

Abstract
A copper complex, [CuI(tmpa)(MeCN)]+, effectively reductively couples NO(g) at RT in methanol (MeOH), giving a structurally characterized hyponitrito-dicopper( II) adduct. Hydrogen-bonding from MeOH is critical for the hyponitrite complex formation and stabilization. This complex exhibits the reverse redox process in aprotic solvents, giving CuI + NO(g), leading to CuI-mediated NO(g)-disproportionation. The relationship of this chemistry to biological iron and/or copper mediated NO(g) reductive coupling to give N2O(g) is discussed.
The interactions and reactivity of redox active metal ions with nitrogen oxides (NOx) have widespread implications/importance in environmental, practical, and biological chemistries. Iron and copper provide for facile redox shuttling and one and/or the other in the presence of proton sources chemically or enzymatically facilitates reactions such as reduction of nitrite (NO2−) to nitrogen monoxide (NO(g); nitric oxide), reductive coupling of NO(g) to nitrous oxide (N2O(g)), and reduction of N2O(g) to N2(g).1
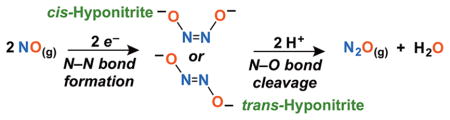
Biological roles of NO(g) include it as a signaling agent, yet, it can be toxic under oxidative conditions; NO(g) is utilized in the human immune response, produced in elevated levels to effect invading pathogen biomolecule destruction (e.g., by oxidation or nitration).2 Fighting back, bacteria/pathogens can upregulate NO-reductase (NOR) activity to eliminate toxic NO(g) by its reductive coupling to give benign N2O(g).3 Bacterial NOR’s1d,4 possess heme/nonheme diiron active sites, effecting the coupling through a putative hyponitrito (N2O22−) intermediate; 5 the metal ion centers provide electrons (diagram above). Evolutionarily related heme-Cu oxidases (HCO’s) effect this reaction, albeit with lower efficiency.6
Our own interests encompass hemes and/or Cu,2a,7 and in this report we focus on the latter. In addition to HCO NO(g) reductive coupling, copper nitrite reductases may also couple excess NO(g) present to give N2O(g).1a Further, in the presence of O2(g), Cu–NO reactivity can lead to peroxynitrite (O=NOO−).2b,8 Also, copper(I) complexes are efficient at NO(g)-disproportionation, 3NO(g) + CuI → N2O(g) + CuII(NO2−), possibly also involving hyponitrite-type intermediates. 1b,9 As well, copper ion loaded zeolites are effective catalysts to decompose NOx to N2(g) and O2(g); automobile catalytic converters may utilize Cu as a catalyst for NO(g) reduction to N2O(g).10
With this background, we highlight the need to elucidate fundamental aspects of NO(g) reductive coupling, details/requirements for (i) N–N bond formation and (ii) N–O bond cleavage for hyponitrite formation and N2O(g) elimination, respectively. Even though hyponitrite intermediates have been proposed/observed within enzymatic systems, details pertaining to their binding mode within the binuclear active site, protonation state(s), and the metal oxidation states, all remain elusive.11 In this report, employing the copper(I) complex [CuI(tmpa)(MeCN)](B(C6F5)4) (MeCN = CH3CN; TMPA = tris(2-pyridylmethyl)amine), we describe (i) the first example of a copper(I) complex whose NO(g) reactivity leads to efficient reductive coupling to give the hyponitrite complex [{CuII(tmpa)}2(μ-N2O2 2−)]2+, (ii) the first structurally characterized Cu-only hyponitrite complex, and (iii) the effective reversal of NO(g) reductive coupling occurring for the hyponitrite complex in tetrahydrofuran (THF), i.e., formation of a putative {[CuI(tmpa)]+ + NO(g)} solution mixture, followed by NO(g)-disproportionation (Figure 1).
Figure 1.

Divergent synthetic routes to [{CuII(tmpa)}2(μ-N2O22−)]2+ in MeOH, and its unique redox behavior in THF, effecting CuI-mediated NO(g)-disproportionation.
When excess NO(g) is bubbled through a RT MeOH solution of [CuI(tmpa)(MeCN)]+, the initial pale yellow solution immediately transforms to forest green giving electronic absorption features: λmax = 310 (ε = 3800 M−1 cm−1), 675 (ε = 250 M−1 cm−1), and 870 nm (ε = 340 M−1 cm−1) (Figure 2). An EPR spectrum of the green solution reveals a classic reverse axial pattern (g⊥ > g||) indicating a dz2 ground state, with a single type of mononuclear copper(II) environment occurring in a trigonal bipyramidal (TBP) geometry; this also indicates that the copper(II) ions are electronically site-isolated, i.e., no magnetic/electronic interaction between the two copper(II) ions occurs. Upon solvent removal, this new complex was obtained in near stoichiometric yields (>97%), and it is the hyponitrito complex [{CuII(tmpa)}2(μ-N2O22−)](B(C6F5)4)2·2H2O·2MeOH (eq 1).12
Figure 2.

Top: Electronic absorption spectrum resulting from the NO(g) reactivity with 1 mM [CuI(tmpa)(MeCN)]+ in MeOH at RT. Bottom: EPR spectra (experimental (green) and simulated (red)) of [{CuII(tmpa)}2(μ-N2O22−)]2+ (2 mM in MeOH/EtOH = 1:1 glass) at 20 K (g⊥ = 2.18, g|| = 1.99; A⊥ = 82 G, A|| = 90 G).
| (1) |
With the intention of further supporting our complex formulation, we probed the CuI/NO(g) stoichiometry; [CuI (tmpa)(MeCN)]+ was titrated with varying amounts of NO(g) from a NO(g)-saturated MeOH solution {[NO(g)] = 14.5 mM at RT}.12 The data clearly indicates that the CuI/NO(g) stoichiometry, as measured in this manner, lies between 1 and 1.5, very close to the expected 1:1 stoichiometry.12,13 It is presumed that NO(g) reaction with [CuI(tmpa)(MeCN)]+ initially gives a CuI–NO adduct, perhaps like that previously reported on (but in EtCN),14 prior to NO(g)-coupling in the present case to give the hyponitrito-dicopper(II) complex (eq 1).5
[{CuII(tmpa)}2(μ-N2O2 2−)]2+ can also be generated by a metathesis reaction involving the CuII–OH complex, [CuII(tmpa)(OH)]+, generated and isolated by reaction of tetrabutylammonium hydroxide with [CuII(tmpa)(MeCN)]-(ClO4)2 in MeCN at RT (νO–H = 3533 cm−1).12 Freshly generated [CuII(tmpa)(OH)]+ mixing with 1/2 equiv Ag2N2O2 in MeOH, with sonication, gave a color change from light green to forest green. The UV–vis changes correspond with those seen for the hyponitrito-dicopper(II) complex obtained via the {[CuI + NO(g)} reaction.12 Isolation of the solid product and redissolution into a MeOH/EtOH = 1:1 solution also gives the same EPR spectrum (20 K).12
X-ray quality crystals of [{CuII(tmpa)}2(μ-N2O22−)](ClO4)2 could be obtained from MeOH at −35 °C, and the structure of the dicationic portion is shown in Figure 3.12 The complex possesses a dicopper-bridged η1-O-trans-hyponitrite species with a Cu···Cu distance of 5.5648(7) Å; the dication possesses a centrosymmetric structure. Each copper(II) ion is pentacoordinate, with the four N atoms from the TMPA chelate plus the anionic hyponitrito O atom (O1 or O1′; Figure 3). However, there also exists a clear interaction of Cu ions with the distal N atom of the N2O22− moiety, (Cu–N = 2.851(2) Å), i.e., Cu1 interacts with N5′, with Cu1′ interacting with N5 (Figure 3, shown in broken lines). See the SI (page S10) for information and arguments in support of this point. We hypothesize that this weak interaction transforms into a “true” metal–ligand bond when [{CuII(tmpa)}2(μ-N2O22−)]2+ is put into aprotic solvents (vide inf ra; Scheme 1).
Figure 3.

X-ray diffraction ORTEP diagram (50% probability thermal ellipsoids) of [{CuII(tmpa)}2(μ-N2O22−)]2+ along with the atom numbering scheme. Hydrogen atoms and noncoordinating perchlorate counteranions are omitted for clarity.12
Scheme 1.

Proposed Decay of [{CuII(tmpa)}2(μ-N2O22−)]2+ when Placed in THF, Giving a Final Product Mixture Consisting of [CuI(tmpa)]+, [CuII(tmpa)(NO2)]+, and N2O(g)
Thus, the copper(II) ions in this structure are distorted octahedrally ligated (six-coordinate), which contrasts greatly with other many well characterized [CuII(tmpa)(X)]n+ species such as those with X = Cl− (n = 1),15 X = nitrito (NO2−, Obound; n = 1)16 and X = NCCH3 (n = 2).17 Recall that methanolic solutions of [{CuII(tmpa)}2(μ-N2O22−)]2+ possess a well-defined TBP coordination geometry, as distinctly and unambiguously observed from frozen solution EPR spectra (vide supra). In fact, this hyponitrito-dicopper(II) complex, [{CuII(tmpa)}2(μ-N2O22−)]2+, is only stable in alcoholic solvents; we thus conclude that the latter observation indicates that MeOH hydrogen-bonds to the N2O22− ligand of the complex, probably with the lone pairs on the central N atoms. Such an interaction has recently been suggested by Liaw and co-workers18 although in that case H-bonding to the CoII–NO O atom is proposed, which aids the formation of a hyponitrite metastable species. Such H-bonding in solution would “relieve” the structurally observed weak interaction between N5 or N5′ with the copper(II) ions, leaving only the O1-to-Cu1 (and O1′-to-Cu1′) bond interaction in addition to N4 ligation from TMPA, allowing for strict TBP coordination (as expected from EPR spectra).
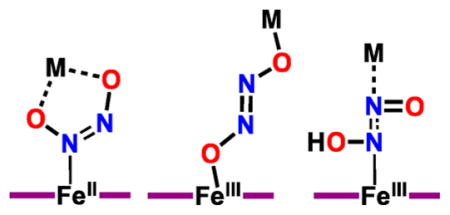
Further aspects of the solid-state structure of [{CuII(tmpa)}2(μ-N2O22−)]2+ are as follows: (a) The N–N and N–O distances of the trans-hyponitrite moiety are 1.257(3) Å and 1.361(2) Å, respectively, which compare well with the N=N double bond (1.256(2) Å) and N–O single bond (1.3622(11) Å) distances of trans-sodium hyponitrite.5c (b) The Cu···Cu distance of 5.565 Å is much shorter compared with the Fe–Fe distance (6.7 Å) in [(OEP)FeIII-ONNOFeIII( OEP)] (OEP = octaethylporphyrinate), with extended hyponitrito; O,O′ bonding to iron(III) occurs, but there are no Fe···N(hyponitrite) interactions.20c (c) The Cu···Cu distance here is virtually identical to that (5.566 Å) of the Agapie’s19 copper–yttrium complex with bridged copper-bound hyponitrite adduct. (d) A number of differing structures have been proposed based on computational analyses for HCO, NOR or other protein hyponitrite species (see diagram below; M = Fe or Cu).4e,6d,20
When solid samples of [{CuII(tmpa)}2(μ-N2O22−)]2+ (as (B(C6F5)4− or ClO4− salts) are dissolved in THF, or excess THF is added to the complex already dissolved in MeOH, a reaction takes place, and a mixture of products are formed (Figure 1), which we hypothesize to arise from a rearrangement of electron density on the bridging N2O22− ligand,21 followed by transformations leading to the products seen from reaction of [CuI(tmpa)]+ + NO(g) in nonalchoholic solvents, namely NO(g)-disproportionation, 3NO(g) + CuI → N2O(g) + CuII–NO2−. We speculate that either THF effects a “reverse” reaction to give a {[CuI(tmpa)]+ + NO(g)} solution, or [{CuII(tmpa)}2(μ-N2O22−)]2+ reacts to give N2O(g) and the CuII-nitrito complex by an as yet undetermined mechanism. However, because there is only one equiv of NO(g) per Cu(I) ion present, excess {[CuI(tmpa)]+ is left over, giving the mixture of products described by Scheme 1.
Experiments carried out on the final product solution confirm the identity, yields and relative-yields of the transformation of [{CuII(tmpa)}2(μ-N2O22−)]2+ originally formed by the {CuI + NO(g)} reaction (vide supra) (with stoichiometry of Scheme 1). (a) Cooling the decay product mixture in THF to −80 °C and bubbling with O2(g) led to the formation of the well-known trans-peroxo-dicopper(II) complex, [{CuII(tmpa)}2(μ-O22−)]2+;22 with known molar absorptivities, one could relate back to the finding of a 96% yield of [CuI(tmpa)]+.12,22 (b) Previously published methodologies16 were followed to determine and quantify the nitrite content in the final decay mixture, giving >85% yields,12 and (c) the headspace of the decay product mixture was sampled by gas chromatographic analysis for the identification and quantitation of product N2O(g); yields of ~85% were obtained.12 The decay of [{CuII(tmpa)}2(μ-N2O22−)]2+ originally formed by the {CuII–OH + Ag2N2O2} method (vide supra), gave 96% [CuI(tmpa)]+, > 85% yield of nitrite and 98% yield N2O(g),12 again all based on the Scheme 1 reaction stoichiometry.
Finally, to further verify the solvent effect for the {[CuI(tmpa)]+ + NO(g)} reaction, we carried out separate experiments. When [Cu(tmpa)(MeCN)]+ is subjected to excess NO(g) in THF solution at RT, the pale yellow solution immediately turns to an olive green color exhibiting the UV–vis features corresponding to the nitrito complex [CuII(tmpa)-(NO2)]+ (96% yield) λmax = 303 (ε = 3360 M−1 cm−1), 420 (ε = 1340 M−1 cm−1) nm. N2O(g) is also produced, so we can conclude that NO(g)-disproportionation occurs.1b As described in detail above, in MeOH as solvent, the hyponitrito-dicopper ( II) complex forms. It is remarkable how the reactivity between [CuI(tmpa)]+ and NO(g) modify to such great extent merely by altering the solvent properties.
In summary, herein we report the first Cu-only system that mediates the reductive coupling of NO(g), stoichiometrically yielding the corresponding hyponitrito-dicopper(II) complex in MeOH at RT. We suggest that H-bonding to the N2O2 2− ligand is key to its stabilization and this course of reaction. Unlike other synthetic bioinspired examples,5a,7b,19,23 this reaction does not require any low-temperature conditions, and is independent of the equiv of added NO(g). The new hyponitrito complex possesses versatile synthetic pathways, and is characterized by X-ray crystallography. Dissolution in aprotic solvents triggers its decay with essentially the reversal of NO(g) coupling, leading to products of CuI-mediated NO(g)-disproportionation. We should also note that addition of HCl to [{CuII(tmpa)}2(μ-N2O22−)]2+ in MeOH gives rise to the release of N2O(g).12 Such unique redox behavior involving copper and NO(g) is unprecedented, and may have implications in metalloenzyme related NO(g)/NOx biochemistry. A deeper understanding is needed as pertains to the NO(g)-coupling mechanism (N–N bond formation), the oxidation and/or protonation state of the N2O2-type intermediate formed,6a,9,11,24 and the mechanism of (N–O bond cleavage) N2O(g) formation.
Supplementary Material
Acknowledgments
The research support of the National Institutes of Health (GM 060353) is gratefully acknowledged.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b07808.
Synthetic and analytical details (methodologies and UV–vis, EPR, NMR, and FT-IR spectra; gas chromatograms); X-ray diffraction data collection details (PDF) Data for C36H36Cl2Cu2N10O10 (CIF)
References
- 1.(a) Averill BA. Chem Rev. 1996;96:2951. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]; (b) Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem Rev. 2002;102:1201. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]; (c) Ford PC, Lorkovic IM. Chem Rev. 2002;102:993. doi: 10.1021/cr0000271. [DOI] [PubMed] [Google Scholar]; (d) Moënne-Loccoz P. Nat Prod Rep. 2007;24:610. doi: 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ford PC. Inorg Chem. 2010;49:6226. doi: 10.1021/ic902073z. [DOI] [PubMed] [Google Scholar]; (f) Timmons AJ, Symes MD. Chem Soc Rev. 2015;44:6708. doi: 10.1039/c5cs00269a. [DOI] [PubMed] [Google Scholar]; (g) Merkle AC, Lehnert N. Dalton Trans. 2012;41:3355. doi: 10.1039/c1dt11049g. [DOI] [PubMed] [Google Scholar]
- 2.(a) Schopfer MP, Wang J, Karlin KD. Inorg Chem. 2010;49:6267. doi: 10.1021/ic100033y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Szabo C, Ischiropoulos H, Radi R. Nat Rev Drug Discov. 2007;6:662. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]; (c) Barry SM, Kers JA, Johnson EG, Song L, Aston PR, Patel B, Krasnoff SB, Crane BR, Gibson DM, Loria R, Challis GL. Nat Chem Biol. 2012;8:814. doi: 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Radi R. Acc Chem Res. 2013;46:550. doi: 10.1021/ar300234c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Justino MC, Ecobichon C, Fernandes AF, Boneca IG, Saraiva LM. Antioxid Redox Signal. 2012;17:1190. doi: 10.1089/ars.2011.4304. [DOI] [PubMed] [Google Scholar]; (b) Kakishima K, Shiratsuchi A, Taoka A, Nakanishi Y, Fukumori Y. Biochem Biophys Res Commun. 2007;355:587. doi: 10.1016/j.bbrc.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 4.(a) Watmough NJ, Field SJ, Hughes RJL, Richardson DJ. Biochem Soc Trans. 2009;37:392. doi: 10.1042/BST0370392. [DOI] [PubMed] [Google Scholar]; (b) Shiro Y. Biochim Biophys Acta, Bioenerg. 2012;1817:1907. doi: 10.1016/j.bbabio.2012.03.001. [DOI] [PubMed] [Google Scholar]; (c) Al-Attar S, de Vries S. FEBS Lett. 2015;589:2050. doi: 10.1016/j.febslet.2015.06.033. [DOI] [PubMed] [Google Scholar]; (d) Hayashi T, Lin MT, Ganesan K, Chen Y, Fee JA, Gennis RB, Moënne-Loccoz P. Biochemistry. 2009;48:883. doi: 10.1021/bi801915r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Bhagi-Damodaran A, Petrik I, Lu Y. Isr J Chem. 2016;56:773. doi: 10.1002/ijch.201600033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Arikawa Y, Onishi M. Coord Chem Rev. 2012;256:468. [Google Scholar]; (b) Wright AM, Hayton TW. Inorg Chem. 2015;54:9330. doi: 10.1021/acs.inorgchem.5b00516. [DOI] [PubMed] [Google Scholar]; (c) Arulsamy N, Bohle DS, Imonigie JA, Sagan ES. Inorg Chem. 1999;38:2716. [Google Scholar]
- 6.(a) Pinakoulaki E, Ohta T, Soulimane T, Kitagawa T, Varotsis C. J Am Chem Soc. 2005;127:15161. doi: 10.1021/ja0539490. [DOI] [PubMed] [Google Scholar]; (b) Varotsis C, Ohta T, Kitagawa T, Soulimane T, Pinakoulaki E. Angew Chem, Int Ed. 2007;46:2210. doi: 10.1002/anie.200602963. [DOI] [PubMed] [Google Scholar]; (c) Pinakoulaki E, Varotsis C. J Inorg Biochem. 2008;102:1277. doi: 10.1016/j.jinorgbio.2008.01.014. [DOI] [PubMed] [Google Scholar]; (d) Ohta T, Soulimane T, Kitagawa T, Varotsis C. Phys Chem Chem Phys. 2015;17:10894. doi: 10.1039/c5cp01013f. [DOI] [PubMed] [Google Scholar]
- 7.(a) Moënne-Loccoz P, Richter O-MH, Huang H-w, Wasser IM, Ghiladi RA, Karlin KD, de Vries S. J Am Chem Soc. 2000;122:9344. [Google Scholar]; (b) Wang J, Schopfer MP, Puiu SC, Sarjeant AAN, Karlin KD. Inorg Chem. 2010;49:1404. doi: 10.1021/ic901431r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Schopfer MP, Mondal B, Lee DH, Sarjeant AAN, Karlin KD. J Am Chem Soc. 2009;131:11304. doi: 10.1021/ja904832j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Pacher P, Beckman JS, Liaudet L. Physiol Rev. 2007;87:315. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Su J, Groves JT. Inorg Chem. 2010;49:6317. doi: 10.1021/ic902157z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metz S. Inorg Chem. 2017;56:3820. doi: 10.1021/acs.inorgchem.6b02551. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kwak JH, Tran D, Szanyi J, Peden CHF, Lee JH. Catal Lett. 2012;142:295. [Google Scholar]; (b) Gao F, Mei D, Wang Y, Szanyi J, Peden CHF. J Am Chem Soc. 2017;139:4935. doi: 10.1021/jacs.7b01128. [DOI] [PubMed] [Google Scholar]; (c) Granger P, Parvulescu VI. Chem Rev. 2011;111:3155. doi: 10.1021/cr100168g. [DOI] [PubMed] [Google Scholar]; (d) Mohiuddin AKM. Adv Mater Res. 2015;1115:462. [Google Scholar]
- 11.(a) Yi J, Morrow BH, Campbell ALOC, Shen JK, Richter-Addo GB. Chem Commun. 2012;48:9041. doi: 10.1039/c2cc34655a. [DOI] [PubMed] [Google Scholar]; (b) Daskalakis V, Ohta T, Kitagawa T, Varotsis C. Biochim Biophys Acta, Bioenerg. 2015;1847:1240. doi: 10.1016/j.bbabio.2015.06.014. [DOI] [PubMed] [Google Scholar]
- 12.See Supporting Information.
- 13.NO(g)-saturated solutions used likely lose some NO(g) into the headspace of the Ar-filled cuvette containing [CuI(tmpa)(MeCN)]+ during each experiment, while NO(g) equilibrates between the liquid–gas interface. Thus, the actual CuI:NO(g) stoichiometry could be slightly smaller than the experimentally observed ratio.
- 14.Paul PP, Karlin KD. J Am Chem Soc. 1991;113:6331. [Google Scholar]
- 15.Karlin KD, Hayes JC, Juen S, Hutchinson JP, Zubieta J. Inorg Chem. 1982;21:4106. [Google Scholar]
- 16.Hematian S, Siegler MA, Karlin KD. J Am Chem Soc. 2012;134:18912. doi: 10.1021/ja3083818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox S, Nanthakumar A, Wikström M, Karlin KD, Blackburn NJ. J Am Chem Soc. 1996;118:24. [Google Scholar]
- 18.Chuang CH, Liaw WF, Hung CH. Angew Chem, Int Ed. 2016;55:5190. doi: 10.1002/anie.201512063. [DOI] [PubMed] [Google Scholar]
- 19.Lionetti D, de Ruiter G, Agapie T. J Am Chem Soc. 2016;138:5008. doi: 10.1021/jacs.6b01083. [DOI] [PubMed] [Google Scholar]
- 20.(a) Blomberg MRA, Siegbahn PEM. J Comput Chem. 2016;37:1810. doi: 10.1002/jcc.24396. [DOI] [PubMed] [Google Scholar]; (b) Blomberg MRA. Biochemistry. 2017;56:120. doi: 10.1021/acs.biochem.6b00788. [DOI] [PubMed] [Google Scholar]; (c) Xu N, Campbell ALO, Powell DR, Khandogin J, Richter-Addo GB. J Am Chem Soc. 2009;131:2460. doi: 10.1021/ja809781r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.(a) Hyponitrite N-ligation, proposed for hemes,21b are possible, although the calculations of Richter-Addo and Lehnert suggest it to be higher energy than that for O-coordination. Berto TC, Xu N, Lee SR, McNeil AJ, Alp EE, Zhao J, Richter-Addo GB, Lehnert N. Inorg Chem. 2014;53:6398. doi: 10.1021/ic5002573.
- 22.Tyeklár Z, Jacobson RR, Wei N, Murthy NN, Zubieta J, Karlin KD. J Am Chem Soc. 1993;115:2677. [Google Scholar]
- 23.Collman JP, Yang Y, Dey A, Decréau RA, Ghosh S, Ohta T, Solomon EI. Proc Natl Acad Sci U S A. 2008;105:15660. doi: 10.1073/pnas.0808606105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.(a) Even the N2O21− radical form of the intermediate in NO reductive coupling has been observed spectroscopically24b,c or proposed.6,9Brozek CK, Miller JT, Stoian SA, Dincä M. J Am Chem Soc. 2015;137:7495. doi: 10.1021/jacs.5b03761.Poskrebyshev GA, Shafirovich V, Lymar SV. J Am Chem Soc. 2004;126:891. doi: 10.1021/ja038042l.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.