant decrease of in vitro bioactivity of both the rough and purified zwitterionic PCB–insulin, but great decrease on the PEG–insulin was observed. The purified PEG–insulin lost ≈27% activity compared with native insulin.
The obtained PCB–insulin was lastly evaluated for its in vivo pharmacological activity. This was done through i.v. injection of PCB–insulin to STZ induced diabetic mice and then monitoring the blood glucose concentration at various time points following the injection. We initially injected free insulin, as well as PCB–insulin and PEG–insulin rough products without any purification (containing both conjugated and unconjugated insulin), to the mice, at the dose of 10 nmol insulin or its conjugate per kg (for native insulin dose, 10 nmol kg−1 = 1.5 IU kg−1). It was found that blood glucose lowering abilities of PEG–insulin and native insulin, as reflected by the relative intensity of blood glucose concentration, are almost similar (Figure 3C). However, remarkably PCB–insulin shows significantly higher blood glucose lowering ability than native insulin (Figure 3C). We further injected free insulin, as well as purified PCB–insulin and PEG–insulin samples to the mice at the dose of 10 nmol kg−1. The purified PEG–insulin did not show any better glucose lowering effect than native insulin (Figure 3D), similar to the result in Figure 3C. But the purified PCB–insulin sample had even higher blood glucose lowering ability than native insulin (Figure 3D), and then its nonpurified form as well (by comparing Figure 3C,D). The in vivo pharmacological activity of insulin samples was evaluated by quantifying the area above the curve (AAC) of the relative intensity of blood glucose concentration. The pharmacological activity of PEG–insulin is about 97.5% comparing with that of free insulin, while PCB–insulin is about 124.3%.
Despite many possible factors proposed, long half-life in blood circulation has been considered to be the most important reason responsive to the high pharmacological activity of proteins.[3a,15] We attribute the increased in vivo pharmacological activity of PCB–insulin to its prolonged blood circulation lifetime plus the retained in vitro bioactivity. PEG–insulin also showed a longer blood circulation character, but because of the loss in in vitro bioactivity its overall in vivo pharmacological activity did not show improvement over native insulin.
Zwitterionic chemical structure of PCB and nonionic structure of PEG result in significant hydrophobicity difference between the two types of polymers: PCB is superhydrophilic while PEG is amphiphilic.[16] The reason for PCB conjugation to retain in vitro bioactivity of the modified insulin might share a similar mechanism to a previously reported PCB conjugated enzyme system. PCB conjugation to α-chymotrypsin did not sacrifice the protein’s catalytic activity, which has been attributed to the superhydrophilic nature of PCB polymer that did not interfere with the hydrophobic–hydrophobic, substrate–binding site interaction.[3a] By contrast, PEG weakened such substrate–binding interaction because of its amphiphilic nature and imposed steric hindrance.[3a] Retained bioactivity of PCB–insulin can be similarly explained by the unimpeded insulin ligand-receptor interaction.
It should be noted that our coupling condition was controlled to have one PCB polymer chain to be conjugated preferentially at LysB29 of insulin to produce PCB–insulin of even higher in vivo pharmacological activity than native insulin. Nevertheless, when PCB was conjugated to most of the three amines of insulin (Figure S1, Supporting Information), there was a drastic loss of in vitro bioactivity (Figure S2A, Supporting Information) and the in vivo pharmacological activity as well (Figure S2B, Supporting Information), potentially due to the occupation of Gly A1 near to the insulin’s active site.
To summarize, our finding indicates that one-PCB-chain modification per insulin obtained from simple, regular conjugation chemistry shows improved PK, retained in vitro bioactivity and remarkably increased in vivo pharmacological activity of lowering blood glucose. The reported PCB–insulin distinguishes itself from nearly all current polymer–protein conjugates which inevitably suffer from bioactivity loss upon polymer conjugation.
Supplementary Material
Acknowledgments
This work was supported by the faculty start-up fund at Wayne State University, Chemical Engineering and Materials Science, and partially supported by the Juvenile Diabetes Research Foundation (1-SRA-2015-41-A-N, and 1-SRA-2016-270-A-N), and the US National Institute of Diabetes And Digestive And Kidney Diseases of the National Institutes of Health (DP2DK111910).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
References
- 1.Pelegri-O’Day EM, Lin EW, Maynard HD. J Am Chem Soc. 2014;136:14323. doi: 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]
- 2.Harris JM, Chess RB. Nat Rev Drug Discovery. 2003;2:214. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 3.a) Keefe AJ, Jiang S. Nat Chem. 2012;4:59. doi: 10.1038/nchem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parrott MC, DeSimone JM. Nat Chem. 2012;4:13. doi: 10.1038/nchem.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Veronese FM, Mero A. Biodrugs. 2008;22:315. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]; d) Chapman AP. Adv Drug Delivery Rev. 2002;54:531. doi: 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]; e) Rodriguez-Martinez JA, Rivera-Rivera I, Sola RJ, Griebenow K. Biotechnol Lett. 2009;31:883. doi: 10.1007/s10529-009-9947-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Antos JM, Francis MB. Curr Opin Chem Biol. 2006;10:253. doi: 10.1016/j.cbpa.2006.04.009. [DOI] [PubMed] [Google Scholar]; b) de Graaf AJ, Kooijman M, Hennink WE, Mastrobattista E. Bioconjugate Chem. 2009;20:1281. doi: 10.1021/bc800294a. [DOI] [PubMed] [Google Scholar]; c) Sletten EM, Bertozzi CR. Angew Chem, Int Ed. 2009;48:6974. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gao WP, Liu WG, Christensen T, Zalutsky MR, Chilkoti A. Proc Natl Acad Sci USA. 2010;107:16432. doi: 10.1073/pnas.1006044107. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Xia Y, Tang SC, Olsen BD. Chem Commun. 2013;49:2566. doi: 10.1039/c3cc38976f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Caparrotta TM, Evans M. Diabetes, Obes Metab. 2014;16:388. doi: 10.1111/dom.12196. [DOI] [PubMed] [Google Scholar]; b) Owens RA, Lockwood JF, Dunbar JD, Zhang C, Ruan X, Kahl SD, Qian HR, Beals JM. Diabetologia. 2012;55:S55. [Google Scholar]
- 6.a) Jiang SY, Cao ZQ. Adv Mater. 2010;22:920. doi: 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]; b) Ueda T, Oshida H, Kurita K, Ishihara K, Nakabayashi N. Polym J. 1992;24:1259. [Google Scholar]; c) Xu Y, Takai M, Ishihara K. Biomacromolecules. 2009;10:267. doi: 10.1021/bm801279y. [DOI] [PubMed] [Google Scholar]; d) Oda H, Konno T, Ishihara K. Biomaterials. 2013;34:5891. doi: 10.1016/j.biomaterials.2013.04.015. [DOI] [PubMed] [Google Scholar]; e) Seo JH, Matsuno R, Lee Y, Konno T, Takai M, Ishihara K. Acta Biomater. 2011;7:1477. doi: 10.1016/j.actbio.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Cao ZQ, Li YT, Ella-Menye JR, Bai T, Jiang SY. ACS Nano. 2012;6:6681. doi: 10.1021/nn301159a. [DOI] [PubMed] [Google Scholar]
- 8.Cao ZQ, Zhang L, Jiang SY. Langmuir. 2012;28:11625. doi: 10.1021/la302433a. [DOI] [PubMed] [Google Scholar]
- 9.a) Zhang P, Sun F, Tsao C, Liu SJ, Jain P, Sinclair A, Hung HC, Bai T, Wu K, Jiang SY. Proc Natl Acad Sci USA. 2015;112:12046. doi: 10.1073/pnas.1512465112. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Liu RY, Shi YJ, Zhang ZZ, Zhang X. Theranostics. 2015;5:583. doi: 10.7150/thno.11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.a) Hoeg-Jensen T, Havelund S, Nielsen PK, Markussen J. J Am Chem Soc. 2005;127:6158. doi: 10.1021/ja051038k. [DOI] [PubMed] [Google Scholar]; b) Hoeg-Jensen T, Ridderberg S, Havelund S, Schaffer L, Balschmidt P, Jonassen I, Vedso P, Olesen PH, Markussen J. J Pept Sci. 2005;11:339. doi: 10.1002/psc.624. [DOI] [PubMed] [Google Scholar]; c) Chou DHC, Webber MJ, Tang BC, Lin AB, Thapa LS, Deng D, Truong JV, Cortinas AB, Langer R, Anderson DG. Proc Natl Acad Sci USA. 2015;112:2401. doi: 10.1073/pnas.1424684112. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Uchio T, Baudyš M, Liu F, Song SC, Kim SW. Adv Drug Delivery Rev. 1999;35:289. doi: 10.1016/s0169-409x(98)00078-7. [DOI] [PubMed] [Google Scholar]; e) Jeong SY, Kimn SW, Eenink MJ, Feijen J. J Controlled Release. 1984;1:57. [Google Scholar]; f) Baudyš M, Uchio T, Mix D, Kim SW, Wilson D. J Pharm Sci. 1995;84:28. doi: 10.1002/jps.2600840108. [DOI] [PubMed] [Google Scholar]
- 11.a) Hu CMJ, Zhang L, Aryal S, Cheung C, Fang RH, Zhang LF. Proc Natl Acad Sci USA. 2011;108:10980. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang W, Zhang L, Wang SL, White AD, Jiang SY. Biomaterials. 2009;30:5617. doi: 10.1016/j.biomaterials.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 12.a) Hinds KD, Kim SW. Adv Drug Delivery Rev. 2002;54:505. doi: 10.1016/s0169-409x(02)00025-x. [DOI] [PubMed] [Google Scholar]; b) Fuertges F, Abuchowski A. J Controlled Release. 1990;11:139. [Google Scholar]
- 13.a) Bhattacharya D, Basu S, Mandal PC. J Photochem Photobiol B. 2000;59:54. doi: 10.1016/s1011-1344(00)00137-8. [DOI] [PubMed] [Google Scholar]; b) Goedert M. Nat Rev Neurosci. 2001;2:492. doi: 10.1038/35081564. [DOI] [PubMed] [Google Scholar]
- 14.a) Melberg SG, Johnson WC. Proteins. 1990;8:280. doi: 10.1002/prot.340080309. [DOI] [PubMed] [Google Scholar]; b) Brems DN, Alter LA, Beckage MJ, Chance RE, Dimarchi RD, Green LK, Long HB, Pekar AH, Shields JE, Frank BH. Protein Eng. 1992;5:527. doi: 10.1093/protein/5.6.527. [DOI] [PubMed] [Google Scholar]; c) Bohm G, Muhr R, Jaenicke R. Protein Eng. 1992;5:191. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- 15.Sato M, Sadamoto R, Niikura K, Monde K, Kondo H, Nishimura SI. Angew Chem, Int Ed. 2004;43:1516. doi: 10.1002/anie.200353058. [DOI] [PubMed] [Google Scholar]
- 16.Cao Z, Jiang S. Nano Today. 2012;7:404. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.