

Abstract
Cytosine derivative dysregulations represent important epigenetic modifications whose impact on the clinical outcome in chronic lymphocytic leukemia (CLL) is incompletely understood. Hence, global levels of 5-methylcytosine (5-mCyt), 5-hydroxymethylcytosine (5-hmCyt), 5-carboxylcytosine (5-CaCyt) and 5-hydroxymethyluracil were tested in purified B cells from CLL patients (n = 55) and controls (n = 17). The DNA methylation ‘writers’ (DNA methyltransferases [DNMT1/3A/3B]), ‘readers’ (methyl-CpG-binding domain [MBD2/4]), ‘editors’ (ten-eleven translocation [TET1/2/3]) and ‘modulators’ (SAT1) were also evaluated. Accordingly, patients were stratified into three subgroups. First, a subgroup with a global deficit in cytosine derivatives characterized by hyperlymphocytosis, reduced median progression free survival (PFS = 52 months) and shorter treatment free survival (TFS = 112 months) was identified. In this subgroup, major epigenetic modifications were highlighted including a reduction of 5-mCyt, 5-hmCyt, 5-CaCyt associated with DNMT3A, MBD2/4 and TET1/2 downregulation. Second, the cytosine derivative analysis revealed a subgroup with a partial deficit (PFS = 84, TFS = 120 months), mainly affecting DNA demethylation (5-hmCyt reduction, SAT1 induction). Third, a subgroup epigenetically similar to controls was identified (PFS and TFS > 120 months). The prognostic impact of stratifying CLL patients within three epigenetic subgroups was confirmed in a validation cohort. In conclusion, our results suggest that dysregulations of cytosine derivative regulators represent major events acquired during CLL progression and are independent from IGHV mutational status.
Keywords: chronic lymphocytic leukemia, methylation, hydroxymethylation, TET, SAT1
INTRODUCTION
The clinical outcome of chronic lymphocytic leukemia (CLL) is heterogeneous with some patients remaining asymptomatic for decades, while others progress rapidly and need therapy soon after diagnosis [24]. Due to the development of novel targeted therapies in CLL, the overall survival of the patients has increased [3].In contrast, the criteria for treatment initiation have not evolved in the last decades alluding to the fact that determining the delay from diagnosis to treatment initiation represents the gold standard for evaluating disease progression [11]. Therefore, when using the treatment free survival (TFS) as end point, a long list of prognostic factors has been established such as the mutational status of the immunoglobulin heavy chain variable region (IGHV), cytogenetic alterations, CD38, and more recently, epigenetic modifications, which have been proposed to be the most accurate factor for disease progression [32, 33, 42]. Moreover, the notion that epigenetic factors are relevant for CLL is further supported by the observation that CLL tumor cells carry less genetic mutations than other adult cancers [30].
Epigenome-wide association studies (EWAS) have contributed to better understanding the significance of DNA methylation in CLL by showing: (1) global DNA hypomethylation combined with local hypermethylation in CLL B cells as observed in other cancers [22]; (2) a link between the methylome profile heterogeneity, an increased genetic complexity and adverse clinical outcome, thus suggesting some epigenetic modifications as an unfavorable and heritable trait [29, 35]; (3) a distinct DNA methylation signature between the two molecular IGHV subtypes in relation to naïve B cells and unmutated (UM)-CLL, or memory B cells and mutated (M)-CLL [27]; (4) that epigenetic patterns are similar between peripheral blood and lymph node compartments [4], while in contrast, the transcriptome is influenced by the B-cell receptor (BCR) activation in the microenvironment of the lymph node [13]; and finally (5) aberrant DNA methylation and specific chromatin accessible region overlap with gene loci encoding for risk factors [44]. In the CLL prone-mice model Eµ-TCL1, development of the disease is preceded by the appearance of important epigenetic modifications [7]. It has also been shown that mice with biallelic loss of the DNA methyltransferase (DNMT)3A develop a lymphoproliferative disease similar to CLL [17].
In general, DNA methylation is considered to be a tightly regulated and stable epigenetic mark. Therefore, modifications in DNA methylation have a major impact in cancer and certainly, CLL. Multiple pathways control DNA methylation. Biochemically, DNA methylation refers to the addition of a methyl subgroup to the fifth carbon of a cytosine by DNMT1/3A/3B resulting in generation of 5-methylcytosine (5-mCyt), and then, methyl-CpG binding domain (MBD) proteins are recruited to regulate chromatin behavior and gene expression. DNMTs and MBDs expression patterns were studied in CLL to a certain extent, mainly revealing that the mechanisms are complex and that other regulators are involved [26]. An active DNA demethylation process was recently described involving the Ten-Eleven Translocation (TET) enzyme family (TET1/2/3), which oxidizes 5-mCyt into 5-hydroxymethylcytosine (5-hmCyt) and subsequently, in a less efficient fashion, into 5-formylcytosine (5-fCyt) and 5-carboxylcytosine (5-CaCyt) with the use of α-ketoglutarate (α-KG), molecular oxygen and iron as cofactors [47]. TET expression varies in CLL patients but paradoxically it is lower expression that has been reported for patients with a reduced TFS [50], while others have reported an overexpression in CLL B cells compared to controls [19]. DNA methylation and active DNA demethylation processes are not restricted to DNMTs and TETs, respectively, as other models have demonstrated that 5-hmC can be enzymatically deaminated by activation-induced deaminase (AID)/apolipoprotein B editing complex (APOBEC) to 5-hydroxymethyluracil (5-hmU) which can then be processed by various DNA glycosylases, including MBD4, generating an unmodified cytosine [21, 31, 37]. Other events can modify methylation levels, such as an increase in spermidine/spermine N1-acetyltransferase (SAT1) which can promote an excessive consumption of S-adenosyl methionine (SAM), the methyl donor molecule leading to global DNA hypomethylation [5, 23, 34].
In the present study, we measured cytosine derivatives and associated them with the clinical outcome to define three subgroups with distinct disease courses. We also investigated DNA methylation regulators according to the 5-Cyt derivative profile in order to better understand key factors regulating DNA methylation and active DNA demethylation in CLL during disease progression.
RESULTS
CLL population
The study cohort consists of 55 untreated patients diagnosed with CLL according to the WHO classification criteria [46]. As listed in Table 1, the median age at diagnosis was 65 years (range 43–84) with a majority of CLL patients with Binet stage A disease 34/55 (60.7%),followed by Binet stage B 19/55 (33.9%) and C 3/55 (5.5%). The median progression free survival (PFS) and TFS of the studied cohort were 104 months and 120 months, respectively. The major prognostic markers associated with disease progression were analyzed including lymphocytosis, lymphocyte doubling time (LDT), IGHV mutational status, CD38 and cytogenetic factors. Patients were categorised into cytogenetic risk groups according to the following classification: low risk group - isolated del(13q); intermediate risk group - trisomy 12 or normal karyotype and fluorescence in situ hybridization (FISH) and high risk group - del(11q), del(17p) or complex karyotype [41].
Table 1. Characteristics of the patients.
| Characteristics | Value |
|---|---|
| Sex (male/female) - number of patients | 37/19 |
| Age at diagnosis - median [range] | 65 [43–84] |
| Age at study entry - median [range] | 72 [55–88] |
| Binet stage - number of patients (%) | |
| A | 34/55 (60.7%) |
| B | 19/55 (33.9%) |
| C | 3/55 (5.4%) |
| Lymphocytosis (Giga/L) - median (± SEM) | 51.6 (± 6.53) |
| Lymphocyte doubling time - median (months)* | 24 |
| IGHV mutational status - number of patients (%) | |
| Unmutated (≥ 98% homology) | 10/48 (20.8%) |
| Mutated (< 98% homology) | 38/48 (79.2%) |
| CD38 > 30% - number of patients (%) | 14/55 (25%) |
| Cytogenetic risk category - number of patients (%) | |
| Low risk - Isolated del(13q) | 17/55 (30.4%) |
| Intermediate risk- Trisomy 12, normal karyotype and FISH | 15/55 (26.8%) |
| High risk - del(11q), del(17p), complex karyotype | 12/55 (21.4%) |
| Progression free survival - median (months)* | 104 |
| Treatment free survival - median (months)* | 120 |
Abbreviations: IGHV, immunoglobulin heavy chain variable region; FISH, fluorescence in situ hybridization; *, Kaplan-Meyer survival analysis.
Cytosine (5-Cyt) derivatives and clinical outcome in CLL
As the DNA methylation and active DNA demethylation processes are dynamic and suspected to vary during disease evolution in CLL [35], four adapted enzyme linked immunosorbent assay (ELISA) techniques were performed in order to study the relationship between global DNA methylation (5-mCyt), on the one hand, and the active demethylation pathway (5-hmCyt, 5-CaCyt and 5-hmU), on the other hand. The ELISAs were performed on genomic DNA extracted from purified B cells of 55 CLL patients (Figure 1 - left part, Table 1) and patients were dichotomized into high versus low levels using the profile likelihood method in a Cox regression model of TFS for optimal cut-off identification.
Figure 1. Global levels of cytosine derivatives in chronic lymphocytic leukemia (CLL) patients.

(A) 5-methylcytosine (5-mCyt), (B) 5-hydroxymethylcytosine (5-hmCyt), (C) 5-carboxylcytosine (5-CaCyt), and (D) 5-hydroxymethyluracil (5-hmU) were determined in purified B cells from 55 untreated CLL patients. ELISA results plots (left) for each 5-Cyt derivative and Kaplan-Meier plots depicting the progression free survival (PFS, middle) and treatment free survival (TFS, right) probability over time from diagnosis. ELISA results are expressed as indexes using a DNA reference sample for normalization (see Materials and methods section). The optimal cut-off, represented by dashed lines on the ELISA plots (left), was calculated using the profile likelihood method in a Cox regression model of TFS. Statistical significance was determined by log-rank test and P < 0.05 are indicated; ns, not significant.
Regarding PFS (Figure 1 - middle part), the Kaplan-Meier log-rank analysis revealed significant differences for the two active DNA demethylation marks 5-hmCyt (P = 0.02) and 5-CaCyt (P = 0.02). When TFS was taken into consideration (Figure 1 - right part), the impact of the DNA methylation marks, 5-mCyt, was more important (median TFS was 108 months in low 5-mCyt CLL group versus > 120 months in high 5-mCyt CLL group, P = 0.003), followed by the active DNA demethylation marks, 5-hmCyt (112 months versus > 120 months, P = 0.02) and 5-CaCyt (112 months versus > 120 months, P = 0.01). For 5-hmU, differences did not reach significance regarding PFS and TFS. Accordingly, the removal of the hydroxymethyl group via 5-hmU seems not to be a highly implicated pathway in the dysregulated dynamics of active DNA demethylation in CLL. Therefore, the latter parameter was no longer considered for further analysis.
Interconnection between DNA methylation and active DNA demethylation processes in CLL B cells
Based on the observation that an association was observed between PFS and the dysregulation of the active DNA demethylation process (5-hmCyt and/or 5-CaCyt low), on the one hand, and between TFS and a reduction in the three 5-Cyt derivatives, on the other hand, we hypothesized that both mechanisms are interconnected in CLL B cells. Therefore, we further segregated CLL patients according to the combined analysis of 5-mCyt, 5-hmCyt and 5-CaCyt. Three subgroups were revealed as follows: CLL patients with a global deficiency in 5-Cyt derivatives (Global, n = 22), CLL patients with 1 or 2 deficient 5-Cyt derivatives (Partial, n = 18) and CLL patients with a 5-Cyt derivative content similar to normal B cells (Normal, n = 15) (Figure 2A). Comparison with control B cells (B cells) from 17 subjects (Figure 2B), revealed for the subgroup with a global deficit the diminishing of 5-mCyt (index: 0.83 ± 0.17 in the CLL Global subgroup versus 1.07 ± 0.05 in control B cells, P < 10-4), 5-hmCyt (index: 0.17 ± 0.04 in the CLL Global subgroup versus0.23 ± 0.01 in control B cells, P < 10–4) and 5-CaCyt (index: 0.54 ± 0.10 in the CLL Global subgroup versus 0.69 ± 0.02 in control B cells, P < 10–4). In the subgroup with a partial deficit, decrease was restricted to 5-hmCyt (index: 0.20 ± 0.03 in the CLL Partial subgroup versus 0.23 ± 0.01 in control B cells, P = 0.01), while no differences were reported for CLL patients from the Normal subgroup compared to control B cells. Of note, 5-mCyt, 5-hmCyt and 5-CaCyt levels were highly correlated (P < 0.0001 for all; Figure 2C), which highlights the interconnections between the different processes.
Figure 2. Analysis of cytosine (5-Cyt) derivatives in chronic lymphocytic leukemia (CLL) B cells defines distinct subgroups.

(A) The graphical representation indicates the presence of three subgroups by comparing CLL B cells and control B cells with high and low levels of global 5-methylcytosine (5-mCyt), 5-hydroxymethylcytosine (5-hmCyt), and 5-carboxylcytosine (5-CaCyt) as determined in Figure 1. The 3 subgroups: global deficiency in all 5-Cyt derivatives (global, n = 22), partial deficiency in 1 or 2 of the 5-Cyt derivatives (partial, n = 18), and 5-Cyt derivative normal content (normal, n=15) similar to healthy control B cells (B cells, n = 17). (B) 5-Cyt derivative levels according to the 3 CLL subgroups and control B cells. The median value is indicated (line), and statistical differences of the comparison with control B cells are indicated as follows: (***) P < 0.001; (**) 0.001 < P < 0.01; (*) 0.01< P < 0.05; ns, not significant. (C) Linear regression analysis between 5-mCyt, 5-hmCyt and 5-CaCyt. The P and r2 values are indicated.
Epigenetic regulator expression is independent from IGHV mutational status and the levels are similar to memory B cells
In order to explain differences between the three 5-Cyt derivative subgroups, we first suspected an innate process due to important variations during germinal B cell differentiation [28]. For getting insight into the mechanisms controlling DNA methylation and active DNA demethylation in CLL, DNA methylation ‘writers’, DNMT1/3A/3B; ‘readers’, MBD2/4; ‘editors’, TET1/2/3; and the ‘modulator’ SAT1, were tested by real time quantitative (RTq)-polymerase chain reaction (PCR) on purified peripheral blood naïve B cells, CD5 B1 cells and memory B cells from 6 healthy controls and B cells from 26 CLL patients for whom we have also determined the IGHV mutational status (Figure 3). DNMT3B, which was expressed at low levels in the CLL and control B cell subsets, was excluded from further analysis.
Figure 3. Epigenetic regulator expression varies according to B cell development.
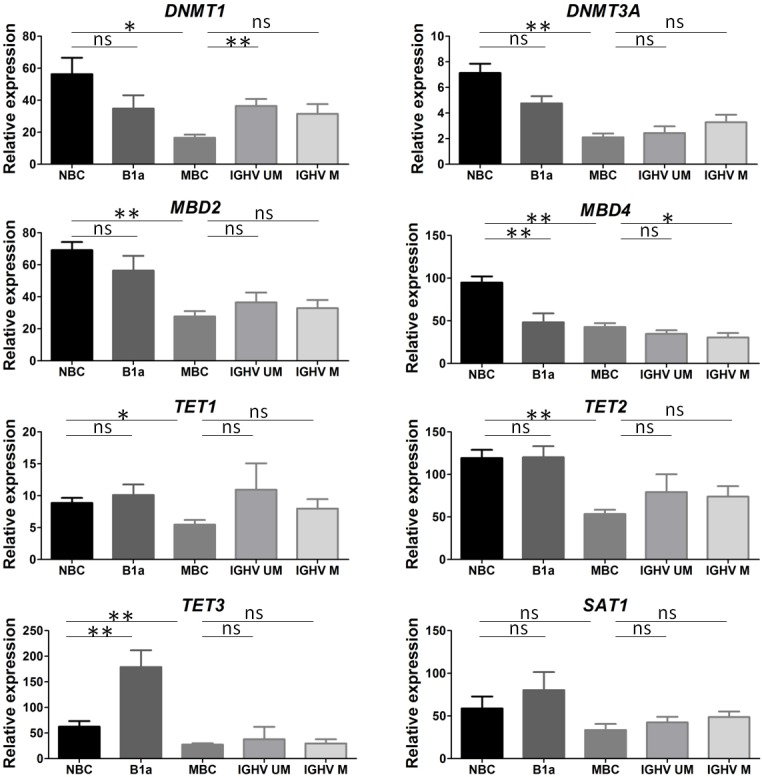
Naïve B cells (NBC), CD5+ B1 cells (B1a) and memory B cells (MBC) were purified from peripheral blood of 6 healthy controls. Chronic lymphocytic leukemia (CLL) patients tested for immunoglobulin heavy chain variable region (IGHV) mutational status were divided into unmutated IGHV gene (UM > 98% homology from germline sequences) and mutated IGHV gene (M < 98% homology). Epigenetic regulators (DNMT1/3A, MBD2/4, TET1/2/3, SAT1) were tested by real time PCR for 6 IGHV UM and 20 IGHV M CLL patients and expressed relative to GAPDH. Values are represented as means ± SEM. The level of significance indicated by asterisks represents the comparison with NBC or MBC, and statistical differences are indicated as follows: (**) 0.001 < P < 0.01; (*) 0.01 < P < 0.05; ns, not significant.
Except for SAT1 which was stable, the transition from naïve B cells to memory B cells was associated with a downregulation of epigenetic regulators (P < 0.05 for all). When naïve B cells and CD5+ B1a cells were taken into consideration for analysis, the main differences were related to MBD4 downregulation (P = 0.02) and TET3 upregulation (P = 0.004) in CD5+ B1a cells.
Next, CLL B cells were subdivided into two groups according to their IGHV mutational status as they originate either from B cells which have not undergone differentiation in the germinal centers (IGHV UM), or from post-germinal center B cells (IGHV M). In contrast to control B cells, for tumor B cells the hypothesis of epigenetic modifications related to the IGHV mutational status has to be rejected based on several observations. First, the distribution of IGHV UM and M CLL patients was similar between the three 5-Cyt derivative subgroups (5/14 in the Global subgroup, 2/14 in the Partial subgroup, and 3/10 in the Normal subgroup) (Table 2),and no difference was observed with regards to the 5-Cyt derivatives according to the IGHV mutational status (Supplementary Figure 1). Second (Figure 3), the transcript levels for the 8 epigenetic regulators were similar between IGHV UM and M CLL B cells. Moreover and third, in both IGHV UM and M CLL B cells, the epigenetic regulator profiles were close to the memory B cell profile. Indeed, when comparing CLL B cells to memory B cells, differences were related to the IGHV UM CLL group for DNMT1 (P = 0.0025) and TET2 (P = 0.04); and to the IGHV M CLL group for MBD4 (P = 0.03).
Table 2. The relevance of clinicobiological prognostic factors according to the three cytosine derivative subgroups.
| Prognostic factors and clinical outcome | Global | Partial | Normal | Statistics (p) |
|---|---|---|---|---|
| Age at diagnosis - median [range] | 67.5 [53–78] | 65.5 [54–84] | 56 [43–79] | ns |
| Age at study entry - median [range] | 72.5 [59–87] | 72.5 [61–88] | 68 [55–83] | ns |
| Binet stage - number of patients (%) | ns | |||
| A | 10/22 (45.5%) | 12/18 (66.7%) | 12/15 (80%) | |
| B and C | 12/22 (54.5%) | 6/18 (33.3%) | 3/15 (20%) | |
| Lymphocytosis (Giga/L) - median (± SEM) | 81.6 (± 12.06) | 33 (± 6.31) | 42.9 (± 8.68) | 0.001 |
| Lymphocyte doubling time from diagnosis (months) * | 19 | 36 | 36 | 0.01 |
| IGHV mutational status - number of patients (%) | ns | |||
| Unmutated (≥ 98% homology) | 5/19 (10.42%) | 2/16 (4.17%) | 3/13 (6.25%) | |
| Mutated (< 98% homology) | 14/19 (29.17%) | 14/16 (29.17%) | 10/13 (20.83%) | |
| Cytogenetics - number of patients (%) | 0.005 | |||
| Low risk - Isolated del(13q) | 2/15 (9.5%) | 10/17 (58.8%) | 5/11 (38.5%) | |
| Intermediate risk - Trisomy 12, normal karyotype and FISH | 7/15 (33.3%) | 5/17 (29.4%) | 2/11 (15.4%) | |
| High risk - del(11q), del(17p), complex karyotype | 6/15 (28.6%) | 2/17 (11.8%) | 4/11 (30.8%) | |
| CD38 > 30% - number of patients (%) | 7/22 (31.8%) | 5/18 (27.8%) | 2/15 (13.3%) | ns |
| 5-mCyt Index - median (± SEM) | 0.83 (± 0.04) | 1.03 (± 0.04) | 1.12 (± 0.03) | p < 0.0001 |
| 5-hmCyt Index - median (± SEM) | 0.17 (± 0.008) | 0.20 (± 0.007) | 0.24 (± 0.005) | p < 0.0001 |
| 5-CaCyt Index - median (± SEM) | 0.54 (± 0.02) | 0.66 (± 0.02) | 0.74 (± 0.01) | p< 0.0001 |
| Progression free survival - median (months)* | 52 | 84 | Undefined | 0.01 |
| Treatment free survival - median (months)* | 112 | 120 | Undefined | 0.01 |
Abbreviations: ns, not significant; IGHV, immunoglobulin heavy chain variable region; FISH, fluorescence in situ hybridization; 5 mCyt, 5-methylcytosine; 5-hmCyt, 5-hydroxymethylcytosine; 5-CaCyt, 5-carboxylcytosine; *, Kaplan-Meyer survival analysis. Statistical analysis was assessed using One-Way Anova test, Fisher’s exact test, or Log-rank analysis.
Epigenetic regulator expression according to the cytosine derivative subgroups
As the epigenetic profiles are not related to the IGHV mutational status, we proceeded in the analysis of the mechanisms regulating DNA methylation and active DNA demethylation in the Global (n = 9) and Partial (n = 11) subgroups by comparing them to the Normal subgroup (n = 13) (Figure 4A, 4B).
Figure 4. Distinct epigenetic regulator profiles according to the 3 cytosine derivative subgroups.
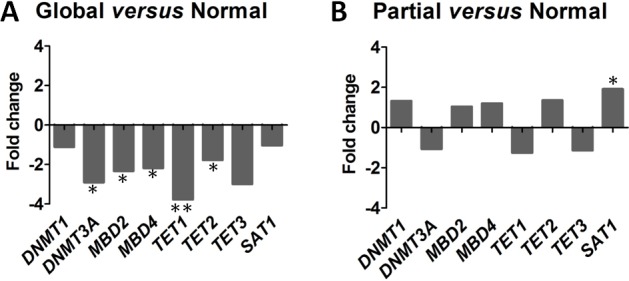
Epigenetic regulators are tested by real time PCR and expressed relative to GAPDH: the DNA methylation ‘writers’, DNMT1/3A; ‘readers’, MBD2/4; ‘editors’, TET1/2/3; and the ‘modulator’ SAT1. (A) Fold changes of epigenetic regulators between the chronic lymphocytic leukemia (CLL) global subgroup (n = 9) compared to the CLL normal subgroup (n = 13). (B) Fold changes of epigenetic regulators between the CLL Partial subgroup (n = 11) compared to the CLL Normal subgroup. Statistical differences between Global and Partial subgroups versus Normal subgroup were assessed using the non-parametric Mann-Whitney test and P < 0.05 are indicated as follows: (***) P < 0.001; (**) 0.001 < P < 0.01; (*) 0.01 < P < 0.05.
When using the non-parametric Mann-Whitney U test to compare the subgroups, patients from the subgroup with a global deficiency were characterized by a reduction in the ‘writer’ DNMT3A (P = 0.01), the ‘readers’ MBD2 (P = 0.03), MBD4 (P = 0.03) and the ‘editors’ TET1 (P = 0.007) and TET2 (P = 0.01). In contrast, the subgroup with a partial deficiency differed from the Normal subgroup through only one epigenetic regulator, SAT1, which was overexpressed (P = 0.02; fold change × 1.9).We conclude from this experiment that a defect of both DNA methylation and active DNA demethylation (Global subgroup) is associated with a downregulation of most of the key epigenetic regulators, while for the Partial subgroup the process is more complex as only the overexpression of the DNA methylation ‘modulator’ SAT1 was observed.
Epigenetic regulator expression during disease progression
Next, we further tested epigenetic regulator mRNA expression levels at two time points in patients with progressive disease (stage Binet A and stage Binet B/C, n = 7) and patients with stable disease (Binet stage A at the two time points, n = 6). The second time point corresponds to patients included in the training cohort and the mean interval between the two time points was 18 months (range 12-36 months). Among the 8 epigenetic regulators tested (Figure 5), a statistically significant modification between the two time points was observed in progressive disease patients for DNMT3A (downregulation, P = 0.03), TET2 (upregulation, P = 0.03) and SAT1 (upregulation, P = 0.01). In the stable disease group, differences concerned SAT1, which was, contrarily to the progressive disease patients, downregulated (P = 0.03). Altogether, these results indicated that transcriptome variations reported during CLL evolution include at least three key epigenetic regulators: DNMT3A, TET2 and SAT1 [56].
Figure 5. Epigenetic regulator variations during disease progression.
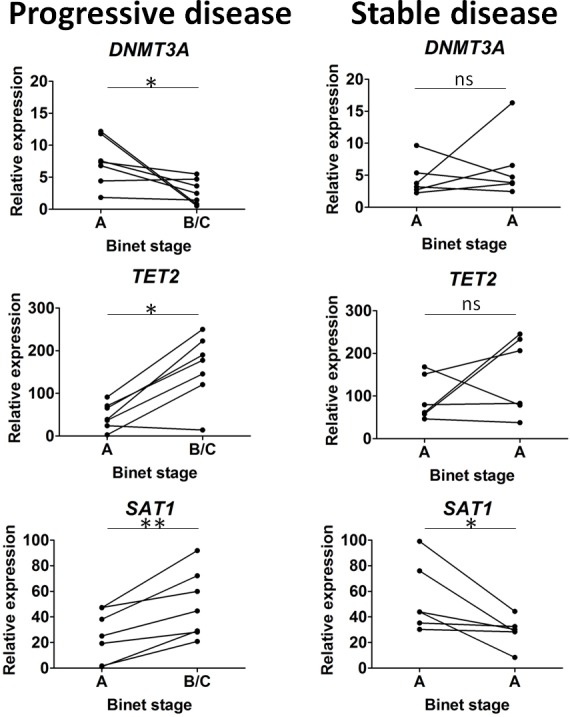
Sequential samples were selected either from chronic lymphocytic leukemia (CLL) patients evolving from Binet stage A to Binet stage B/C (progressive disease, n = 7), or from CLL patients in Binet stage A at the 2 time points (stable disease, n = 6). Among the 8 epigenetic regulators tested, only DNMT3A, TET2 and SAT1 presented significant differences and are represented in the figure. Statistical differences are indicated by asterisks: (**) 0.001 < P < 0.01; (*) 0.01 < P < 0.05, ns, not significant.
CLL patient subgroups and clinical outcome
We next analyzed the relationship between established prognostic markers and the three 5-Cyt derivative subgroups identified (Table 2).
Regarding the CLL subgroup with a global deficiency, a more aggressive profile was highlighted, as patients were characterized by an elevated lymphocytosis (81.6 ± 56.6 Giga/L in the Global subgroup versus 33.0 ± 26.8 Giga/L and 42.9 ± 33.7 Giga/L in the Partial and Normal subgroups, respectively; P = 0.001) and a reduced LDT (19 ± 10.5 months in the Global subgroup versus 36 ± 25 months in the Partial subgroup and 36 ± 23 months in the Normal subgroup; P = 0.01). Additionally, the cytogenetic abnormalities which define the intermediate and high risk CLL patients were better represented in the subgroups with a global and partial 5-Cyt derivative deficit (P = 0.005).
Next, the prognostic power of the 3 defined 5-Cyt derivative subgroups of CLL patients was evaluated first on PFS (Supplementary Figure 2). As depicted in the Kaplan-Meier curves (Supplementary Figure 2A), CLL patients from the Global subgroup had the shortest median PFS of 52 months (Global versus Normal, P = 0.003). The Partial subgroup is intermediate with a median PFS of 84 months, while for the Normal subgroup, the median survival intervals were not reached (> 120 months). In univariate analysis, the following variables were associated with a shorter PFS: elevated SAT1 transcripts (P = 0.04), advanced age at diagnosis (P = 0.009), higher lymphocytosis (P = 0.008), shorter LDT (P = 0.003) and CD38 levels ≥ 30% (P = 0.0007).
Finally, regarding TFS (Figure 6), the median times to initial therapy were 112 months, 120 months and > 120 months for the CLL Global, Partial and Normal subgroups, respectively (Global versus Normal, P = 0.001). In univariate analysis, significant associations with shorter TFS were related to a reduction of DNMT3A (P = 0.05), MBD4 (P = 0.04), TET1 (P = 0.02) and LDT (P = 0.006); and to an increase of SAT1 (P = 0.04), age at diagnosis (P = 0.007), lymphocytosis (P = 0.02) and CD38 (P = 0.001); We also performed a multivariate analysis, and the 5-Cyt derivatives emerged as independent prognostic parameters of TFS.
Figure 6. Prognostic power of epigenetic subgroups and regulators.

Treatment free survival (TFS) Kaplan-Meier curves are depicted for the 3 cytosine derivative subgroups (A), DNMT3A (B), MBD4 (C), TET1 (D), SAT1 (E), age at diagnosis (F), lymphocytosis (G), lymphocyte doubling time from diagnosis (H) and CD38 (I). Epigenetic regulator expression patterns were measured by real time PCR and normalized to GAPDH. The Cox regression model of TFS was used to identify the optimal cut-off level in order to dichotomize CLL patients into high versus low levels, except for CD38. Statistical differences between the curves were calculated using the log-rank test.
Validation with an independent cohort
The ability of stratifying CLL patients in epigenetic subgroups according to the 5-Cyt derivative content was confirmed in a separate group of 56 CLL patients (Table 3). By using the cut-offs previously determined with the Cox regression model for TFS, we classified these cases within a subgroup with a global deficit (n = 22), a subgroup with a partial deficit (n = 26) and a subgroup similar to control B cells (n = 8). As depicted in the Kaplan-Meyer curves from Figure 7A, the subgroup with a global deficit had the shortest median PFS (48 months), followed by the Partial deficit subgroup (90 months), while in the Normal subgroup, the median survival intervals were not reached (> 120 months) (Global versus Normal, P = 0.008). Concerning TFS (Figure 7B), the Global deficit subgroup had the shortest time to treatment initiation (60 months), followed by the Partial deficit subgroup (92 months) and the Normal subgroup (> 120 months) (Global versus Normal, P = 0.007). Herein, the PFS and TFS results on the validation cohort are similar to those obtained in the training series.
Table 3. The relevance of clinicobiological prognostic factors according to the three cytosine derivative subgroups in the validation cohort.
| Prognostic factors and clinical outcome | Global | Partial | Normal | Statistics (p) |
|---|---|---|---|---|
| Age at diagnosis - median [range] | 62.5 [43–77] | 62.5 [33–83] | 67.4 [60–77] | ns |
| Age at study entry - median [range] | 67.4 [45–85] | 66.9 [33–87] | 74.5 [61.89] | ns |
| Binet stage - number of patients (%) | ns | |||
| A | 11/22 (50%) | 15/26 (57.7%) | 8/8 (100%) | |
| B and C | 11/22 (50%) | 11/26 (42.3%) | 0/8 (0%) | |
| Lymphocytosis (Giga/L) - median (±SEM) | 37.36 (± 11.96) | 28.01 (± 5.01) | 17.82 (± 3.55) | 0.03 |
| Lymphocyte doubling time from diagnosis (months)* | 12 | 14.5 | 48 | ns |
| IGHV mutational status - number of patients (%) | ns | |||
| Unmutated (≥ 98% homology) | 5 (10.42%) | 2 (4.17%) | 3 (6.25%) | |
| Mutated (< 98% homology) | 14 (29.17%) | 14 (29.17%) | 10 (20.83%) | |
| Cytogenetic risk category - number of patients (%) | 0.005 | |||
| Low risk - Isolated del(13q) | 7/17 (41.2%) | 11/20 (55%) | 5/8 (62.5%) | |
| Intermediate risk- Trisomy 12, normal karyotype and FISH | 0/17 (0%) | 5/20 (25%) | 3/8 (37.5%) | |
| High risk - del(11q), del(17p), complex karyotype | 10/17 (58.8%) | 4/20 (20%) | 0/8 (0%) | |
| CD38 > 30% - number of patients (%) | 5/20 (25%) | 7/25 (28%) | 2/8 (25%) | ns |
| 5-mCyt Index - median (± SEM) | 0.88 (± 0.02) | 0.98 (± 0.03) | 1.07 (± 0.03) | p < 0.0001 |
| 5-hmCyt Index - median (± SEM) | 0.18 (± 0.007) | 0.21 (± 0.003) | 0.21 (± 0.003) | p < 0.0001 |
| 5-CaCyt Index - median (± SEM) | 0.60 (± 0.01) | 0.65 (± 0.01) | 0.63 (± 0.01) | ns |
| Progression free survival - median (months)* | 48 | 90 | Undefined | 0.03 |
| Treatment free survival - median (months)* | 60 | 92 | Undefined | 0.03 |
Abbreviations: ns, not significant; IGHV, immunoglobulin heavy chain variable region; FISH, fluorescence in situ hybridization; 5mCyt, 5-methylcytosine; 5-hmCyt, 5-hydroxymethylcytosine; 5-CaCyt, 5-carboxylcytosine; *, Kaplan-Meyer survival analysis. Statistical analysis was assessed using One-Way Anova test, Fisher’s exact test, or Log-rank analysis.
Figure 7. Prognostic power of epigenetic subgroups for the validation cohort.

Progression free survival (PFS) (A) and treatment free survival (TFS) (B) Kaplan-Meier curves are depicted for the 3 cytosine derivative subgroups (Global: n = 22; Partial: n = 26; Normal: n = 8) in the validation cohort. Statistical differences between the curves were calculated using the log-rank test.
The relationship between the 5-Cyt subgroups and CLL established prognostic markers was also analyzed for the validation cohort (Table 3). In the subgroups of patients with a global and a partial deficit, the proportion of Binet stage B and C disease is significantly higher compared to the Normal subgroup (P = 0.03). The more aggressive profile for these two subgroups was further confirmed by increased lymphocytosis (P = 0.03) and by the greater proportion of the CLL patients presenting a high cytogenetic risk (P = 0.005). Altogether, these data validate the ability of a 5-Cyt derivative content stratification to predict CLL outcome.
DISCUSSION
In this study, 5-Cyt derivatives were characterized in purified CLL B cells from 55 untreated patients in the training cohort and 56 patients in the validation cohort, in order to better understand the dysregulated epigenetic mechanisms with repercussions on disease progression. Results generated from the present study indicated that: (1) 5-Cyt derivative levels in CLL B cells have a critical impact on disease progression and initial treatment delay in CLL; (2) DNA methylation and active demethylation data analysis classifies CLL patients into 3 subgroups: a worst prognosis subgroup of patients with a defect of both DNA methylation and active DNA demethylation (Global subgroup) associated with a downregulation of most of the key epigenetic regulators, the Partial subgroup, with an altered active DNA demethylation process associated with SAT1 overexpression, and the Normal subgroup which is epigenetically similar to controls and has a stable disease course; (3) disease progression is associated with dynamic modifications in epigenetic regulator expression, particularly DNMT3A, TET2 and SAT1.
In terms of prognosis, determining global 5-Cyt derivatives related to DNA methylation (5-mCyt) and active DNA demethylation (5-hmCyt and 5-CaCyt) represent an accurate tool for evaluating disease progression. However, it should be kept in mind that global 5-Cyt derivative studies using ELISA are only able to reveal important quantitative variations but not qualitative variations in contrast to EWAS studies that are able to demonstrate that epigenetic differences are more marked in the promoters in IGHV UM than what is observed in IGHV M CLL B cells [27, 39]. Aberrant promoter DNA methylation changes have been established in CLL, resulting in silencing of tumor suppressor genes (e.g. Wnt pathway regulators) and reactivation of genes involved in apoptosis, cell proliferation and prognostic markers (e.g. NFATc1 and LPL). Testing global DNA methylation in the repetitive elements Alu, LINE-1 and satellite DNA sequences (Sat-α) in CLL, Hoxha et al. have observed a link between global DNA hypomethylation and disease progression, a lower TFS, a shorter telomere length and an increase in chromosome instability. Contrarily, no associations were reported with IGHV mutational status, CD38 and ZAP70 expression, which is consistent with our observations [12, 20]. Furthermore, and similar to our study, previous analyses have failed to show a correlation between DNMT levels and global DNA methylation levels, on one hand, and between TET and global DNA hydroxymethylation levels, on the other hand [12, 50]. The lack of association could be most likely due to complex mechanisms (see below).
Exploring transcript levels in CLL is challenging as no consensus has emerged to define the normal counterpart of CLL B cells. Peripheral blood total B cells, peripheral blood B cell subsets (naïve, memory), CD5+ B1a cells from umbilical cord blood, peripheral blood or tonsil and CD5 transfected B cell lines have been used in order to uncover CLL specific modifications [15, 16, 43].This key point may explain, in part, the conflicting results reported in CLL when exploring DNMT [26, 27] and TET expression [19, 50]. Based on the well described process of epigenetic specialization during B cell lineage development [36], CLL B cells were subdivided according to their IGHV mutational status and compared to naïve B cells, CD5+ B1a cells and memory B cells. Regarding the epigenetic regulator profiles, results from our study revealed important changes during normal B cell differentiation and a more homogeneous profile in CLL B cells. Overall, the CLL epigenetic regulator profile has also been shown to be more related to memory B cells and independent from the IGHV mutational status. This is consistent with the observation that gene expression profiles resemble more the typical features of memory B cells [25] and with the report that the IGHV M CLL B cell transcriptome progressively evolves to a sub-network similar to that of the IGHV UM CLL B cell transcriptome before therapy [8]. To further explore an acquired process, epigenetic regulators were tested at two time points during disease progression from Binet stage A to Binet stage B/C. Remarkably such analysis revealed that disease evolution affects pathways controlling both DNA methylation (DNMT3A, SAT1) and active DNA demethylation (TET2). However, upstream pathways controlling DNA methylation and active DNA demethylation have not yet been deciphered.
Regarding DNA methylation, the DNA methylation ‘writer’ DNMT3A is the gene significantly downregulated in the 5-Cyt Global subgroup, which is in agreement with the report that DNMT3A is in the top 1% of the genes downregulated in CLL [49]. It has been further documented in the DNMT3A knockout (KO) CLL-mice model that DNMT3A gene expression controls global DNA methylation patterns in haematopoietic stem cells, and in addition, DNMT3A was shown to be critical for B cell and CD8+ T cell development [18, 40, 45]. Therefore, it is not surprising that an epigenetic reprogramming is described in CD8+ T cells from CLL patients, which is associated with an inverted CD4/CD8 ratio and a poor outcome [53]. In CLL B cells from patients with aggressive disease, mutated NOTCH1 and TCL1 can act as DNA methylation inhibitors by interacting with DNMT3A at the protein level [2, 38]. Such an interaction was not explored in our study as patients with the most aggressive profile (Global subgroup) were characterized by DNMT3A downregulation. We also showed that a downregulation of DNMT3A occurs during disease progression and that low levels of DNMT3A were associated with an early treatment initiation. In addition to DNMT3A, we further analyzed the expression of SAT1, which is part of a complex system, causing excessive consumption of SAM, the 5-Cyt methyl donor molecule [5, 34]. We found that SAT1 was upregulated when the patients progress from Binet stage A to Binet stage B/C, and contrarily, a downregulation was observed in patients with stable disease. In addition, SAT1 was the only factor increased when comparing the 5-Cyt Partial subgroup with the Normal subgroup. Thus, it is not unexpected that elevated levels of SAT1 were associated with a lower PFS and TFS, suggesting therefore that monitoring SAT1 could be helpful in order to predict disease outcome.
Concerning active DNA demethylation and patients from the Partial subgroup, we uncovered a previously unappreciated role of active DNA demethylation intermediates in CLL outcome. A dysregulated active DNA demethylation process in CLL is further supported by the report of Hernández-Sánchez et al. [19], and by our observation that TET2 levels were increased in patients with progressive disease. However, according to Van Damme et al. [50], it was a reduction of TET2 that was associated with reduced PFS in CLL. We failed to confirm such an assertion but report an association between lower expression of TET1 and reduced PFS. We also observed in the most aggressive disease profile, the Global subgroup, that TET2 was downregulated together with DNMT3A, TET1 and MBD2/4 leading to the triple deficit in 5-mCyt, 5-hmCyt and 5-CaCyt. One explanation is that distinct epigenetic profiles can lead to lymphoproliferative disease and disease progression in CLL, as supported by knock-in (KI) or KO experiments. TET2 overexpression in KI mice was shown to induce B cell reprogramming [10, 14], while the loss of TET2 induces myeloid malignancies such as chronic myelomonocytic leukemia in KO mice [6]. The TET1/2 double-KO (DKO) mice develop B cell malignancies, and in two-thirds of the cases, the immunophenotype is close to CLL B cells [55]. Cells from TET1/2 DKO mice present an increase in 5-mCyt and a reduction in 5-hmCyt content [52]. In humans, TET1 and TET2 have been previously shown to be concomitantly downregulated in B cell acute lymphoblastic leukemia and to have overlapping functions in B cell development and leukemogenesis [55]. The Global subgroup of CLL patients, who are characterized by TET1/2 downregulation associated with DNMT3A downregulation, display decrease of all 5-Cyt derivatives. The impact of the dual gene loss DNMT3A/TET2 has also been investigated recently, and it induces the development of multiple lymphoid diseases [54]. In CLL patients from the Global group, further experiments are required to better understand how the tumor repressors DNMT3A, TET1/2 but also MBD2/4 are dysregulated, and the consequences of this repression on disease progression.
In conclusion, our findings suggest that dysregulated DNA methylation and active DNA demethylation in CLL B cells have a critical impact on disease progression and their association represents a major prognostic factor. This prognostic factor is independent from the other prognostic factors tested such as IGHV and CD38. Accordingly, stratifying CLL patients in epigenetic subgroups according to the 5-cyt derivative content, rather than to the analysis of the epigenetic regulators, can be helpful for improving CLL prognostic accuracy.
MATERIALS AND METHODS
Cells and sample preparation
Blood samples were collected from 55 untreated patients diagnosed with CLL according to the World Health Organisation (WHO) classification [46], and 17 healthy volunteers at the Brest University Medical School Hospital. In addition to the training cohort which includes the second time point samples from the longitudinal study, samples were collected for the first time point of the longitudinal study (n = 13) and for the validation cohort (n = 56). Consent was obtained from all individuals and the protocol approved by the Ethical Board at the Brest University Medical School Hospital (OFICE, November 26th, 2015; CRB Brest, collection 2008–214), in accordance with the Declaration of Helsinki.
Lymphocytes were isolated from peripheral blood mononuclear cells (PBMC) by Ficoll-Hypaque density gradient centrifugation (Eurobio, Courtaboeuf, France) and tumor B cells were further enriched using the Pan B-cell Isolation Kit (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). For control B cells, several purification protocols were conducted using either (1) the Pan B-cell Isolation Kit; the Naïve B Cell Isolation Kit II (non-target cells magnetically labeled with biotin-conjugated antibodies [Abs] against CD2, CD14, CD16, CD27, CD36, CD43, CD235a) and (3) the Switched Memory B Cell Isolation Kit (non-target cells magnetically labeled with biotin-conjugated Abs against CD2, CD14, CD16, CD36, CD43, CD235a, IgM, IgD) all from Miltenyi Biotec GmbH. For B1a cell subset isolation, purified CD19+ B cells from controls were further sorted on the basis of CD19 and CD5 surface expression with a MoFlow XDP cell sorter (Beckman Coulter, Brea, CA). Cell purity was assessed by fluorescence-activated cell sorting (FACS) analysis and was over 95% for B cells (CD19+), naïve B cells (CD19+, IgD+, CD27-) and switched memory B cells (CD19+, CD27+, IgD-). Purity of B1a cells (CD19+ CD5+) was > 98%. All FACS Abs used were purchased from Beckman Coulter.
DNA sample preparation and global DNA 5-mCyt, 5-hmCyt, 5-CaCyt and 5-hmU levels assessment by ELISA
DNA was extracted from purified B cells using the Biosprint 15 DNA Blood Kit (Qiagen, Hilden, Germany). Next, DNA was quantified and its purity assessed using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). An ELISA previously developed in the laboratory was used and adapted to measure global 5-mCyt, 5-hmCyt, 5-CaCyt and 5-hmU [48]. Briefly, high affinity microplates (Thermo 269620, Thermo Fisher Scientific) were pre-coated 90 min at room temperature (RT) with 100μl poly-L-lysine 0.01% (Sigma-Aldrich, St. Louis, MO) to attach DNA. Next, DNA samples adjusted at 2 ng/μl (for 5-mCyt and 5-hmCyt) and 1.5 ng/μl (for 5-CaCyt and 5-hmU) in carbonate/bicarbonate buffer 0.1M pH 9.6 were denatured at 95°C for 6 minutes, kept on ice 5–10 minutes and then 100 μl dispensed in each well, in duplicate. The plates were next incubated overnight at 4°C, three washes with phosphate buffer saline (PBS)-Tween 0.01% were performed and 200μl of PBS with bovine serum albumin (BSA) 1% were added in each well as a blocking solution. After 1 hour incubation at RT and extensive washing, 100 μl of mouse IgG anti-5-mCyt (diluted 1:5000 in PBS-BSA 1%), mouse IgG anti-5-hmCyt (1:1000), rabbit IgG anti-5-CaCyt (2μg/ml) or goat IgG anti-5-hmU (1:1000) were added and plates were incubated 2 hours at RT. All anti-cytosine derivative Abs were purchased from Abcam (Cambridge, UK). After 6 washes, 100 μl of alkaline phosphate-labelled goat anti-mouse, goat anti-rabbit or bovine anti-goat IgGs (Jackson Laboratory, Bar Harbor, ME), diluted at 1:5000 in PBS-BSA 1% were added and the plate was incubated for 1 hour at RT. After 3 washes, color was developed with 100μl p-nitrophenyl-phosphate (Sigma-Aldrich) diluted in carbonate/bicarbonate buffer 0.1M pH 9.6. Plates were kept at 37°C for 4 hours, and optical density (OD) determined at 405 nm using a Titertek Multiscan microplate reader (Flow laboratories, Rockville, MD). Each sample was tested in duplicate and non-specific background OD (duplicate wells without DNA) was subtracted from the corresponding test sample. For normalization, a reference sample (salmon sperm gDNA - Sigma Aldrich) was included on each plate and indexes calculated for 5-mCyt, 5-hmCyt, 5-CaCyt and 5-hmU using the ratio between the patient OD and the reference sample OD at 200ng/well (for 5-mCyt, 5-hmCyt) and 100 ng/well (for 5-CaCyt, 5-hmU).
Mutational status of IGHV
According to the BIOMED-2 consortium guidelines [51], the IGHV gene mutation status was determined by sequencing after conducting a PCR multiplex amplification. Briefly, for multiplex PCR, 100 ng of genomic DNA, 0.25 µl of Ampli Taq Gold DNA Polymerase (Applied Biosystems, Foster City, CA), 10pmol of each primer, 0.2 mM dNTP Mix, 1.5 mM MgCl2, 1× PCR Buffer II, were adjusted to 50 µl with DNase/RNase free ultrapure distilled water. Next, PCR products were visualized on 2% agarose gel, and purified with ExoSAP-IT PCR product cleanup kit (Affymetrix, High Wycombe, UK). Finally, amplicons were sequenced with a Big Dye Terminator v3.1 cycle sequencing kit (Applied Biosystems). Results were analyzed with the database IMGT/HighV-Quest (The international ImMunoGeneTics information system, Montpellier) and a homology sequence > 98% defined an UM status [1].
RNA sample preparation, reverse transcription and RTq-PCR.
RNA was extracted from purified B cells using the RNeasy Plus Micro Kit (Qiagen). Quantification and purity were assessed using the NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific). Next, RNA (300 ng) was reverse transcribed into cDNA using the Super Script III enzyme and random primers (Invitrogen Life Sciences, Carlsbad, CA). RTq-PCR was carried out in 20 µl mixtures containing 6 µl of template cDNA diluted 1/12 with DNase/RNase free ultrapure distilled water, 1X Power SYBR® Green PCR Master Mix (Applied Biosystems) and 250nM of each primer (Table 4) using Applied Biosystems® QuantStudio™ 7 Flex Real-Time PCR System. The PCR conditions were the same for all genes. All assays included a negative control, which consisted of the reaction mixture with no template. Comparison of cycle thresholds was completed with the 2ΔΔCT method using GAPDH as an endogenous control.
Table 4. Primer sequences of genes used for real time quantitative PCR.
| Symbol | Gene description | Forward primer | Reverse primer |
|---|---|---|---|
| GAPDH | glyceraldehyde-3-phosphate dehydrogenase | TGCCCTCAACGACCACTTT | GGTCCAGGGGTCTTACTCCTT |
| DNMT1 | DNA methyltransferase 1 | CCTGTACCGAGTTGGTGATGGT | CCTTCCGTGGGCGTTTC |
| DNMT3A | DNA methyltransferase 3 alpha | CTCCTGTGGGAGCCTCAATGTTACC | CAGTTCTTGCAGTTTTGGCACATTCC |
| DNMT3B | DNA methyltransferase 3 beta | ACCACCTGCTGAATTACTCACGC | GATGGCATCAATCATCACTGGATT |
| MBD2 | methyl-CpG binding domain protein 2 | CCATGGAACTACCCAAAGGTCTT | CAGCAGATAAAAGGGTCTCATCATT |
| MBD4 | methyl-CpG binding domain 4, DNA glycosylase | TCTAGTGAGCGCCTAGTCCCAG | TTCCAATTCCATAGCAACATCTTCT |
| TET1 | tet methylcytosine dioxygenase 1 | AATGGAAGCACTGTGGTTTG | ACATGGAGCTGCTCATCTTG |
| TET2 | tet methylcytosine dioxygenase 2 | AATGGCAGCACATTGGTATG | AGCTTCCACACTCCCAAACT |
| TET3 | tet methylcytosine dioxygenase 3 | TTGCGTCGAACAAATAGTGG | CCCGTGTAGATGACCTTCTC |
| SAT1 | spermidine/spermine N1-acetyltransferase 1 | GGTTGCAGAAGTGCCGAAAG | GTAACTTGCCAATCCACGGG |
Statistical analysis
The profile likelihood method using a Cox regression model of PFS and TFS was used in univariate analysis to determine the optimal threshold and stratify patients into two groups, and in multivariate analysis to compare risk factors. This analysis was computed using the Survival and SurvMisc R packages [9]. TFS and PFS analyses were performed using Kaplan–Meier curves and prognosis differences between groups were assessed with a log-rank test. Continuous data are described as mean ± standard error of the mean (SEM). Differences among groups were analyzed by one-way ANOVA in a non-parametric test, or the Fisher’s exact test for categorical data. Following normality and equality of variance tests, nominal values were compared to controls using the student’s t test or alternatively by using a nonparametric test (Mann-Whitney rank sum test). Paired samples were compared by Wilcoxon signed-rank tests and Pearson’s coefficient r2 for correlation analysis. P values under 0.05 were considered significant. Statistical analyses were performed using GraphPad Prism 7.0 (La Jolla, CA).
SUPPLEMENTARY MATERIALS FIGURES
Acknowledgments
Authors express their thanks to Stephanie Dehayes, Charlotte Laot and Pierre Pochard for technical assistance, to Dr Wesley H. Brooks (University of South Florida, USA) for editorial assistance, and to Simone Forest and Genevieve Michel for secretarial assistance.
Author contributions
CBa, MZ, VC, PFC and YR designed the study. CBa performed the research. NDG performed the cytogenetic studies. CBa, AB, AM and VO performed the clinical data gathering. AT, HS and CBe took care of the patients and validated clinical data accuracy. CBa, CL and YR analyzed the data. CBa and YR prepared the initial draft. The final manuscript was read and approved by all authors.
CONFLICTS OF INTEREST
None.
FUNDING
This study was supported by research funding from the “Association Laurette Fugain” (ALF 2015/03), the “Region Bretagne” and from the “Canceropole Grand Ouest”.
REFERENCES
- 1.Alamyar E, Duroux P, Lefranc MP, Giudicelli V. IMGT((R)) tools for the nucleotide analysis of immunoglobulin (IG) and T cell receptor (TR) V-(D)-J repertoires, polymorphisms, and IG mutations: IMGT/V-QUEST and IMGT/HighV-QUEST for NGS. Methods Mol Biol. 2012;882:569–604. doi: 10.1007/978-1-61779-842-9_32. https://doi.org/10.1007/978-1-61779-842-9_32. [DOI] [PubMed] [Google Scholar]
- 2.Arruga F, Gizdic B, Bologna C, Cignetto S, Buonincontri R, Serra S, Vaisitti T, Gizzi K, Vitale N, Garaffo G, Mereu E, Diop F, Neri F, et al. Mutations in NOTCH1 PEST-domain orchestrate CCL19-driven homing of Chronic Lymphocytic Leukemia cells by modulating the tumor suppressor gene DUSP22. Leukemia. doi: 10.1038/leu.2016.383. 2016 Dec 26. https://doi.org/10.1038/leu.2016.383. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 3.Bagacean C, Zdrenghea M, Tempescul A, Cristea V, Renaudineau Y. Anti-CD20 monoclonal antibodies in chronic lymphocytic leukemia: from uncertainties to promises. Immunotherapy. 2016;8:569–581. doi: 10.2217/imt-2015-0015. https://doi.org/10.2217/imt-2015-0015. [DOI] [PubMed] [Google Scholar]
- 4.Cahill N, Bergh AC, Kanduri M, Goransson-Kultima H, Mansouri L, Isaksson A, Ryan F, Smedby KE, Juliusson G, Sundstrom C, Rosen A, Rosenquist R. 450K-array analysis of chronic lymphocytic leukemia cells reveals global DNA methylation to be relatively stable over time and similar in resting and proliferative compartments. Leukemia. 2013;27:150–158. doi: 10.1038/leu.2012.245. https://doi.org/10.1038/leu.2012.245. [DOI] [PubMed] [Google Scholar]
- 5.Casero RA, Jr, Marton LJ. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat Rev Drug Discov. 2007;6:373–390. doi: 10.1038/nrd2243. https://doi.org/10.1038/nrd2243. [DOI] [PubMed] [Google Scholar]
- 6.Chen E, Schneider RK, Breyfogle LJ, Rosen EA, Poveromo L,Elf S, Ko A, Brumme K, Levine R, Ebert BL, Mullally A. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood. 2015;125:327–335. doi: 10.1182/blood-2014-04-567024. https://doi.org/10.1182/blood-2014-04-567024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen SS, Raval A, Johnson AJ, Hertlein E, Liu TH, Jin VX, Sherman MH, Liu SJ, Dawson DW, Williams KE, Lanasa M, Liyanarachchi S, Lin TS, et al. Epigenetic changes during disease progression in a murine model of human chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2009;106:13433–13438. doi: 10.1073/pnas.0906455106. https://doi.org/10.1073/pnas.0906455106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuang HY, Rassenti L, Salcedo M, Licon K, Kohlmann A, Haferlach T, Foa R, Ideker T, Kipps TJ. Subnetwork-based analysis of chronic lymphocytic leukemia identifies pathways that associate with disease progression. Blood. 2012;120:2639–2649. doi: 10.1182/blood-2012-03-416461. https://doi.org/10.1182/blood-2012-03-416461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, Ghia EM, Zhang L, Bao L, Rassenti LZ, Messer K, Calin GA, Croce CM, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood. 2014;124:546–554. doi: 10.1182/blood-2014-03-559690. https://doi.org/10.1182/blood-2014-03-559690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Di Stefano B, Sardina JL, van Oevelen C, Collombet S, Kallin EM, Vicent GP, Lu J, Thieffry D, Beato M, Graf T. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature. 2014;506:235–239. doi: 10.1038/nature12885. https://doi.org/10.1038/nature12885. [DOI] [PubMed] [Google Scholar]
- 11.Eichhorst B, Robak T, Montserrat E, Ghia P, Hillmen P, Hallek M, Buske C. ESMO Guidelines Committee. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26:v78–84. doi: 10.1016/j.annonc.2020.09.019. https://doi.org/10.1093/annonc/mdv303. [DOI] [PubMed] [Google Scholar]
- 12.Fabris S, Bollati V, Agnelli L, Morabito F, Motta V, Cutrona G,Matis S, Grazia Recchia A, Gigliotti V, Gentile M, Deliliers GL, Bertazzi PA, Ferrarini M, et al. Biological and clinical relevance of quantitative global methylation of repetitive DNA sequences in chronic lymphocytic leukemia. Epigenetics. 2011;6:188–194. doi: 10.4161/epi.6.2.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira PG, Jares P, Rico D, Gomez-Lopez G, Martinez-Trillos A, Villamor N, Ecker S, Gonzalez-Perez A, Knowles DG, Monlong J, Johnson R, Quesada V, Djebali S, et al. Transcriptome characterization by RNA sequencing identifies a major molecular and clinical subdivision in chronic lymphocytic leukemia. Genome Res. 2014;24:212–226. doi: 10.1101/gr.152132.112. https://doi.org/10.1101/gr.152132.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ficz G, Gribben JG. Loss of 5-hydroxymethylcytosine in cancer: cause or consequence? Genomics. 2014;104:352–357. doi: 10.1016/j.ygeno.2014.08.017. https://doi.org/10.1016/j.ygeno.2014.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garaud S, Morva A, Lemoine S, Hillion S, Bordron A, Pers JO, Berthou C, Mageed RA, Renaudineau Y, Youinou P. CD5 promotes IL-10 production in chronic lymphocytic leukemia B cells through STAT3 and NFAT2 activation. J Immunol. 2011;186:4835–4844. doi: 10.4049/jimmunol.1003050. https://doi.org/10.4049/jimmunol.1003050. [DOI] [PubMed] [Google Scholar]
- 16.Garaud S, Taher TE, Debant M, Burgos M, Melayah S, Berthou C, Parikh K, Pers JO, Luque-Paz D, Chiocchia G, Peppelenbosch M, Isenberg DA, Youinou P, et al. CD5 expression promotes IL-10 production through activation of the MAPK/Erk pathway and upregulation of TRPC1 channels in B lymphocytes. Cell Mol Immunol. doi: 10.1038/cmi.2016.42. 2016 Aug 8. https://doi.org/10.1038/cmi.2016.42. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haney SL, Upchurch GM, Opavska J, Klinkebiel D, Appiah AK, Smith LM, Heavican TB, Iqbal J, Joshi S, Opavsky R. Loss of Dnmt3a induces CLL and PTCL with distinct methylomes and transcriptomes in mice. Sci Rep. 2016;6:34222. doi: 10.1038/srep34222. https://doi.org/10.1038/srep34222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haney SL, Upchurch GM, Opavska J, Klinkebiel D, Hlady RA, Suresh A, Pirruccello SJ, Shukla V, Lu R, Costinean S, Rizzino A, Karpf AR, Joshi S, et al. Promoter Hypomethylation and Expression Is Conserved in Mouse Chronic Lymphocytic Leukemia Induced by Decreased or Inactivated Dnmt3a. Cell Reports. 2016;15:1190–1201. doi: 10.1016/j.celrep.2016.04.004. https://doi.org/10.1016/j.celrep.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Sanchez M, Rodriguez AE, Kohlmann A, Benito R, Garcia JL, Risueno A, Ferminan E, De Las Rivas J, Gonzalez M, Hernandez-Rivas JM. TET2 overexpression in chronic lymphocytic leukemia is unrelated to the presence of TET2 variations. Biomed Res Int. 2014;2014:814294. doi: 10.1155/2014/814294. https://doi.org/10.1155/2014/814294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoxha M, Fabris S, Agnelli L, Bollati V, Cutrona G, Matis S, Recchia AG, Gentile M, Cortelezzi A, Morabito F, Bertazzi PA, Ferrarini M, Neri A. Relevance of telomere/telomerase system impairment in early stage chronic lymphocytic leukemia. Genes Chromosomes Cancer. 2014;53:612–621. doi: 10.1002/gcc.22171. https://doi.org/10.1002/gcc.22171. [DOI] [PubMed] [Google Scholar]
- 21.Jacobs AL, Schar P. DNA glycosylases: in DNA repair and beyond. Chromosoma. 2012;121:1–20. doi: 10.1007/s00412-011-0347-4. https://doi.org/10.1007/s00412-011-0347-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kanduri M, Cahill N, Goransson H, Enstrom C, Ryan F, Isaksson A, Rosenquist R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood. 2010;115:296–305. doi: 10.1182/blood-2009-07-232868. https://doi.org/10.1182/blood-2009-07-232868. [DOI] [PubMed] [Google Scholar]
- 23.Karouzakis E, Gay RE, Gay S, Neidhart M. Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2012;64:1809–1817. doi: 10.1002/art.34340. https://doi.org/10.1002/art.34340. [DOI] [PubMed] [Google Scholar]
- 24.Kipps TJ, Stevenson FK, Wu CJ, Croce CM, Packham G, Wierda WG, O’Brien S, Gribben J, Rai K. Chronic lymphocytic leukaemia. Nat Rev Dis Primers. 2017;3:17008. doi: 10.1038/nrdp.2017.8. https://doi.org/10.1038/nrdp.2017.8. [DOI] [PubMed] [Google Scholar]
- 25.Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, Freedman A, Inghirami G, Cro L, Baldini L, Neri A, Califano A, Dalla-Favera R. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194:1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kn H, Bassal S, Tikellis C, El-Osta A. Expression analysis of the epigenetic methyltransferases and methyl-CpG binding protein families in the normal B-cell and B-cell chronic lymphocytic leukemia (CLL) Cancer Biol Ther. 2004;3:989–994. doi: 10.4161/cbt.3.10.1137. [DOI] [PubMed] [Google Scholar]
- 27.Kulis M, Heath S, Bibikova M, Queiros AC, Navarro A, Clot G, Martinez-Trillos A, Castellano G, Brun-Heath I, Pinyol M, Barberan-Soler S, Papasaikas P, Jares P, et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat Genet. 2012;44:1236–1242. doi: 10.1038/ng.2443. https://doi.org/10.1038/ng.2443. [DOI] [PubMed] [Google Scholar]
- 28.Kulis M, Merkel A, Heath S, Queiros AC, Schuyler RP, Castellano G, Beekman R, Raineri E, Esteve A, Clot G, Verdaguer-Dot N, Duran-Ferrer M, Russinol N, et al. Whole-genome fingerprint of the DNA methylome during human B cell differentiation. Nat Genet. 2015;47:746–756. doi: 10.1038/ng.3291. https://doi.org/10.1038/ng.3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, Stevenson K, Sougnez C, Wang L, Li S, Kotliar D, Zhang W,Ghandi M, et al. Locally disordered methylation forms the basis of intratumor methylome variation in chronic lymphocytic leukemia. Cancer Cell. 2014;26:813–825. doi: 10.1016/j.ccell.2014.10.012. https://doi.org/10.1016/j.ccell.2014.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. https://doi.org/10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, Zhang Y, Kohli RM. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat Chem Biol. 2012;8:751–758. doi: 10.1038/nchembio.1042. https://doi.org/10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nabhan C, Raca G, Wang YL. Predicting Prognosis in Chronic Lymphocytic Leukemia in the Contemporary Era. JAMA Oncol. 2015;1:965–974. doi: 10.1001/jamaoncol.2015.0779. https://doi.org/10.1001/jamaoncol.2015.0779. [DOI] [PubMed] [Google Scholar]
- 33.Nabhan C, Raca G, Wang YL. Prognosis in Chronic Lymphocytic Leukemia-Reply. JAMA Oncol. 2015;1:988–989. doi: 10.1001/jamaoncol.2015.2532. https://doi.org/10.1001/jamaoncol.2015.2532. [DOI] [PubMed] [Google Scholar]
- 34.Neidhart M, Karouzakis E, Jungel A, Gay RE, Gay S. Inhibition of spermidine/spermine N1-acetyltransferase activity: a new therapeutic concept in rheumatoid arthritis. Arthritis Rheumatol. 2014;66:1723–1733. doi: 10.1002/art.38574. https://doi.org/10.1002/art.38574. [DOI] [PubMed] [Google Scholar]
- 35.Oakes CC, Claus R, Gu L, Assenov Y, Hullein J, Zucknick M,Bieg M, Brocks D, Bogatyrova O, Schmidt CR, Rassenti L, Kipps TJ, Mertens D, et al. Evolution of DNA methylation is linked to genetic aberrations in chronic lymphocytic leukemia. Cancer Discov. 2014;4:348–361. doi: 10.1158/2159-8290.CD-13-0349. https://doi.org/10.1158/2159-8290.CD-13-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oakes CC, Seifert M, Assenov Y, Gu L, Przekopowitz M, Ruppert AS, Wang Q, Imbusch CD, Serva A, Koser SD, Brocks D, Lipka DB, Bogatyrova O, et al. DNA methylation dynamics during B cell maturation underlie a continuum of disease phenotypes in chronic lymphocytic leukemia. Nat Genet. 2016;48:253–264. doi: 10.1038/ng.3488. https://doi.org/10.1038/ng.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Otani J, Arita K, Kato T, Kinoshita M, Kimura H, Suetake I, Tajima S, Ariyoshi M, Shirakawa M. Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem. 2013;288:6351–6362. doi: 10.1074/jbc.M112.431098. https://doi.org/10.1074/jbc.M112.431098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palamarchuk A, Yan PS, Zanesi N, Wang L, Rodrigues B, Murphy M, Balatti V, Bottoni A, Nazaryan N, Alder H, Rassenti L, Kipps TJ, Freitas M, et al. Tcl1 protein functions as an inhibitor of de novo DNA methylation in B-cell chronic lymphocytic leukemia (CLL) Proc Natl Acad Sci USA. 2012;109:2555–2560. doi: 10.1073/pnas.1200003109. https://doi.org/10.1073/pnas.1200003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pei L, Choi JH, Liu J, Lee EJ, McCarthy B, Wilson JM, Speir E, Awan F, Tae H, Arthur G, Schnabel JL, Taylor KH, Wang X, et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics. 2012;7:567–578. doi: 10.4161/epi.20237. https://doi.org/10.4161/epi.20237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peters SL, Hlady RA, Opavska J, Klinkebiel D, Pirruccello SJ,Talmon GA, Sharp JG, Wu L, Jaenisch R, Simpson MA, Karpf AR, Opavsky R. Tumor suppressor functions of Dnmt3a and Dnmt3b in the prevention of malignant mouse lymphopoiesis. Leukemia. 2014;28:1138–1142. doi: 10.1038/leu.2013.364. https://doi.org/10.1038/leu.2013.364. [DOI] [PubMed] [Google Scholar]
- 41.Puiggros A, Blanco G, Espinet B. Genetic abnormalities in chronic lymphocytic leukemia: where we are and where we go. Biomed Res Int. 2014;2014:435983. doi: 10.1155/2014/435983. https://doi.org/10.1155/2014/435983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Queiros AC, Villamor N, Clot G, Martinez-Trillos A, Kulis M,Navarro A, Penas EM, Jayne S, Majid A, Richter J, Bergmann AK, Kolarova J, Royo C, et al. A B-cell epigenetic signature defines three biologic subgroups of chronic lymphocytic leukemia with clinical impact. Leukemia. 2015;29:598–605. doi: 10.1038/leu.2014.252. https://doi.org/10.1038/leu.2014.252. [DOI] [PubMed] [Google Scholar]
- 43.Renaudineau Y, Hillion S, Saraux A, Mageed RA, Youinou P. An alternative exon 1 of the CD5 gene regulates CD5 expression in human B lymphocytes. Blood. 2005;106:2781–2789. doi: 10.1182/blood-2005-02-0597. https://doi.org/10.1182/blood-2005-02-0597. [DOI] [PubMed] [Google Scholar]
- 44.Rendeiro AF, Schmidl C, Strefford JC, Walewska R, Davis Z, Farlik M, Oscier D, Bock C. Chromatin accessibility maps of chronic lymphocytic leukaemia identify subtype-specific epigenome signatures and transcription regulatory networks. Nat Commun. 2016;7:11938. doi: 10.1038/ncomms11938. https://doi.org/10.1038/ncomms11938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott EW, Simon MC, Anastasi J, Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 46.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. IARC WHO Classification of Tumours. 2008; ISBN: 9283224310. [Google Scholar]
- 47.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930–935. doi: 10.1126/science.1170116. https://doi.org/10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thabet Y, Le Dantec C, Ghedira I, Devauchelle V, Cornec D, Pers JO, Renaudineau Y. Epigenetic dysregulation in salivary glands from patients with primary Sjogren’s syndrome may be ascribed to infiltrating B cells. J Autoimmun. 2013;41:175–181. doi: 10.1016/j.jaut.2013.02.002. https://doi.org/10.1016/j.jaut.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Upchurch GM, Haney SL, Opavsky R. Aberrant Promoter Hypomethylation in CLL: Does It Matter for Disease Development? Front Oncol. 2016;6:182. doi: 10.3389/fonc.2016.00182. https://doi.org/10.3389/fonc.2016.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Damme M, Crompot E, Meuleman N, Maerevoet M, Mineur P, Bron D, Lagneaux L, Stamatopoulos B. Characterization of TET and IDH gene expression in chronic lymphocytic leukemia: comparison with normal B cells and prognostic significance. Clin Epigenetics. 2016;8:132. doi: 10.1186/s13148-016-0298-y. https://doi.org/10.1186/s13148-016-0298-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, Delabesse E, Davi F, Schuuring E, Garcia-Sanz R, van Krieken JH, Droese J, Gonzalez D, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia. 2003;17:2257–2317. doi: 10.1038/sj.leu.2403202. https://doi.org/10.1038/sj.leu.2403202. [DOI] [PubMed] [Google Scholar]
- 52.Wiehle L, Raddatz G, Musch T, Dawlaty MM, Jaenisch R, Lyko F, Breiling A. Tet1 and Tet2 Protect DNA Methylation Canyons against Hypermethylation. Mol Cell Biol. 2015;36:452–461. doi: 10.1128/MCB.00587-15. https://doi.org/10.1128/MCB.00587-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu J, Xu X, Lee EJ, Shull AY, Pei L, Awan F, Wang X, Choi JH, Deng L, Xin HB, Zhong W, Liang J, Miao Y, et al. Phenotypic alteration of CD8+ T cells in chronic lymphocytic leukemia is associated with epigenetic reprogramming. Oncotarget. 2016;7:40558–40570. doi: 10.18632/oncotarget.9941. https://doi.org/10.18632/oncotarget.9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X, Su J, Jeong M, Ko M, Huang Y, Park HJ, Guzman A, Lei Y, Huang YH, Rao A, Li W, Goodell MA. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet. 2016;48:1014–1023. doi: 10.1038/ng.3610. https://doi.org/10.1038/ng.3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao Z, Chen L, Dawlaty MM, Pan F, Weeks O, Zhou Y, Cao Z, Shi H, Wang J, Lin L, Chen S, Yuan W, Qin Z, et al. Combined Loss of Tet1 and Tet2 Promotes B Cell, but Not Myeloid Malignancies, in Mice. Cell Reports. 2015;13:1692–1704. doi: 10.1016/j.celrep.2015.10.037. https://doi.org/10.1016/j.celrep.2015.10.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao Z, Goldin L, Liu S, Wu L, Zhou W, Lou H, Yu Q, Tsang SX, Jiang M, Li F, McMaster M, Li Y, Lin X, et al. Evolution of multiple cell clones over a 29-year period of a CLL patient. Nat Commun. 2016;7:13765. doi: 10.1038/ncomms13765. https://doi.org/10.1038/ncomms13765. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.