

Abstract
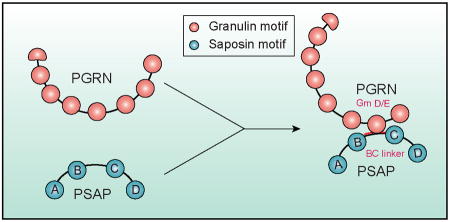
The frontotemporal lobar degeneration (FTLD) protein progranulin (PGRN) is essential for proper lysosomal function. PGRN localizes in the lysosomal compartment within the cell. Prosaposin (PSAP), the precursor of lysosomal saposin activators (saposin A, B, C, D), physically interacts with PGRN. Previously we have shown that PGRN and PSAP facilitate each other's lysosomal trafficking. Here we report that the interaction between PSAP and PGRN requires the linker region of saposin B and C (BC linker). PSAP protein with the BC linker mutated fails to interact with PGRN and target PGRN to lysosomes in the biosynthetic and endocytic pathways. On the other hand, PGRN interacts with PSAP through multiple granulin motifs. Granulin D and E bind to PSAP with similar affinity as full length PGRN. In summary, our data shows that the BC linker of PSAP interacts with multiple granulin motifs in PGRN.
Keywords: Progranulin, Prosaposin, Neuronal Ceroid Lipofuscinosis, Frontotemporal lobar degeneration, Lysosome, Lysosomal storage diseases
Graphical Abstract
The frontotemporal lobar degeneration (FTLD) protein progranulin (PGRN) is essential for proper lysosomal function. Prosaposin (PSAP), the precursor of lysosomal saposin activators (saposin A, B, C, D), physically interacts with PGRN. Here we show that PGRN and prosaposin PSAP interact through the BC linker of PSAP and granulin motifs (primarily granulin D and E) to regulate each other's trafficking and function.
Introduction
Frontotemporal lobar degeneration (FTLD) is the most prevalent early onset dementia after Alzheimer's disease (AD) and accounts for 20-25% of pre-senile dementias (Neary et al., 1998). A large subset of FTLD cases has characteristic ubiquitin-positive inclusions comprised of the protein TDP-43 (FTLD-TDP) (Arai et al., 2006; Neumann et al., 2006). Haploinsufficiency of progranulin (PGRN) due to mutations in the granulin (GRN) gene is one of the major causes of FTLD-TDP (Baker et al., 2006; Cruts et al., 2006; Gass et al., 2006). PGRN is an evolutionarily conserved glycoprotein of 7.5 granulin repeats with multiple functions in the nervous system, including neuronal survival and regulating microglia mediated inflammation (Bateman and Bennett, 2009; Cenik et al., 2012; Nicholson et al., 2012). Recent studies, however, have supported a surprising role of PGRN in the lysosome. While PGRN haploinsufficiency causes FTLD, total loss of PGRN in humans results in neuronal ceroid lipofuscinosis (NCL) (Almeida et al., 2016; Smith et al., 2012), a lysosomal storage disease (Cotman et al., 2013; Kousi et al., 2012). Furthermore, PGRN is transcriptionally co-regulated with a number of essential lysosomal genes by the transcriptional factor TFEB (Belcastro et al., 2011). Finally, within the cell, PGRN localizes to the lysosomal compartment. The VPS10 family trafficking receptor sortilin is highly expressed in neurons and interacts with the C-terminus of PGRN to mediate PGRN lysosomal targeting (Hu et al., 2010; Zheng et al., 2011). More recently, PGRN was also shown to directly bind another lysosomal protein, prosaposin (PSAP), and get a “piggyback” ride to lysosomes through the PSAP receptors M6PR and LRP1 (Zhou et al., 2015). In fibroblasts which do not express sortilin, PGRN lysosomal trafficking is totally dependent on PSAP (Zhou et al., 2015). The interaction between PGRN and PSAP was further confirmed by a large family-based genome-wide association study (GWAS) study, in which polymorphisms in PSAP were shown to affect plasma PGRN levels (Nicholson et al., 2016).
PSAP is the precursor of lysosomal saposin activators (saposin A, B, C and D) essential for glycosphingolipid degradation, and mutations in the saposin motifs are known to cause several distinct lysosomal storage diseases (Matsuda et al., 2007; O'Brien and Kishimoto, 1991; Qi and Grabowski, 2001). The physical interaction of PGRN and PSAP not only facilitates PGRN lysosomal trafficking but also PSAP trafficking, as we have recently shown that PGRN bridges the interaction between PSAP and sortilin to facilitate PSAP uptake and lysosomal delivery in neurons (Zhou et al., 2017b). Furthermore, PGRN haploinsufficiency in FTLD patients impairs PSAP lysosomal trafficking and decreases neuronal saposin level (Zhou et al., 2017b). Thus PGRN and PSAP facilitate each other's lysosomal trafficking, and PGRN-PSAP interaction is important for maintaining proper lysosomal function in the aged brain. In addition, both PGRN and PSAP are also secreted and both have been reported to have neurotrophic functions (Ahmed et al., 2007; Bateman and Bennett, 2009; Cenik et al., 2012; Matsuda et al., 2007; Nicholson et al., 2012; O'Brien and Kishimoto, 1991; Qi and Grabowski, 2001). The interaction of PGRN and PSAP in the extracellular space might also regulate their neurotrophic properties. Thus given the importance of PGRN-PSAP interaction and relevance to FTLD, we decided to map domains involved in PGRN-PSAP binding. Here we report that PSAP interacts with PGRN through granulin motifs in PGRN and the linker region between saposin B and C (BC linker) in PSAP.
Materials and Methods
Antibodies
The following antibodies were used in this study: mouse anti-FLAG (M2) from Sigma (RRID:AB_439685), rabbit anti-human PSAP antibodies from Proteintech Group (RRID:AB_2172462), goat anti-human PGRN (RRID:AB_2114489), and sheep anti-mouse PGRN (RRID:AB_2114504) from R&D systems, rat anti-mouse LAMP1 (1D4B) from BD Biosciences (RRID:AB_2134499). Rabbit anti-human saposin B antibodies (Leonova et al., 1996) is a gift from Dr. Xiaoyang Qi (University of Cincinnati School of Medicine, Cincinnati, OH).
DNA and Plasmids
Full length human PSAP and PGRN expression constructs were previously described (Zhou et al., 2015). FLAG tagged PGRN and PSAP truncation constructs were generated by cloning the PGRN and PSAP fragments into the pSectag2B vector (Invitrogen) with an N-terminal FLAG tag. GST-saposin constructs and his Sumo saposin constructs were generated by cloning saposin and linker regions into pGEX6p-1 and pET his Sumo vector, respectively. AP-granulin constructs were generated by cloning granulins into pAP5 vector (Genhunter). GFP-granulins were generated by inserting GFP and granulin sequences into pSectag2B vector after the signal sequence. PSAP mutant with the BC linker (amino acid sequence: VKEMPMQTLVPAKVASKNVIPALELVEPIKKHEVPAK) replaced by CD linker was generated from hPSAP construct by PCR using following primers (hSap B 3: 5PhosCTCATCACAGAACCCAACC and CD linker hSap C 5: 5Phos ACGCGGCTGCCTGCACTGACCGTTCACGTGACTCAGCCAAAGGACGGTTCTGATGTTTACTGTGAGGTG). His tagged and lentiviral human WT PSAP, human mutant PSAP with the BC linker replaced by CD linker, and human PGRN constructs were generated by cloning corresponding genes into pSectag2B vector (Invitrogen) and into pCDH-puro vector (System bio), respectively. To generate fusion construct of PSAP with the platelet derived growth factor receptor (PDGFR) transmembrane domain, full-length WT and mutant PSAP with the BC linker replaced by CD linker were cloned into the pDisplay vector (Invitrogen). Rat FLAG-RFP-LAMP1 was obtained from Addgene. FLAG tagged SapB-BC linker-TcLAMP1 or BC linker-TcLAMP1 were generated by clone rat transmembrane and cytosolic domains of LAMP1 (Tc) into FLAG-SapB-BC linker and FLAG-BC linker constructs (with stop codon removed) using XhoI and PmeI sites.
Cell Culture
HEK293T (RRID:CVCL_0063, from ATCC), COS-7 (RRID:CVCL_0224, from ATCC) and WT and PSAP-/- fibroblast cells (derived from Psap-/- mice (RRID:IMSR_JAX:002792)) were maintained in Dulbecco's Modified Eagle's medium (Cellgro) supplemented with 10% fetal bovine serum (Gibco) and 1% Penicillin–Streptomycin (Invitrogen) in a humidified incubator at 37°C and 5% CO2. Cells were transiently transfected with polyethyleneimine (PEI) as described (Vancha et al., 2004). Psap-/- fibroblasts were infected with lentivirus expressing WT or mutant PSAP.
Primary cortical neurons were isolated from P0-P1 pups from Grn-/- mice (Jackson laboratories, RRID:IMSR_JAX:013175) using a modified protocol (Beaudoin et al., 2012). Briefly, cortices were rapidly dissected from the brain in 2 mL HBSS supplemented with B27 (ThermoFisher Scientific catalog#17504044) and 0.5 mM L-glutamine (ThermoFisher Scientific catalog#25030024) at 4°C. Meninges were removed before digestion with papain (Worthington LS003119, 2mg/ml in HBSS) and DNaseI (1mg/ml in HBSS, Sigma) for 12 minutes at 37°C. Tissues were then dissociated using fire polished glass pipettes. Cells were spun down and resuspended in Neuroplex medium (Gemini) plus B27 and plated onto poly-lysine (Sigma) coated dishes.
Protein preparation and Western blot analysis
Purification of recombinant his-human WT PSAP, mutant PSAP with BC linker replaced with CD linker, and his-human PGRN proteins, co-immunoprecipitation (IP) assays and Western blots were performed as previously described (Zhou et al., 2015). Gst and his-sumo saposin fusion proteins were purified using Gst or cobalt beads, respectively, from Origami™ B(DE3) strain (Novagen) after IPTG induction.
Cell surface binding assay and binding
AP binding assays were done as previously described (Hu et al., 2010). Briefly, conditioned medium with AP or AP-PGRN/Granulins generated from HEK293T cells were incubated with pDisplay-PSAP-transfected COS-7 cells or for 2 h at room temperature before fixation and heat inactivation of endogenous AP at 65°C overnight. Bound AP to the primary cortical neurons was measured using NIH image software. Followed by measuring the intensity of bound AP signal, the saturation binding curves and scatchard plots were generated, and binding KD were calculated by using GraphPad Prism 5.0 software.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described (Zhou et al., 2015). Briefly the cells were fixed with 4% paraformaldehyde and permeabilized and blocked with blocking buffer (0.05% saponin, 3% BSA in PBS). The cells were incubated with primary antibodies in blocking buffer overnight at 4 °C and CF488A, CF568, or CF660C (Biotum) conjugated secondary antibodies. Images were acquired on a CSU-X spinning disc confocal microscope (Intelligent Imaging Innovations) with an HQ2 CCD camera (Photometrics) using a 100× objective.
Quantitative analysis of lysosomal PGRN and endocytosed PGRN and PSAP
Images were processed and analyzed by ImageJ program as previously described (Zhou et al., 2015). Briefly, the colocalization between PGRN and lysosomal markers, LAMP1, was quantified by using JACoP plugin. To quantify the endocytosed PGRN and PSAP, the entire cell was selected and the fluorescence intensity was measured directly by ImageJ.
Ethical Approval and Consent to participate
All applicable international, national, and/or institutional guidelines for recombinant DNA and cell lines were followed. The work under protocol (MUA#: 15965) were approved by the Cornell University Institutional Biosafety Committee (IBC).
Results
PSAP interacts with granulin motifs in PGRN
To determine the binding site for PSAP in PGRN, we made a series of deletion constructs of individual granulin (Grn) motifs (Fig. 1A). While deletion of both Grn D and Grn E motifs results in a great decrease of the interaction between PGRN and PSAP, further deletion of other granulin motifs only slightly weaken the interaction, suggesting that Grn D and Grn E are the main binding site for PSAP in PGRN (Fig. 1B). To further confirm that PSAP interacts with granulins, we tested the interaction between PSAP and individual granulins tagged with alkaline phosphatase (AP) or GFP. Among the AP-tagged granulins with decent expression, granulin A, B, D and E coimmunoprecipitate with PSAP, but Grn D and E exhibited the strongest binding towards PSAP (Fig. 1C). Grn G, on the other hand, does not show any detectable binding to PSAP (Fig. 1C). Similarly, with GFP-tagged granulins, granulins D and E showed the strongest interaction followed by granulin B (Fig. 1D). Taken together, these results suggest that PGRN binds to PSAP through multiple granulin motifs with highest affinity to Grn D and E.
Figure 1. PSAP interacts with granulin motifs.

(A) Domain structure of human PGRN (aa 1-593). (B) HEK293T cells were co-transfected with FLAG tagged PGRN truncation constructs and untagged human PSAP as indicated. Cells were lysed two days after transfection and the lysates were immunoprecipitated with anti-FLAG antibodies. The IP products were analyzed by Western blot using anti-FLAG and anti-PSAP antibodies (RRID:AB_2172462). (C) Conditioned medium from HEK293T expressing different AP-fusion proteins were incubated with FLAG beads only or FLAG beads with FLAG-PSAP recombinant proteins. The amount of AP proteins co-immunoprecipitated with FLAG-PSAP was analyzed by Western blot. (D) Conditioned medium from HEK293T expressing different GFP-fusion proteins were mixed with 1μg purified recombinant FLAG-PSAP. Immunoprecipitations were then performed with anti-GFP antibodies and the IP products were analyzed by Western blot.
To determine the relative affinity for the PSAP and PGRN/Grn binding, we anchored PSAP to the cell surface by fusing it to the transmembrane domain of PDGF receptor and measured the binding affinity using an alkaline phosphatase (AP) based cell surface binding assay (Hu et al., 2010). AP-Grn D and AP-Grn-E bind to PDGFR fused PSAP on the cell surface with a Kd around 20nM, similar to that of full length PGRN (Fig. 2), confirming that these two granulin motifs are the main binding sites for PSAP in PGRN.
Figure 2. Granulin D and E bind to PSAP with similar affinity as full length PGRN.

(A) Conditioned medium containing AP, AP-PGRN, AP-Grn D, or AP-Grn E were incubated with COS-7 cells transfected with PSAP fused to PDGFR. Cells were fixed and AP binding was visualized with AP substrates. Scale bar=100μm. (B) AP–PGRN binding to PSAP-PDGFR expressing COS-7 cells measured as a function of AP–PGRN concentration. (C-E) Scatchard plot of AP-PGRN (C), AP-GRN D (D) or AP-GRN E (E) binding to PSAP-PDGFR expressing COS-7 cells. KD, mean ± sem, n = 4.
PSAP interacts with PGRN through the linker region between saposin B and C (BC linker)
To map the binding site for PGRN in PSAP, we created a series of PSAP deletion constructs and tested their interaction with PGRN. While deletion of saposin D (ABC) has no effect on the PSAP-PGRN interaction, further deletion of saposin C and the linker region between saposin B and C (AB) results in loss of the PGRN-PSAP interaction (Fig. 3A). The construct containing saposin B, the BC linker, and saposin C (BC), binds to PGRN as strong as full-length PSAP (Fig. 3B). This suggests that PGRN interacts with saposin C or the linker region between B and C. However, saposin C itself is not sufficient to interact with PGRN (Fig. 3B). Addition of the BC linker region to saposin C (L+C) allows full binding to PGRN despite very weak expression, suggesting that the BC linker region is the binding site for PGRN in PSAP (Fig. 3B). This is further supported with purified recombinant saposin proteins from bacteria. None of the recombinant saposin domains interact with PGRN, but fusion of the BC linker region with saposin C allows strong binding to PGRN (Fig. 3C). To determine if the BC linker alone is able to bind with PGRN, we fused either saposin B plus BC linker or BC linker alone with the transmembrane and cytoplasmic domains of LAMP1 (TcLAMP1). The co-immunoprecipitation result shows that FLAG-tagged BC linker-TcLAMP1 was able to co-immunoprecipitate PGRN as efficiently as FLAG tagged saposin B plus BC linker and FLAG tagged full-length PSAP (Fig. 3D), suggesting that the BC linker alone is sufficient for PGRN binding.
Figure 3. PSAP interacts with PGRN through the BC linker.

(A) HEK293T cells were co-transfected with FLAG tagged PGRN and full length his-PSAP or PSAP truncation mutants as indicated. Cells were lysed two days after transfection and the lysates were immunoprecipitated with anti-FLAG antibodies. The IP products were analyzed by Western blot using anti-FLAG and anti-PSAP antibodies (RRID:AB_2172462). (B) HEK293T cells were co-transfected with untagged PGRN and FLAG tagged PSAP fragments as indicated. Cells were lysed two days after transfection and the lysates were immunoprecipitated with anti-FLAG antibodies. (C) Recombinant Gst- or his-sumo tagged saposins purified from bacteria were incubated with FLAG beads only or FLAG beads with FLAG-PGRN recombinant proteins. The amount of saposin co-immunoprecipitated with FLAG-PGRN was analyzed by Western blot. (D) HEK293T cells were co-transfected with untagged PGRN and FLAG tagged full-length PSAP, full-length LAMP1, saposin B with BC linker fused with transmembrane and cytoplasmic domain of LAMP1 (Tc-LAMP1) and BC linker fused with Tc-LAMP1 as indicated. Cells were lysed two days after transfection and the lysates were immunoprecipitated with anti-FLAG antibodies and immunoblotted with anti-PGRN and anti-FLAG antibodies.
To further prove that the BC linker is the binding site for PGRN in PSAP, we generated a PSAP mutant construct in which the BC linker is replaced by the linker between saposin C and D (CD linker) (Fig. 4A). This replacement totally abolished the binding between PSAP and PGRN in the co-immunoprecipitation (Fig. 4A) and cell surface binding assays (Fig. 4B), further supporting that the BC linker is required for binding between PSAP and PGRN.
Figure 4. PSAP mutant with the BC linker replaced failed to interact with PGRN.

(A) HEK293T cells were co-transfected with FLAG tagged wild type (WT) or mutant (mut) PSAP and untagged human PGRN as indicated. Media was collected and cells were lysed three days after transfection. The lysates and media were separately immunoprecipitated with anti-FLAG antibodies. (B) Conditioned medium containing AP or AP-PGRN were incubated with COS-7 cells transfected with WT or mutant PSAP fused to PDGFR. Cells were fixed and AP binding were visualized with AP substrates. Scale bar=100μm.
PSAP mutant that fails to bind to PGRN cannot deliver PGRN to lysosomes
Previously, we showed that PSAP mediates sortilin-independent delivery of PGRN into lysosomes (Zhou et al., 2015). In fact, since sortilin is not expressed in fibroblasts, lysosomal trafficking of PGRN is totally abolished in PSAP deficient fibroblasts (Zhou et al., 2015). We predicted that the PSAP mutant with its PGRN binding site mutated cannot facilitate PGRN lysosomal trafficking. Indeed, the PSAP mutant with the BC linker replaced by the CD linker fails to rescue the PGRN trafficking defect in PSAP deficient fibroblasts, although lysosomal trafficking of the PSAP mutant itself is not impaired (Fig. 5A, 5B). PSAP also facilitates lysosomal delivery of PGRN from the extracellular space. However, this is also abolished by mutation of the BC linker region of PSAP. While recombinant wild type PSAP facilitates neuronal uptake and lysosomal delivery of recombinant PGRN protein lacking the sortilin binding site (Zheng et al., 2011), the PSAP mutant with the BC linker replaced fails to do so (Fig. 5C-5E).
Figure 5. PSAP mutants fail to deliver PGRN to lysosomes.

(A) PSAP-/- mouse fibroblasts were infected with lentivirus expressing WT or mutant PSAP with the BC linker replaced by CD linker. Cells were fixed and stained with rabbit anti-human saposin B, sheep anti-mouse PGRN and rat anti-LAMP1 antibodies. Scale bar=10 μm. (B) The colocalization of PGRN with lysosomal marker LAMP1 in (A) were quantified using Image J. n=3, ***, p<0.001. (C) Grn-/- cortical neurons were incubated with C-terminally FLAG his tagged human PGRN which does not bind to sortilin (5ug/ml) together with recombinant WT PSAP or mutant PSAP with the BC linker replaced by CD linker (5ug/ml). 12 hours later, cells were fixed and stained with rabbit anti-human saposin B, goat anti-human PGRN and rat anti-LAMP1 antibodies. Scale bar=10 μm. (D, E) The endocytosed PGRN (D) and PSAP (E) for the experiment in (C) were quantified by using Image J. n=3; ns, no significance; ***, p<0.001. One way ANOVA.
Discussion
Our previous studies have shown that PGRN and PSAP form a complex both intracellularly and extracellularly and facilitate each other's lysosomal delivery (Zhou et al., 2015; Zhou et al., 2017b). Since both PGRN and PSAP are essential for proper lysosomal function and loss of function in either gene causes lysosomal storage diseases, more studies on the interaction of PGRN and PSAP will help us develop strategies to enhance the function of PGRN and PSAP. This will be of therapeutic interest for FTLD, since haploinsufficiency of PGRN is a leading cause of FTLD and impaired PSAP lysosomal trafficking is observed in FTLD patients with GRN mutations (Zhou et al., 2017b). Future studies to design or screen small molecules to enhance the binding affinity between these two proteins might be therapeutic interest. Thus it is important for us to understand the molecular mechanisms involved in the physical interaction between PGRN and PSAP.
In agreement with our previous finding that saposins do not bind to PGRN (Zhou et al., 2015), here we show that PGRN interacts with the linker between saposin B and C domain through multiple granulin motifs. Granulin D and E bind to PSAP with similar affinity as full length PGRN. How the BC linker region interacts with granulin motifs is still unclear and structural studies will be useful to further understand this interaction. We have failed to further narrow down the binding site in the 37 amino acid BC linker to a shorter peptide (data not shown). But most likely PSAP-PGRN binding involves an interaction between a peptide (BC linker) and a folded domain (granulins).
PSAP is known to undergo processing into saposins during its trafficking to lysosomes (Matsuda et al., 2007; O'Brien and Kishimoto, 1991; Qi and Grabowski, 2001). Additionally, PGRN is hypothesized to be processed into granulin peptides in the lysosome (Cenik et al., 2012). Both PGRN and PSAP have been shown to interact with cathepsin D (Beel et al., 2017; Gopalakrishnan et al., 2004; Laurent-Matha et al., 2002; Zhou et al., 2017a) and cathepsin D is known to cleave PSAP during trafficking (Gopalakrishnan et al., 2004). Thus it is possible that binding of PGRN to the BC linker region of PSAP will affect the kinetics of PSAP, and maybe also PGRN processing. Since granulin peptides, especially granulin D and E, bind to PSAP with the similar affinity as full length PGRN, granulins might interact with full length PSAP or its processing intermediates with BC linker still attached. The relative rates of PGRN and PSAP processing during trafficking and in the lysosome might determine the interaction modes between the final products and processing intermediates of these two proteins. Further studies on these aspects of PGRN-PSAP biology will allow a better understanding on how these two proteins interact and regulate each other's functions and help develop therapeutic means to enhance their activities.
Acknowledgments
This work is supported by funding to F.H. from Weill Institute for Cell and Molecular Biology, the Association of Frontotemporal Dementia (AFTD), and NINDS (R01NS088448-01) and by funding to X. Z. from the Weill Institute Fleming Postdoctoral Fellowship.
Abbreviations
- FTLD-TDP
frontotemporal lobe degeneration with ubiquitin and TDP-43 positive inclusions
- ALS
amyotrophic lateral sclerosis
- TDP-43
TAR DNA-binding protein-43
- PGRN
progranulin
- PSAP
prosaposin
- SAP
saposin
- kDa
kilo Dalton
- WT
wild type
Footnotes
Conflict of interest: none.
References
- Ahmed Z, Mackenzie IR, Hutton ML, Dickson DW. Progranulin in frontotemporal lobar degeneration and neuroinflammation. J Neuroinflammation. 2007;4:7. doi: 10.1186/1742-2094-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida MR, Macario MC, Ramos L, Baldeiras I, Ribeiro MH, Santana I. Portuguese family with the co-occurrence of frontotemporal lobar degeneration and neuronal ceroid lipofuscinosis phenotypes due to progranulin gene mutation. Neurobiol Aging. 2016 doi: 10.1016/j.neurobiolaging.2016.02.019. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245–1254. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- Beaudoin GM, 3rd, Lee SH, Singh D, Yuan Y, Ng YG, Reichardt LF, Arikkath J. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Beel S, Moisse M, Damme M, De Muynck L, Robberecht W, Van Den Bosch L, Saftig P, Van Damme P. Progranulin functions as a cathepsin D chaperone to stimulate axonal outgrowth in vivo. Hum Mol Genet. 2017 doi: 10.1093/hmg/ddx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcastro V, Siciliano V, Gregoretti F, Mithbaokar P, Dharmalingam G, Berlingieri S, Iorio F, Oliva G, Polishchuck R, Brunetti-Pierri N, et al. Transcriptional gene network inference from a massive dataset elucidates transcriptome organization and gene function. Nucleic Acids Res. 2011;39:8677–8688. doi: 10.1093/nar/gkr593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B, Sephton CF, Kutluk Cenik B, Herz J, Yu G. Progranulin: a proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J Biol Chem. 2012;287:32298–32306. doi: 10.1074/jbc.R112.399170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotman SL, Karaa A, Staropoli JF, Sims KB. Neuronal ceroid lipofuscinosis: impact of recent genetic advances and expansion of the clinicopathologic spectrum. Curr Neurol Neurosci Rep. 2013;13:366. doi: 10.1007/s11910-013-0366-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan MM, Grosch HW, Locatelli-Hoops S, Werth N, Smolenova E, Nettersheim M, Sandhoff K, Hasilik A. Purified recombinant human prosaposin forms oligomers that bind procathepsin D and affect its autoactivation. Biochem J. 2004;383:507–515. doi: 10.1042/BJ20040175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vaegter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kousi M, Lehesjoki AE, Mole SE. Update of the mutation spectrum and clinical correlations of over 360 mutations in eight genes that underlie the neuronal ceroid lipofuscinoses. Hum Mutat. 2012;33:42–63. doi: 10.1002/humu.21624. [DOI] [PubMed] [Google Scholar]
- Laurent-Matha V, Lucas A, Huttler S, Sandhoff K, Garcia M, Rochefort H. Procathepsin D interacts with prosaposin in cancer cells but its internalization is not mediated by LDL receptor-related protein. Exp Cell Res. 2002;277:210–219. doi: 10.1006/excr.2002.5556. [DOI] [PubMed] [Google Scholar]
- Leonova T, Qi X, Bencosme A, Ponce E, Sun Y, Grabowski GA. Proteolytic processing patterns of prosaposin in insect and mammalian cells. J Biol Chem. 1996;271:17312–17320. doi: 10.1074/jbc.271.29.17312. [DOI] [PubMed] [Google Scholar]
- Matsuda J, Yoneshige A, Suzuki K. The function of sphingolipids in the nervous system: lessons learnt from mouse models of specific sphingolipid activator protein deficiencies. J Neurochem. 2007;103(Suppl 1):32–38. doi: 10.1111/j.1471-4159.2007.04709.x. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Nicholson AM, Finch NA, Almeida M, Perkerson RB, van Blitterswijk M, Wojtas A, Cenik B, Rotondo S, Inskeep V, Almasy L, et al. Prosaposin is a regulator of progranulin levels and oligomerization. Nat Commun. 2016;7:11992. doi: 10.1038/ncomms11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson AM, Gass J, Petrucelli L, Rademakers R. Progranulin axis and recent developments in frontotemporal lobar degeneration. Alzheimers Res Ther. 2012;4:4. doi: 10.1186/alzrt102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JS, Kishimoto Y. Saposin proteins: structure, function, and role in human lysosomal storage disorders. FASEB J. 1991;5:301–308. doi: 10.1096/fasebj.5.3.2001789. [DOI] [PubMed] [Google Scholar]
- Qi X, Grabowski GA. Molecular and cell biology of acid beta-glucosidase and prosaposin. Prog Nucleic Acid Res Mol Biol. 2001;66:203–239. doi: 10.1016/s0079-6603(00)66030-0. [DOI] [PubMed] [Google Scholar]
- Smith KR, Damiano J, Franceschetti S, Carpenter S, Canafoglia L, Morbin M, Rossi G, Pareyson D, Mole SE, Staropoli JF, et al. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet. 2012;90:1102–1107. doi: 10.1016/j.ajhg.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancha AR, Govindaraju S, Parsa KV, Jasti M, Gonzalez-Garcia M, Ballestero RP. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 2004;4:23. doi: 10.1186/1472-6750-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Brady OA, Meng PS, Mao Y, Hu F. C-terminus of progranulin interacts with the beta-propeller region of sortilin to regulate progranulin trafficking. PLoS ONE. 2011;6:e21023. doi: 10.1371/journal.pone.0021023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Paushter DH, Feng T, Pardon CM, Mendoza CS, Hu F. Regulation of cathepsin D activity by the FTLD protein progranulin. Acta Neuropathol. 2017a doi: 10.1007/s00401-017-1719-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bastos de Oliveira F, Qi X, Brown WJ, Smolka MB, Sun Y, Hu F. Prosaposin facilitates sortilin-independent lysosomal trafficking of progranulin. J Cell Biol. 2015;210:991–1002. doi: 10.1083/jcb.201502029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Sun L, Bracko O, Choi JW, Jia Y, Nana AL, Brady OA, Hernandez JCC, Nishimura N, Seeley WW, et al. Impaired prosaposin lysosomal trafficking in frontotemporal lobar degeneration due to progranulin mutations. Nat Commun. 2017b;8:15277. doi: 10.1038/ncomms15277. [DOI] [PMC free article] [PubMed] [Google Scholar]