

Abstract
Cytotoxic T lymphocyte (CTL)-mediated immune responses are the primary defense mechanism against cancer and infection. CTL epitope peptides have been used as vaccines to boost CTL responses; however, the efficacy of these peptides is suboptimal. Under current vaccine formulation and delivery strategies, these vaccines are delivered into and processed inside antigen presenting cells such as dendritic cells (DCs). However, the intracellular process is not efficient, which at least partially contributes to the suboptimal efficacy of the vaccines. Thus, we hypothesized that directly loading epitopes onto MHC class I complexes (MHC-Is) on the DC surface would significantly improve the efficacy of the epitopes because the direct loading bypasses inefficient intra-DC vaccine processing. To test the hypothesis, we designed an immune-tolerant elastin-like polypeptide (iTEP)-delivered CTL vaccine containing a metalloproteinase-9 (MMP-9) sensitive peptide and an CTL epitope peptide. We found that the epitope was released from this MMP sensitive vaccine through cleavage by DC-secreted MMP-9 outside of the DCs. The released epitopes were directly loaded onto MHC-Is on the DC surface. Ultimately, the MMP sensitive vaccine strikingly increased epitope presentation by DCs by 7-fold and enhanced the epitope-specific CD8+ T cell response by as high as 9.6-fold compared to the vaccine that was uncleavable by MMP. In summary, this novel direct loading strategy drastically boosted vaccine efficacy. This study offered a new avenue to enhance CTL vaccines.
Keywords: iTEP, CTL vaccine, epitope, MHC-I, MMP, DC presentation, direct loading
Introduction
Cytotoxic T lymphocyte (CTL)-mediated immune responses are the primary defense mechanism against cancer and infection.1, 2 Vaccines that are able to boost CTL responses are being intensively pursued in laboratory and clinical settings.2, 3 However, there is a gap between the vaccine development effort and the efficacy of these vaccines. For example, although there have been many cancer CTL vaccine clinical trials, only two cancer vaccines have so far been efficacious enough to be used clinically.4-6 This discrepancy, we believe, can be attributed to the intra-dendritic cell (DC) processing that current vaccination strategies rely on. Current CTL vaccination strategies require the vaccines to be processed through an antigen cross-presentation pathway inside DCs.4, 7, 8 This pathway, however, includes multiple barriers: e.g., vaccine uptake by DCs, undesired destruction of vaccines in lysosomes after the uptake, necessary yet inefficient vaccine translocation from phagolysosomes to cytosol, and release of CTL epitopes (8-9 mer peptides) from the vaccines.8-12 Although various vaccine delivery systems have been developed to overcome these barriers.9, 10, 13-16 the outcome is modest, and the aforementioned barriers still dampen the effectiveness of these vaccines. In consideration of the barriers and limited successes of current vaccination strategies, it is necessary to explore a completely distinct vaccination strategy that bypasses cross-presentation and its associated barriers. Here, we aimed to develop a vaccination strategy that directly loads CTL epitopes onto the MHC class I complexes (MHC-I) of DCs.
The direct loading of free peptides composed of CTL epitopes onto MHC-Is on DC surfaces is powerful in vitro.17, 18 However, peptide application in vivo is impeded because of the peptides' low proteolytic stability and short plasma half-life. iTEPs, immune-tolerant elastin-like polypeptide polymers, functioned as macromolecule carriers to increase the proteolytic stability and plasma half-life of their peptide payloads. iTEPs were successfully applied as CTL epitope carriers and potentiated the vaccine efficacy.15, 19 However, the previous iTEP-delivered CTL vaccines work with the same mechanism as traditional vaccines: vaccines were taken up by DCs and cross-presented to the MHC-I on the cells. To directly load CTL epitopes onto the MHC-I on DCs, it is necessary to control the release of iTEP-delivered CTL epitopes around DCs before uptake. Matrix metalloproteinase (MMPs), a family of zinc endopeptidases-degrading extracellular matrix proteins, captured our attention for the aim of controlled release of CTL epitope in the proximity of DCs. MMPs perform multiple roles in physiological and pathological cellular processes, especially tissue remodeling in morphogenesis, angiogenesis, tissue repair, and metastasis.20-22 They are also involved in immunological processes, such as regulation of bioavailability and activity of cytokines and chemokines, integrity of physical tissue barriers, and immune evasion of tumor cells.23, 24 Among the MMP family, MMP-2 and MMP-9 are gelatinases and share similar substrates. Both human and mouse dendritic cells secrete MMP-2/9 for their migration needs.25-28 Although the two MMPs are not exclusively expressed by DCs, constitutive expression of the MMPs in tissues are generally low or non-existent.29, 30 Thus, the proximity of migrating DCs should have greater MMP activity than other tissues under a normal physiological state, a difference favoring a spatially-controlled epitope release around DCs. Therefore, if the iTEP-delivered epitope vaccine can be digested by DC-secreted MMPs and release the epitopes around the DCs, the direct loading of CTL epitopes onto DC MHC-Is can be accomplished.
Taking use of both iTEP delivery and the DC's MMP activity, in this study, we designed a vaccine that not only delivers to but also releases epitopes around DCs by fusing iTEPs with an MMP-2/9 cleavage site and a model CTL epitope. The MMP cleavage site chosen for this project is a proven substrate of MMP-2/931, 32 and has been used to target tissues having high MMP activity.33-35 We proved that this MMP sensitive vaccine was more potent than a vaccine without MMP cleavage site. DCs secreted MMP-9 to its environment. The DC-secreted MMP-9 controlled extracellular epitope release from the iTEP-delivered vaccine. Subsequently, these epitopes were directly loaded onto the MHC-I by substituting epitopes that were previously on the complexes without being taken up and intracellular processing. This novel vaccine strategy bypasses intra-DC processing required by the traditional vaccine strategy, dramatically increases vaccine efficacy, and will have great potential in CTL vaccine application.
Materials and Methods
Cell Lines
The DC2.4 cell line (H-2Kb) was a gift from Dr. Kenneth Rock (University of Massachusetts, USA). DC2.4 cells were cultured in RPMI-1640 medium supplemented with 10% heat inactivated fetal bovine serum (FBS), 2 mM glutamine, 1% non-essential amino acids, 1% HEPES, 50 μM β-mercaptoethanol, 100 units/mL penicillin, and 100 μg/mL streptomycin (ThermoFisher Scientific, USA). The B3Z T-cell hybridoma, which is specific for H-2Kb, OVA257-264 (SIINFEKL, also known as pOVA), was kindly provided by Dr. Nilabh Shastri (University of California, USA). B3Z cells were cultured in RPMI-1640 medium supplemented with 10% heat inactivated FBS, 2 mM glutamine, 1 mM pyruvate, 50 μM β-mercaptoethanol, 100 units/mL penicillin, and 100 μg/mL streptomycin (ThermoFisher Scientific, USA). The Raw264.7 cells were from ATCC. Raw264.7 cells were cultured in the same medium of culturing DC2.4 cells.
Generation of Bone Marrow Dendritic Cells (BMDCs)
BMDCs were generated according to a protocol described previously with minor modifications.36 Femoral and tibial bone marrow cells were obtained from 8-week-old female C57BL/6 mice by the protocol approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Utah. Approximately 1 × 107 bone marrow cells were seeded on 10 cm tissue culture dish into 10 mL of RPMI-1640 medium containing 10% FBS, 100 units/mL penicillin, and 100 μg/mL streptomycin. The cells were cultured at 37 °C and 5% CO2. After overnight depletion of adherent cells, suspension cells were seeded on a new dish with the same medium and 20ng/mL murine GM-CSF (Gold Biotechnology, USA). Fresh medium with murine GM-CSF were added every 2–3 days. Suspension and loosely attached cells were harvested on the seventh day of cultivation.
Collection of cell-cultured conditional medium (designated as DC-cultured medium)
When DC2.4 cells, Raw264.7 cells or BMDCs grew to 90% confluence in growth media containing 10% FBS on 10cm dishes, the culture media were changed to media without FBS. After 48 h, the supernatants of the cell culture were collected and concentrated using Amicon Ultra-4 filter units (10,000 MW, Millipore, USA). The proteins in the concentrated samples were quantified by UV 280 using NANODROP 2000 (Thermo Scientific, USA). The collected cell-cultured conditional media were saved at -80°C for later use.
Construction of the expression plasmids for the iTEP vaccines
The genes encoding iTEP, iTEP-pOVA, and iTEP-sMMP-pOVA were constructed in a modified pET25b (+) vector using a previously described PRe-RDL method.19, 37 Specifically, genes encoding the peptides GVLPGVG, GAGVPG, ESIINFEKL, and PLGLAGSIINFEKL were generated by annealing sense and antisense oligonucleotides and then inserting the product into the vector at the BseRI site. Then, the iTEP gene was polymerized by the PRe-RDL method until a desired length of the iTEP gene was achieved. The iTEP sequences contain 35 repeats of GAGVPG and 16 repeats of GVLPGVG. Genes encoding pOVA (ESIINFEKL) or sMMP-pOVA (PLGLAGSIINFEKL) were inserted into the C-terminus of iTEP in a similar manner. The sequences of the oligonucleotides used to construct these genes are listed in Table S1. After DH5α had been transformed with the resulting expression vectors for amplification, the lengths of the coding genes were confirmed by an XbaI and BamHI double digestion, followed by agarose gel analysis. The coding genes were also verified by DNA sequencing (Genewiz, USA).
Production and purification of the iTEP vaccines
iTEP and iTEP fusions were produced from BL21 competent cells and purified as previously described.19 Endotoxin was removed by treating with 1% Triton X-114 three times, as previously described.38, 39 Then, the Triton X-114 was removed using Amicon Ultra-15 (10k) centrifugal filters (Millipore, USA). The residual endotoxin in the samples was measured by using Limulus Amebocyte Lysate (LAL) PYROGENT Single Test Vials (Lonza, Allendale, NJ, USA). All samples had endotoxin levels below 0.1EU per 100μg protein in 100μL.
Characterization of the thermally induced, reversible, inverse phase transition of the iTEP and iTEP vaccines
The phase transitions of the iTEP or iTEP fusions were characterized by turbidity changes in sample solutions as a function of temperature. Briefly, the OD350 of a sample was dynamically recorded using a UV-visible spectrophotometer equipped with a multi-cell thermoelectric temperature controller (Cary 300, Varian Instruments, Walnut Creek, CA), during which the sample (25 μM, with 1M NaCl) was heated from 20 to 80 °C and then cooled to 20 °C at a rate of 1 °C/min. The maximum first derivative of the turbidity curve of a sample was identified. The transition temperature (Tt) of the sample was the temperature that corresponded to the maximum derivative.
Fluorescent labeling of iTEPs
iTEP-pOVA and iTEP-sMMP-pOVA were labeled with NHS-Fluorescein following a protocol described by the manufacturer's instructions. Briefly, 10mg of iTEPs were reacted with 1mg of NH-Fluorescein in 0.1 M sodium bicarbonate buffer, pH 8.3 at a final volume of 1mL. The reaction was carried out with continuous stirring in the dark and at room temperature for 2 h. The protein-dye conjugates were purified using a PD-10 desalting column (GE Health Care Life Sciences) followed by Amicon Ultra-15 (10k) centrifugal filters (Millipore). The fluorescein conjugates were measured for their absorbances at 205 nm (A205) and 493 nm (A493, λmax for NHS-Fluorescein). The concentrations of iTEP fusion and the degree of labeling (DOL) were calculated based on these equations:
where ε is the extinction coefficient in cm−1 M−1. Both iTEPs have one lysine (K) and one asparagine (N) in their sequences. iTEP-pOVA has two glutamic acids (E) while iTEP-sMMP-pOVA has one. NHS-Fluorescein couples principally with the α-amines at the N-terminals and the ε-amines of lysine side chains.40 The labeling efficiency of NHS ester to either N or E is quite low in our reaction condition. Actually, the labeling efficiency to both polypeptides are very similar. The DOLs for both iTEPs were around 8-10%. Then they were all adjusted to 8% for cell uptake experiments.
Enzymatic cleavage assay
NHS-Fluorescein labeled iTEP-pOVA or iTEP-sMMP-pOVA was used as the substrate for the MMP enzymatic cleavage. The substrates were incubated with recombinant active MMP-2, MMP-9, or DC-cultured conditional medium at indicated concentrations for 16 h at 37°C. The resulting mixtures were then subjected to 4-15% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), separating cleaved small molecule pOVAs from big molecule iTEPs. For the inhibitor experiments, DC-cultured conditional media were treated with different inhibitors at indicated concentrations for 2 h before adding the labeled iTEP substrates. Fluorescent images were taken using FluoChem FC2 (Alpha Innotech Corp.). The band intensities were quantified by ImageJ software (NIH). The results are expressed as fold change compared to MMP activity without inhibitor.
Presentation of pOVA delivered by iTEP vaccine
The DC presentation of pOVA was studied as previously described.19 Briefly, 1.5 × 105 DC2.4 cells/well were set in 48-well plates and cultured with iTEP, iTEP vaccines, or pOVA peptide, as indicated, for 16 h at 37°C and 5% CO2. The cells were then washed and stained by PE-tagged monoclonal antibody 25-D1.16 (Biolegend, San Diego, CA, 1:100 dilution) before flow cytometry analysis. The data were processed using FlowJo × software. The mean fluorescence intensity (MFI) of treated cells was normalized to the MFI of untreated cells.
Activation of B3Z hybridoma (CD8+ T) cells
This assay was performed using a previously described protocol with modifications.19 Briefly, 1×105 DC2.4 cells/well were set to 96-well plates. Then the DCs were incubated with iTEPs at 37 °C for 4 h or with iTEPs pretreated with DC-cultured conditional media at 4 °C for 30 min, as indicated. After wash, the DCs were fixed on the plate with 1% paraformaldehyde for 15 min at room temperature. The plate was washed 5 times with B3Z culture medium. Then, 1×105 B3Z cells/well were added into the wells and co-cultured with DC2.4 cells for 24 h. The cells were washed and lysed with 100 μL lysis buffer (PBS containing 100 mM 2-mercaptoethanol, 9 mM MgCl2, and 0.125% NP-40) together with 0.15 mM chlorophenol red β-galactoside substrate (Sigma, St. Louis, MO, USA). After a 2-h incubation at 37 °C, the reaction was stopped with 50 μL of 15 mM EDTA and 300 mM glycine. The OD570 of the solutions was measured, and OD630 was used as a reference when using an Infinite M1000 PRO plate reader (Tecan Trading AG, Switzerland).
Gelatin Zymography
Zymography was performed following a previously described protocol with a few modification.41 In brief, 4-15% SDS-polyacrylamide gel was prepared by copolymerizing with 0.1% gelatin from porcine skin (Sigma, St Louis, MO). Recombinant active MMP-2, MMP-9, or DC-cultured conditional media at indicated concentrations were loaded onto the gel without boiling and electrophoresed under non-reducing condition. The gel was washed with 2.5% (v/v) Triton X-100 at room temperature on a shaker for 0.5 h to remove the SDS. Then, the gel was placed in developing buffer (50 mM Tris [pH8.0] and 5mM CaCl2, 10mM NaCl) for 0.5 h at room temperature. The gel was then incubated in fresh developing buffer for 24 h at 37 °C to allow proteinase digestion of the substrate. The developed gel was rinsed with water and stained with 0.1% Coomassie brilliant blue (Sigma) followed by destaining with H2O until clear zones became evident. The clear bands on a blue background showed the positions of the enzyme activity of gelatinases. The band intensities were quantified by ImageJ software (NIH). The protein standard in the gel is ColorPlus™ Prestained Protein Marker (NEB, P7709S) which includes a mixture of purified proteins covalently coupled to colored dye. Unlike MMPs digesting gelatin, the proteins in the standard did not digest gelatin and showed faint bands under the blue background of coomassie blue-stained gelatin gel.
To verify that the bands of proteolytic activity were MMPs, inhibition studies were conducted. The MMP inhibitors, 1,10-phenanthroline (20mM, Sigma) or EDTA (5mM) were added to the samples and incubated for 0.5 h at 37°C before loading to the gelatin gel for the zymography analysis. PMSF (5 mM), a serine protease inhibitor, was used as a negative control to verify the MMP activity of the samples. In another set experiment, the samples without inhibitor were loaded onto gelatin gel for electrophoresis. The gel was incubated in 2.5% Triton X-100 and then developing buffer containing GM6001 (10μM). The band intensities were quantified by ImageJ software (NIH). The results are expressed as fold change compared to MMP activity without inhibitor.
Microfluometric assay to detect uptake of iTEP vaccine by DCs
The assays were conducted using previous protocols with a few modifications.42 1.0 × 105 DC2.4 cells/well in a 96-well plate were incubated with fluorescein-labeled iTEPs at 37°C and 5% CO2 for various times. After incubation, the cells were washed with PBS once before adding trypan blue (250 μg/mL, pH 4.4) to quench the extra fluorescence. The trypan blue was removed after 1 min, and cells were washed three times with PBS. The cells were then lysed with 1% TritonX-100 for 10 min, and the cell lysates were transferred to a new 96-well black plate. The fluorescence intensity of the cell lysates was measured using an Infinite M1000 PRO plate reader (Tecan Trading AG, Switzerland) at 494 nm excitation and 518 nm emission. The uptake intensity was normalized by minus binding intensity derived from 10 min incubation. Standard curve of iTEP concentration vs fluorescence intensity was generated. Then the up taken amount of iTEPs were calculated using standard curve, normalized uptake intensity, and volume of the samples. The results were expressed as the percentage of the input dose of iTEP vaccine:
Statistical analysis
Data were analyzed for statistical significance using an unpaired Student's t-test. GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA) was used for statistical analysis and figure construction. A P value <0.05 was statistically significant.
Results
Generation and characterization of iTEP-sMMP-pOVA, an MMP-2/9 sensitive vaccine
We designed a CTL vaccine which can bypass the requirement of cytosolic delivery and cross-presentation of the CTL epitope by DCs. The idea to drive this vaccine design was to generate an MMP-sensitive vaccine carrier from which the CTL epitope can be released from the carrier by DC-secreted MMP. Therefore, the released epitope may be directly loaded onto the MHC-I on DC surfaces (Figure 1). Toward this end, an iTEP polymer [M(GAGVPG)35(GVLPGVG)16GC, Table 1] was used as a vaccine carrier. A substrate peptide of MMP-2/9, [sMMP: PLGLAG], together with a model CTL epitope [pOVA: SIINFEKL] were fused to the C-terminus of the iTEP. The newly synthesized iTEP polymer was named iTEP -sMMP-pOVA (sequences are in Table 1). As a control, an iTEP-pOVA fusion which was devoid of sMMP was also designed (sequences are in Table 1). The iTEP carrier and iTEP vaccines were produced from E.coli and purified through temperature and salt dependent phase transition. Their purity and sizes were analyzed through an SDS-PAGE gel separation (Table 1 and Figure 2A).
Figure 1.

The iTEP-delivered, MMP-9-sensitive vaccine that enables direct epitope-loading. The vaccine, which contains a MMP-9 cleavage site, is able to take advantage of MMP-9 ((shown as red scissors) secreted from and surrounding DCs to release CTL epitopes. The MMP-9 cleave the epitopes from the vaccines and facilitate an accumulation of the epitopes around DCs. The direct epitope loading occurs when these epitopes, at high concentration, replace the predecessor epitopes bound with MHC-I on DC surfaces.
Table 1. Physicochemical properties of iTEP polymers evaluated in this study.
| Nomenclature | Amino acid sequence | MW [kDa] | a Protein purity (%) | b Heating Tt | c Cooling Tt |
|---|---|---|---|---|---|
| iTEP | M(GAGVPG)35(GVLPGVG)16GG | 24.89 | 97.94 | 56.27 ± 1.88 | 49.47 ± 1.71 |
| iTEP-pOVA | M(GAGVPG)35(GVLPGVG)i6GE SIINFEKL | 25.90 | 96.60 | 54.07 ± 1.95 | 47.27 ± 1.67 |
| iTEP-sMMP-pOVA | M(GAGVPG)35(GVLPGVG)16GP LGLAGSIINFEKL | 26.28 | 90.21 | 54.58 ±2.06 | 45.03 ± 1.86 |
Protein purity was determined using samples separated by SDS-PAGE gel and stained by CuCI2 followed by Image J quantification.
Heating Tt and
Cooling Tt were measured for samples at 25 μM, with IMNaCI, pH 7.4 PBS.
Figure 2.
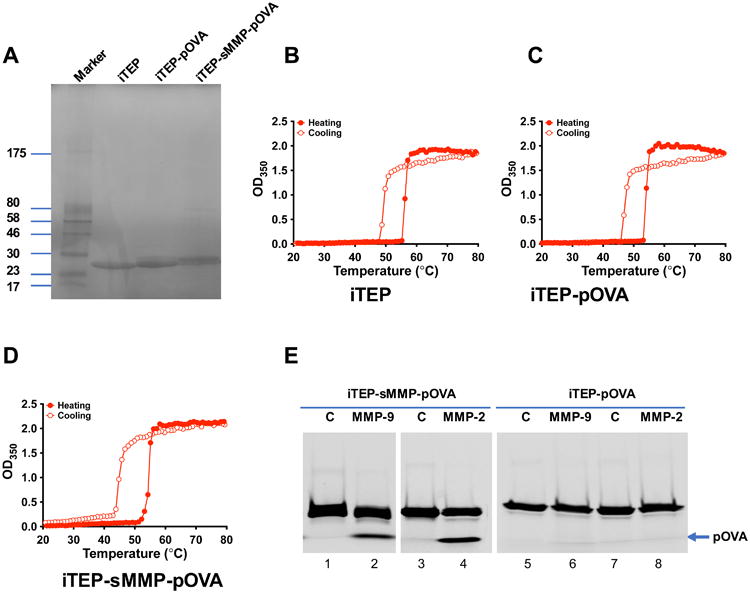
Characterization of iTEP-derived iTEP vaccines and their response to MMPs. (A) SDS-PAGE gel showing the identity and purity of iTEP and iTEP-derived vaccines. (B–D) Turbidity profiles (OD350) of iTEP (B), iTEP-pOVA (C), and iTEP-sMMP-pOVA (D) when 25 μM samples were heated and then cooled between 20 °C and 80 °C in PBS and 1M NaCl. (E) Enzymatic digestion of iTEP-pOVA and iTEP-sMMP-pOVA by recombinant active MMP-2 or MMP-9. iTEP vaccines were labeled with NHS-Fluorescein and treated with enzymes at a 1:1000 ratio of enzyme to substrate at 37 °C for 16 h. The enzyme-digested products were resolved by SDS-PAGE, and the image was recorded under UV. The position of the fragment of pOVA generated by MMP-2/9 is shown by an arrow.
The thermally induced, reversible phase transition feature of the iTEP and iTEP vaccines were characterized through optical density measurements. Because all the iTEPs in this study did not display a phase transition in the tested temperature range (20–80 °C) in PBS (data not shown), 1M of NaCl was included in the solutions to achieve a thermally-induced phase transition. The results showed that at 25 μM, all iTEP and iTEP fusions displayed the thermally induced, reversible phase transition feature. The heating transition temperature (Tt) of the three iTEP polymers were similar, which were around 54-56 °C. The cooling Tts of the three iTEPs were also similar and around 45-49 °C (Table 1 and Figure 2B-2D). These results suggested that fusing of pOVA or sMMP-pOVA peptides minimally influence the phase transition feature of iTEP and do not affect the hydrophobicity of iTEP.
To investigate whether pOVA peptide can be cleaved from the sMMP containing vaccine by MMP digestion, the iTEP vaccines were evaluated by enzymatic cleavage using recombinant MMP-2 and MMP-9. In order to separate and detect cleaved pOVA from the original uncleaved iTEP vaccine, iTEP-pOVA and iTEP-sMMP-pOVA were first covalently labelled with NHS-Fluorescein at a degree of labeling (D.O.L) of 0.08 dye/protein. Then the labelled iTEPs were treated with MMPs, and the cleavage products were resolved through SDS-PAGE. As shown in Figure 2E, after treated with MMP-9, full length iTEP-sMMP-pOVA was cleaved to generate a peptide showing a low molecular weight band in lane 2. Comparison with free pOVA run on the same SDS-PAGE indicated that this enzyme digestion product is pOVA peptide (Figure S1). Treatment of MMP-2 had a similar effect, which also cleaved iTEP-sMMP-pOVA to release free pOVA (Figure 2E, lane 4). However, treatment of MMP-9 or MMP-2 did not cleave iTEP-pOVA, an iTEP vaccine without an MMP cleavage site (Figure 2E, lane 6 and 8 respectively). Therefore, iTEP-sMMP-pOVA was specifically responsive to and cleavable by MMP-2/9.
iTEP-sMMP-pOVA is cleaved by DC-secreted MMP-9 to release pOVA
It was reported that human and mouse DCs secreted both MMP-2 and MMP-9.25-28 We proposed that DCs secrete MMPs to their extracellular environment to control cleavage of iTEP-sMMP –pOVA and release of pOVA. To explore if this is the case, we first measured the MMP secretion from both primary DC (BMDC of C57BL/6 mice) and DC cell line (DC2.4 derived from C57BL/6 mice). The supernatants of the cell cultures were collected and concentrated as DC-cultured medium and loaded on a gelatin gel for gelatin zymography analysis. With the analysis, we observed a 92 kDa clear zone in the DC medium from either BMDC or DC2.4 (Figure 3A, lane 1 for BMDC, and lane 2 or 5 for DC2.4). This clear zone ran to the same position of MMP-9 proenzyme which had been proved to be secreted by Raw264.7 cells (Figure 3A, lane 4).43, 44 Interestingly, the amount of MMP-9 proenzyme secreted by BMDC was 26.38 fold by DC2.4 cells, suggesting that the primary cells are potent and that iTEP-sMMP-pOVA has a high possibility to encounter MMP-9 when it is delivered to the proximity of DCs in vivo. The active MMP-2 was also resolved in the gelatin gel as a control to show its smaller MW (62 kDa) and gelatin activity (Figure 3A, lane 3). The gelatin digestion ability of DC-secreted MMP-9 was specific because it was inhibited by MMP inhibitors such as 1,10 phenanthroline, EDTA, GM6001, but not non-MMP inhibitors such as PMSF, a serine kinase inhibitor (Figure 3B). With these data, we concluded that DCs greatly secrete MMP-9 but only a small amount of MMP-2.
Figure 3.
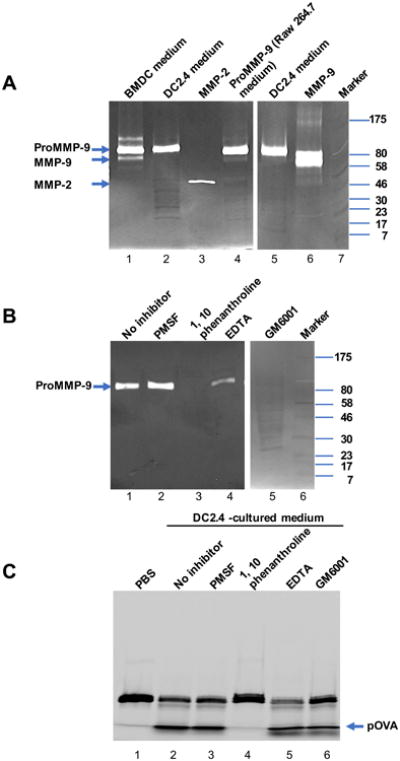
DCs secrete MMP-9 to cleave iTEP-sMMP-pOVA and release pOVA epitope. (A) Gelatin zymography analysis showing that like Raw264.7 cells, DC2.4 cells and BMDCs from C57BL/6 mouse greatly secreted MMP-9 but not MMP-2. 50 μg protein from DC2.4 cell medium, 2.5 μg protein from BMDC medium, 12.5 μg protein from Raw264.7 cell medium, 20 ng MMP-9, or 10 ng MMP-2 were resolved by SDS-PAGE containing 0.1% gelatin. (B) Gelatin zymography analysis of MMP-9 secreted by DC2.4 cells with different inhibitors. Lane2-4, 50 μg protein from DC2.4 cell medium was treated with PMSF, 1,10 phenothroline, or EDTA for 30 min before loading to the gelatin gel. Lane 5, 50 μg protein from DC2.4 cell medium without treatment was resolved by gelatin gel and incubated with developing buffer containing 10 μM GM6001 before Coomassie brilliant blue staining. (C) Effect of inhibitors on enzymatic digestion of iTEP-sMMP-pOVA by DC-cultured medium. DC-cultured medium was pretreated with inhibitors as in (B) at 37 °C for 2 h. Then fluorescein-labeled iTEP-sMMP-pOVA was added and incubated at 37 °C for 16 h. The enzyme-digested products were resolved by SDS-PAGE, and the image was recorded under UV. The position of the fragment of pOVA generated by MMP-2/9 is shown by an arrow.
To study whether the DC-cultured medium is active to cleave iTEP-sMMP-pOVA and release pOVA extracellularly, we incubated fluorescein-labeled iTEP-sMMP-pOVA in the DC-cultured medium and examined the products of enzymatic digestion through separation of SDS-PAGE. We found that the DC medium cleaved iTEP-sMMP-pOVA to create free pOVAs like the recombinant MMP-9 or MMP-2 did (lane 2 in Figure 3C verses lane 2 and 4 in Figure 2E). The enzymatic activity of DC-cultured medium was MMP specific because the activity can be inhibited by inhibitors specific to MMP but not by other inhibitors. 1, 10 phenenthroline, a broad inhibitor of zinc metallopeptidases and MMPs, completely inhibited cleavage of iTEP-sMMP-pOVA by DC-cultured medium (lane 4 of Figure 3C). EDTA, a metal chelator, and GM6001, a potent inhibitor of collagenases and gelatinases, also significantly inhibited digestion of iTEP-sMMP-pOVA by DC-cultured medium, with only 77.83% and 65.93% of pOVA production compared to the digestion without inhibitor, respectively (Figure 3C, lane 5 and 6). Contrariwise, PMSF, a non-MMP inhibitor, did not affect the activity of the DC-cultured medium at all (Figure 3C, lane 3). Thus, DC cells secreted MMP-9 proenzyme into the environment around; the activated extracellular MMP-9 specifically digested iTEP-sMMP-pOVA containing an MMP cleavage site to release the pOVA peptide outside of the cells.
MMP-sensitive vaccines drastically promote epitope presentation by DCs
To analyze if adding of MMP cleavage site increases the biological activity of iTEP vaccine, we compared DC presentation and the downstream antigen-specific CD8+ T cell activation induced by the iTEP vaccines. The presentation of pOVA by DCs was detected by an antibody recognizing the pOVA/ H-2Kb complex. After being cultured with iTEP vaccines, DC2.4 cells, murine DC cells containing H-2Kb on their surface,45 presented pOVA with different efficiency. The histogram of flow cytometry analysis showed that treatment with iTEP-sMMP-pOVA significantly increased presentation of pOVA by DC than with iTEP-pOVA (Figure 4A). Quantification of mean fluorescence intensity (MFI) revealed that the DCs incubated with iTEP-sMMP-pOVA presented a 7-fold higher amount of pOVA epitopes than DCs with treatment of iTEP-pOVA (Figure 4B). The treatment with iTEP carrier without epitope was used as a negative control. The peptide pOVA pulse directly bound to H-2Kb of DCs with more than a 10-fold increase in presentation and served as a positive control in this assay (Figure 4A and 4B).
Figure 4.
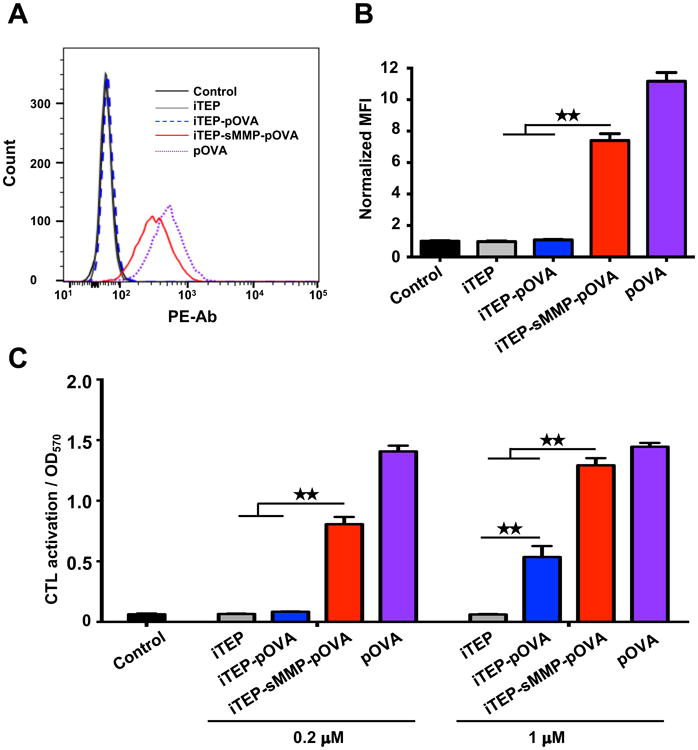
iTEP-sMMP-pOVA more effectively presents pOVA to DCs than iTEP-pOVA. (A) Histograms of flow cytometric analysis. DC2.4 cells were stained with anti-mouse pOVA bound H-2Kb after the cells were incubated with 5 μM iTEPs or pOVA. (B) Mean fluorescence intensity (MFI) of H-2Kb-pOVA on DC2.4 cells after different treatments. (C) Stimulation of B3Z cells via presentation of pOVA by DC2.4 cells which were pre-incubated with different reagents at 0.2 μM or 1 μM. The data in all panels are representative of 3 independent experiments. Bars, mean+SD, n=3. Analysis of variance (Student's t-test). ★★P<0.01.
To examine the biological significance of the above presentation results, we conducted a B3Z assay. B3Z is a CD8+ T-cell hybridoma clone which T cell receptors are restricted to the pOVA:H-2Kb complex presented by DCs. When B3Z cells are activated by the association between their T cell receptors and the pOVA:H-2Kb on DCs, they are able to produce β-galactosidase46 that can be quantified by a β-galactosidase assay. Thus, the production amount of β-galactosidase can be used to infer the activation levels of B3Z cells, which, in turn, is used to measure biologically significant presentation of pOVA by DCs47, 48. In this study, we compared the activation of B3Z cells after we incubated them with DC2.4 cells. The DCs were priorly incubated with different vaccines. Similar to the response of DC presentation, B3Z activation trigged by the two iTEP vaccines were different. At low concentration (0.2 μM), iTEP-sMMP-pOVA had a significant, 12-fold increase in B3Z activation, while iTEP-pOVA did not change the B3Z cell response (Figure 4C). At high concentration stimulation (1 μM), iTEP-sMMP-pOVA resulted in an even more significant increase, which was 21-fold of the iTEP carrier treatment and close to the response of pOVA, the positive control. iTEP-pOVA also increased B3Z activation by almost 10-fold at 1 μM, but this response was still 2.5-fold lower than that of iTEP-sMMP-pOVA at the same concentration (Figure 4C). Thus, the data of both DC presentation and antigen-specific CD8+ T cell activation proved that iTEP vaccines with an MMP cleavage site are much more efficient than ones without an MMP site.
pOVAs released from iTEP-sMMP-pOVA directly load onto MHC class I complexes for antigen presentation
As stated above, we found that iTEP-sMMP-pOVA drastically increased DC presentation and antigen-specific CD8+ T cell activation. The reasons for the enhanced DC presentation might be multifaceted: 1. direct loading of epitopes onto MHC-I on the DC surface, 2. increased uptake by DCs, and/or 3. increased intra-DC processing and cross-presentation. To find out which one is the major reason for iTEP-sMMP-pOVA's high potency in DC presentation, we first compared uptake of iTEP-sMMP-pOVA and iTEP-pOVA by DCs. DC2.4 cells were incubated with fluorescein-labeled iTEP vaccines and the amount of the ingested vaccine by the cells was measured with time extension. The results demonstrated that the uptake percentage of these two vaccines by DC2.4 cells were similar after 10 min, 30 min, and 1 h of incubation at 37 °C. Even though the uptake of iTEP-sMMP-pOVA was higher than iTEP-pOVA after 2 h of incubation, the change was subtle and then there was no difference after 4 h of incubation (Figure 5A). Therefore, overall there was no obvious difference between the uptake efficiency of these two vaccines by DCs and cellular uptake did not contribute for iTEP-sMMP-pOVA's high potency in antigen presentation.
Figure 5.

pOVAs released from iTEP-sMMP-pOVA directly load onto DC for antigen presentation which is dependent on DC-secreted MMP-9. (A) Comparison of uptake of iTEP-pOVA vs iTEP-sMMP-pOVA by DC cells. DC2.4 cells were incubated with 10 μM of fluorescein-labeled iTEP vaccines in FBS free medium at 37 °C from 10 min to 4 h. The normalized cellular fluorescence intensity was recorded and compared. (B) Stimulation of B3Z cells via direct loading of pOVA on H-2Kb of DC 2.4 cells. The iTEP vaccines were incubated with RPMI medium without FBS, supernatant of DC-cultured medium without column concentration, or activated MMP-9 at 37 °C for 16 h before loading to DC2.4 cells on ice. After 30 min of ice incubation, the DC2.4 cells were washed, fixed, and then mixed with B3Z cells for B3Z activation. (C). Effect of MMP inhibitors on direct loading of pOVA released from iTEP-sMMP-pOVA. The experiment procedure was similar to (B) except that DC-cultured medium was treated with inhibitors at 37 °C for 2 h before incubation with iTEP-sMMP-pOVA. The data in all panels are representative of 3 independent experiments. Bars, mean+SD, n=3. Analysis of variance (Student's t-test). ★ P<0.05. ★★ P<0.01.
Then we examined the second possibility: direct loading of the pOVA epitope released from iTEP vaccines onto MHC-I of DCs. For this end, we loaded the pOVA epitopes, which were released from DC-cultured medium-pretreated iTEP vaccines, to the DC2.4 cells at 4 °C for just 30 min. This condition completely inhibited the cellular uptake of iTEP vaccine and resulted in direct loading only (Figure S2). We then evaluated the direct loading through measuring B3Z activation by the pOVA-loaded DC2.4 cells because the activation of the antigen specific CD8+ cells is the direct consequence of DC presentation through the complex of epitope and MHC-I. The results showed that iTEP-sMMP-pOVA and iTEP-pOVA induced similar B3Z activation when they were pretreated with medium without any supplement. Interestingly, iTEP-sMMP-pOVA resulted in significantly, 3 fold more increase of B3Z activation when the vaccine was pretreated with DC-cultured medium or MMP-9 enzyme. Contrarily, iTEP-pOVA did not increased B3Z activation when it was with the same pretreatments (Figure 5B). These results suggested that DC-secreted MMP-9 results in significantly more direct loadings of pOVAs from iTEP-sMMP-pOVA than from iTEP-pOVA.
The iTEP-sMMP-pOVA-stimulated direct loading was MMP dependent. When 1,10 phenanthroline or GM6001 was added in the DC-cultured medium, the activated B3Z response was significantly inhibited to 25% and 47% of the uninhibited sample, respectively. However, PMSF did not affect the B3Z activation (Figure 5C). On the other side, all the inhibitors had no effect on iTEP-pOVA-induced presentation (Figure S3). Therefore, combining all the collected data, we concluded that pOVA released from iTEP-sMMP-pOVA through DC-secreted MMP-9 is responsible for the direct loading of epitopes on MHC-I and enhanced antigen presentation, even though we could not exclude the existence of the third possibility, that increased intra-DC processing and cross-presentation by iTEP-sMMP-pOVA might also contribute.
Discussion
According to traditional wisdoms on CTL vaccine processing, these vaccines are internalized and translocated to the cytosol of DCs where CTL epitopes are released from the vaccines. Later, the epitopes are cross-presented together with MHC-I by DCs to CD8+ T cells, which activates the T cells and initiate CTL responses. Up to date, most CTL vaccine carriers are developed to facilitate the vaccine uptake and translocation processes, which, in turn, enhances the presentation of CTL epitopes and CTL responses.49-51 Here, we developed a vaccine delivery system that enabled a direct loading of CTL epitopes to MHC-I on DC surface. This direct loading method bypassed the cytosolic delivery and cross-presentation steps and was believed more efficient than previous CTL vaccine carriers. The direct loading method utilized MMP-9 secreted by DCs to release epitope from the delivery system. Our data showed that the direct loading method was superior to those cytosolic vaccine delivery methods in promoting epitope presentation by DCs.
The idea of direct epitope loading was inspired by the observation that there are a significant amount of empty MHC-I on DC surface that are ready to accommodate CTL epitopes (20,000 to 60,000 empty MHC-I per DC2.4 cell per hour).52 In a typical MHC-I antigen cross-presentation pathway, one CTL epitope is first loaded onto one MHC-I in endoplasmic reticulum of DCs: the epitope sits in the binding cleft of the MHC-I and forms non-covalent interactions with the MHC-I; then, this epitope/MHC-I complex is transported to DC surface.53 However, epitopes that bind with MHC-I with a low affinity may dissociate from the MHC-I on DC surface, which generates empty MHC-I.52, 54 The binding affinity between CTL epitopes and MHC-I is governed by an intrinsic binding specificity between the two. For a given allele of MHC-I, only epitope with a certain sequence motif can bind with the MHC-I strongly 55. For example, for the H-2Kb allele, the MHC-I of DC2.4 cells, the preferred motif is that the c-terminus of the epitope should be a leucine residue; the number 5th residue from the N-terminus of the epitope should be either a phenylalanine or a tyrosine residue.55 The sequence of pOVA, SIINFEKL, matches the motif perfectly. Consequently, pOVA binds with H-2Kb tightly with an affinity as of 215.07 nM. In general, the epitope/MHC-I complex with an affinity smaller than 500nM is recognized as a tight complex. 52, 56 According to these insights of epitope loading, when a high affinity epitope is released from its carrier around DCs, the epitope has a good chance to bind with empty MHC-I on DC surface and form a stable complex.
The improved epitope presentation of iTEP-sMMP-pOVA was mainly due to direct epitope loading. This conclusion is based on two observations that 1) DC internalized comparable amount of iTEP-sMMP-pOVA and iTEP-pOVA (Figure 5A), and 2) when antigen presentation experiment was conducted at a condition that vaccine internalization is halted (Figure 5B), iTEP-sMMP-pOVA led to greater epitope presentation than iTEP-pOVA. Because the internalization is halted and only the direct epitope loading is allowed, we can attribute the different presentation results between the two vaccines to the different direct loading efficiency of the two vaccines. Thus, we infer that iTEP-sMMP-pOVA is more efficient in direct loading. As of different presentation results between iTEP-sMMP-pOVA over iTEP-pOVA observed at 37 °C, there might be an additional cause other than the different direct loading efficiency: the MHC-I antigen cross-presentation pathway favors iTEP-sMMP-pOVA over iTEP-pOVA. Indeed, some intracellular metallopeptidases were found to facilitate epitope release from protein antigens in the MHC-I pathway.57 Some of these metallopeptidases are able to cleave pOVA from iTEP-sMMP-pOVA because they recognize sMMP.58 Taken together, iTEP-sMMP-pOVA may utilize two mechanisms to improve antigen presentation, the direct loading of epitopes and the metallopeptidase-mediated, intra-DC epitope release.
This concept of controlled release of epitopes delivered by an MMP sensitive carrier has an extra benefit when delivering tumor-related epitope vaccines. Some tumor cells secrete high levels of MMP-2/9 when they metastasize and invade LNs.59, 60 These tumor cell-secreted MMPs can be leveraged to release the epitopes around tumor cells in LNs. These epitopes may be subsequently loaded onto tumor cells, predisposing the cells to epitope-directed immune attack: the epitope-specific CTLs eliminate the tumor cells which were loaded with the epitopes.
Our data showed that the direct loading method was superior to cytosolic vaccine delivery methods in vitro. To translate this advantage into strong CTL responses in vivo, few additional requirements have to be fulfilled: first, MMP-sensitive vaccines like iTEP-sMMP-pOVA need to accumulate around DCs so that DC-secreted MMP-9 is available to cleave CTL epitopes from the vaccines; second and conversely, the vaccines should have minimal exposure to heathy cells that secrete MMP-9 because such exposure may lead to direct epitope loading to these healthy cells. Such loading falsely masks these healthy cells by the epitopes. Then, these cells could be recognized by the immune system as infected or cancerous cells and hence predisposed for epitope-directed immune destruction.61, 62 Thus, one of future developments to furnish the direct loading method is to target these direct loading carriers to the proximity of DCs.
In consideration of the importance and current challenges of CTL vaccines, the main contribution of this study is that it explores a novel and direct epitope-loading strategy to unprecedentedly strengthen CTL vaccine-induced responses. This novel yet straightforward strategy may bring about an unprecedented impact on these vaccines, allowing them to play a more decisive role in the immunotherapy and immune prevention of cancer, infections, and other diseases. This new strategy may also be utilizable for delivering chemicals to the proximity of DCs.
Supplementary Material
Acknowledgments
The authors thank Drs. Kenneth Rock and Nilabh Shastri for providing cell lines, Mr. James Marvin for technique support in flow cytometry analysis. The flow cytometry work was supported by the University of Utah Flow Cytometry. The research work was supported by the University of Utah Start-up Fund to M. C., the Huntsman Cancer Institute Pilot Grant 170301 to M. C., and NIH CA EB024083 to M. C.
References
- 1.Chen Daniel S, Mellman I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013;39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Robinson HL, Amara RR. T cell vaccines for microbial infections. Nat Med. 2005;11(4 Suppl):S25–32. doi: 10.1038/nm1212. [DOI] [PubMed] [Google Scholar]
- 3.Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunological reviews. 2011;239(1):27–44. doi: 10.1111/j.1600-065X.2010.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klebanoff CA, Acquavella N, Yu Z, Restifo NP. Therapeutic cancer vaccines: are we there yet? Immunological Reviews. 2011;239(1):27–44. doi: 10.1111/j.1600-065X.2010.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pinto LA, Edwards J, Castle PE, Harro CD, Lowy DR, Schiller JT, Wallace D, Kopp W, Adelsberger JW, Baseler MW, Berzofsky JA, Hildesheim A. Cellular immune responses to human papillomavirus (HPV)-16 L1 in healthy volunteers immunized with recombinant HPV-16 L1 viruslike particles. J Infect Dis. 2003;188(2):327–38. doi: 10.1086/376505. [DOI] [PubMed] [Google Scholar]
- 6.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Investigators IS. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 7.Murphy KP. Janeway's Immunobiology. 8th. Garland Science; 2012. Antigen presentation to T lymphocytes. [Google Scholar]
- 8.Joffre OP, Segura E, Savina A, Amigorena S. Cross-presentation by dendritic cells. Nat Rev Immunol. 2012;12(8):557–69. doi: 10.1038/nri3254. [DOI] [PubMed] [Google Scholar]
- 9.Jia F, Liu X, Li L, Mallapragada S, Narasimhan B, Wang Q. Multifunctional nanoparticles for targeted delivery of immune activating and cancer therapeutic agents. J Control Release. 2013;172(3):1020–34. doi: 10.1016/j.jconrel.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 10.Hu Y, Litwin T, Nagaraja AR, Kwong B, Katz J, Watson N, Irvine DJ. Cytosolic delivery of membrane-impermeable molecules in dendritic cells using pH-responsive core-shell nanoparticles. Nano letters. 2007;7(10):3056–64. doi: 10.1021/nl071542i. [DOI] [PubMed] [Google Scholar]
- 11.Kreutz M, Tacken PJ, Figdor CG. Targeting dendritic cells--why bother? Blood. 2013;121(15):2836–44. doi: 10.1182/blood-2012-09-452078. [DOI] [PubMed] [Google Scholar]
- 12.Brossart P, Bevan MJ. Presentation of Exogenous Protein Antigens on Major Histocompatability Complex Class I Molecules by Dendritic Cells: Pathway of Presentation and Regulation by Cytokines. Blood. 1997;90(4):1594–1599. [PMC free article] [PubMed] [Google Scholar]
- 13.Li WA, Mooney DJ. Materials based tumor immunotherapy vaccines. Curr Opin Immunol. 2013;25(2):238–45. doi: 10.1016/j.coi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho S, Dong S, Parent KN, Chen M. Immune-tolerant elastin-like polypeptides (iTEPs) and their application as CTL vaccine carriers. Journal of Drug Targeting. 2016;24(4):328–339. doi: 10.3109/1061186X.2015.1077847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong S, Xu T, Zhao P, Parent KN, Chen M. A Comparison Study of iTEP Nanoparticle-Based CTL Vaccine Carriers Revealed a Surprise Relationship between the Stability and Efficiency of the Carriers. Theranostics. 2016;6(5):666–78. doi: 10.7150/thno.14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu Z, Ramishetti S, Tseng YC, Guo S, Wang Y, Huang L. Multifunctional nanoparticles co-delivering Trp2 peptide and CpG adjuvant induce potent cytotoxic T-lymphocyte response against melanoma and its lung metastasis. Journal of Controlled Release. 2013;172(1):259–265. doi: 10.1016/j.jconrel.2013.08.021. [DOI] [PubMed] [Google Scholar]
- 17.Porgador A, Gilboa E. Bone marrow-generated dendritic cells pulsed with a class I-restricted peptide are potent inducers of cytotoxic T lymphocytes. J Exp Med. 1995;182(1):255–60. doi: 10.1084/jem.182.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macatonia SE, Taylor PM, Knight SC, Askonas BA. Primary stimulation by dendritic cells induces antiviral proliferative and cytotoxic T cell responses in vitro. The Journal of Experimental Medicine. 1989;169(4):1255–1264. doi: 10.1084/jem.169.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cho S, Dong S, Parent KN, Chen M. Immune-tolerant elastin-like polypeptides (iTEPs) and their application as CTL vaccine carriers. Journal of drug targeting. 2015:1–12. doi: 10.3109/1061186X.2015.1077847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sternlicht MD, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zitka O, Kukacka J, Krizkova S, Huska D, Adam V, Masarik M, Prusa R, Kizek R. Matrix metalloproteinases. Curr Med Chem. 2010;17(31):3751–68. doi: 10.2174/092986710793213724. [DOI] [PubMed] [Google Scholar]
- 22.Chambers AF, Matrisian LM. Changing views of the role of matrix metalloproteinases in metastasis. Journal of the National Cancer Institute. 1997;89(17):1260–70. doi: 10.1093/jnci/89.17.1260. [DOI] [PubMed] [Google Scholar]
- 23.Parks WC, Wilson CL, Lopez-Boado YS. Matrix metalloproteinases as modulators of inflammation and innate immunity. Nature reviews Immunology. 2004;4(8):617–29. doi: 10.1038/nri1418. [DOI] [PubMed] [Google Scholar]
- 24.Nissinen L, Kahari VM. Matrix metalloproteinases in inflammation. Biochimica et biophysica acta. 2014;1840(8):2571–80. doi: 10.1016/j.bbagen.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 25.Yen JH, Khayrullina T, Ganea D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood. 2008;111(1):260–70. doi: 10.1182/blood-2007-05-090613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kouwenhoven M, Ozenci V, Tjernlund A, Pashenkov M, Homman M, Press R, Link H. Monocyte-derived dendritic cells express and secrete matrix-degrading metalloproteinases and their inhibitors and are imbalanced in multiple sclerosis. Journal of neuroimmunology. 2002;126(1-2):161–71. doi: 10.1016/s0165-5728(02)00054-1. [DOI] [PubMed] [Google Scholar]
- 27.Marsac D, Garcia S, Fournet A, Aguirre A, Pino K, Ferres M, Kalergis AM, Lopez-Lastra M, Veas F. Infection of human monocyte-derived dendritic cells by ANDES Hantavirus enhances proinflammatory state, the secretion of active MMP-9 and indirectly enhances endothelial permeability. Virology journal. 2011;8:223. doi: 10.1186/1743-422X-8-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zozulya AL, Reinke E, Baiu DC, Karman J, Sandor M, Fabry Z. Dendritic cell transmigration through brain microvessel endothelium is regulated by MIP-1alpha chemokine and matrix metalloproteinases. J Immunol. 2007;178(1):520–9. doi: 10.4049/jimmunol.178.1.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loffek S, Schilling O, Franzke CW. Series “matrix metalloproteinases in lung health and disease”: Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38(1):191–208. doi: 10.1183/09031936.00146510. [DOI] [PubMed] [Google Scholar]
- 30.Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13(9):649–65. doi: 10.1038/nri3499. [DOI] [PubMed] [Google Scholar]
- 31.Turk BE, Huang LL, Piro ET, Cantley LC. Determination of protease cleavage site motifs using mixture-based oriented peptide libraries. Nature biotechnology. 2001;19(7):661–7. doi: 10.1038/90273. [DOI] [PubMed] [Google Scholar]
- 32.Chen EI, Kridel SJ, Howard EW, Li W, Godzik A, Smith JW. A unique substrate recognition profile for matrix metalloproteinase-2. The Journal of biological chemistry. 2002;277(6):4485–91. doi: 10.1074/jbc.M109469200. [DOI] [PubMed] [Google Scholar]
- 33.Zhu L, Kate P, Torchilin VP. Matrix metalloprotease 2-responsive multifunctional liposomal nanocarrier for enhanced tumor targeting. ACS nano. 2012;6(4):3491–8. doi: 10.1021/nn300524f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mansour AM, Drevs J, Esser N, Hamada FM, Badary OA, Unger C, Fichtner I, Kratz F. A new approach for the treatment of malignant melanoma: enhanced antitumor efficacy of an albumin-binding doxorubicin prodrug that is cleaved by matrix metalloproteinase 2. Cancer research. 2003;63(14):4062–6. [PubMed] [Google Scholar]
- 35.Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics. 2012;2(2):156–78. doi: 10.7150/thno.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Inaba K, Swiggard WJ, Steinman RM, Romani N, Schuler G, Brinster C. Isolation of dendritic cells. Current protocols in immunology / edited by John E Coligan … [et al] 2009;Chapter 3(Unit 3 7) doi: 10.1002/0471142735.im0307s86. [DOI] [PubMed] [Google Scholar]
- 37.McDaniel JR, Mackay JA, Quiroz FG, Chilkoti A. Recursive directional ligation by plasmid reconstruction allows rapid and seamless cloning of oligomeric genes. Biomacromolecules. 2010;11(4):944–52. doi: 10.1021/bm901387t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aida Y, Pabst MJ. Removal of endotoxin from protein solutions by phase separation using Triton X-114. Journal of immunological methods. 1990;132(2):191–5. doi: 10.1016/0022-1759(90)90029-u. [DOI] [PubMed] [Google Scholar]
- 39.Liu S, Tobias R, McClure S, Styba G, Shi Q, Jackowski G. Removal of endotoxin from recombinant protein preparations. Clin Biochem. 1997;30(6):455–63. doi: 10.1016/s0009-9120(97)00049-0. [DOI] [PubMed] [Google Scholar]
- 40.Hermanson GT. Bioconjugate Techniques. Second. Academic Press; New York: 2008. Chapter 2 - The Chemistry of Reactive Groups; pp. 171–172. [Google Scholar]
- 41.Toth M, Sohail A, Fridman R. Assessment of gelatinases (MMP-2 and MMP-9) by gelatin zymography. Methods Mol Biol. 2012;878:121–35. doi: 10.1007/978-1-61779-854-2_8. [DOI] [PubMed] [Google Scholar]
- 42.Wan CP, Park CS, Lau BH. A rapid and simple microfluorometric phagocytosis assay. Journal of immunological methods. 1993;162(1):1–7. doi: 10.1016/0022-1759(93)90400-2. [DOI] [PubMed] [Google Scholar]
- 43.Yoo HG, Shin BA, Park JS, Lee KH, Chay KO, Yang SY, Ahn BW, Jung YD. IL-1beta induces MMP-9 via reactive oxygen species and NF-kappaB in murine macrophage RAW 264.7 cells. Biochemical and biophysical research communications. 2002;298(2):251–6. doi: 10.1016/s0006-291x(02)02431-2. [DOI] [PubMed] [Google Scholar]
- 44.Lee YS, Lan Tran HT, Van Ta Q. Regulation of expression of matrix metalloproteinase-9 by JNK in Raw 264.7 cells: presence of inhibitory factor(s) suppressing MMP-9 induction in serum and conditioned media. Experimental & molecular medicine. 2009;41(4):259–68. doi: 10.3858/emm.2009.41.4.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen Z, Reznikoff G, Dranoff G, Rock KL. Cloned dendritic cells can present exogenous antigens on both MHC class I and class II molecules. J Immunol. 1997;158(6):2723–30. [PubMed] [Google Scholar]
- 46.Karttunen J, Sanderson S, Shastri N. Detection of rare antigen-presenting cells by the lacZ T-cell activation assay suggests an expression cloning strategy for T-cell antigens. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(13):6020–4. doi: 10.1073/pnas.89.13.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shastri N, Gonzalez F. Endogenous generation and presentation of the ovalbumin peptide/Kb complex to T cells. J Immunol. 1993;150(7):2724–36. [PubMed] [Google Scholar]
- 48.Mandl S, Sigal LJ, Rock KL, Andino R. Poliovirus vaccine vectors elicit antigen-specific cytotoxic T cells and protect mice against lethal challenge with malignant melanoma cells expressing a model antigen. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(14):8216–21. doi: 10.1073/pnas.95.14.8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li WA, Mooney DJ. Materials based tumor immunotherapy vaccines. Curr Opin Immunol. 2013;25(2):238–45. doi: 10.1016/j.coi.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cruz LJ, Tacken PJ, Rueda F, Domingo JC, Albericio F, Figdor CG. Targeting nanoparticles to dendritic cells for immunotherapy. Methods Enzymol. 2012;509:143–63. doi: 10.1016/B978-0-12-391858-1.00008-3. [DOI] [PubMed] [Google Scholar]
- 51.De Koker S, Lambrecht BN, Willart MA, van Kooyk Y, Grooten J, Vervaet C, Remon JP, De Geest BG. Designing polymeric particles for antigen delivery. Chem Soc Rev. 2011;40(1):320–39. doi: 10.1039/b914943k. [DOI] [PubMed] [Google Scholar]
- 52.Eisen HN, Hou XH, Shen C, Wang K, Tanguturi VK, Smith C, Kozyrytska K, Nambiar L, McKinley CA, Chen J, Cohen RJ. Promiscuous binding of extracellular peptides to cell surface class I MHC protein. Proc Natl Acad Sci USA. 2012;109(12):4580–5. doi: 10.1073/pnas.1201586109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy K. Chapter 6: Antigen presentation to T Lymphocytes. Janeway's Immunobiology. (8th) 2012:201–216. [Google Scholar]
- 54.Fahnestock ML, Tamir I, Narhi L, Bjorkman PJ. Thermal stability comparison of purified empty and peptide-filled forms of a class I MHC molecule. Science. 1992;258(5088):1658–62. doi: 10.1126/science.1360705. [DOI] [PubMed] [Google Scholar]
- 55.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351(6324):290–6. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 56.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, Buus S, Nielsen M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nandan AN, K M. Microbial Aminopeptidases. Vol. 21 John Fedor; Cambridge, MA: 2016. [Google Scholar]
- 58.Lazaro S, Gamarra D, Del Val M. Proteolytic enzymes involved in MHC class I antigen processing: A guerrilla army that partners with the proteasome. Molecular immunology. 2015;68(2 Pt A):72–6. doi: 10.1016/j.molimm.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 59.Hofmann UB, Eggert AA, Blass K, Brocker EB, Becker JC. Expression of matrix metalloproteinases in the microenvironment of spontaneous and experimental melanoma metastases reflects the requirements for tumor formation. Cancer research. 2003;63(23):8221–5. [PubMed] [Google Scholar]
- 60.Daniele A, Zito AF, Giannelli G, Divella R, Asselti M, Mazzocca A, Paradiso A, Quaranta M. Expression of metalloproteinases MMP-2 and MMP-9 in sentinel lymph node and serum of patients with metastatic and non-metastatic breast cancer. Anticancer Res. 2010;30(9):3521–7. [PubMed] [Google Scholar]
- 61.Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nat Rev Drug Discov. 2007;6(5):404–14. doi: 10.1038/nrd2224. [DOI] [PubMed] [Google Scholar]
- 62.van der Burg SH, Bijker MS, Welters MJ, Offringa R, Melief CJ. Improved peptide vaccine strategies, creating synthetic artificial infections to maximize immune efficacy. Adv Drug Deliv Rev. 2006;58(8):916–30. doi: 10.1016/j.addr.2005.11.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.