

Abstract
Translation elongation factor P (EF-P) in Bacillus subtilis is required for a form of surface migration called swarming motility. Furthermore, B. subtilis EF-P is post-translationally modified with a 5-aminopentanol group but the pathway necessary for the synthesis and ligation of the modification is unknown. Here we determine that the protein YmfI catalyzes the reduction of EF-P-5 aminopentanone to EF-P-5 aminopentanol. In the absence of YmfI, accumulation of 5-aminopentanonylated EF-P is inhibitory to swarming motility. Suppressor mutations that enhanced swarming in the absence of YmfI were found at two positions on EF-P, including one that changed the conserved modification site (Lys 32) and abolished post-translational modification. Thus, while modification of EF-P is thought to be essential for EF-P activity, here we show that in some cases it can dispensable. YmfI is the first protein identified in the pathway leading to EF-P modification in B. subtilis, and B. subtilis encodes the first EF-P ortholog that retains function in the absence of modification.
Keywords: YmfI, EF-P, translation, post-translational modification, swarming motility
INTRODUCTION
The protein Elongation Factor P (EF-P) is a translation elongation factor highly conserved in all living things (eIF-5a in Eurkayotes and aIF-5a in Archaea) (Ganoza, 2002, Lassak, 2016, and Rajkovic, 2017). Recent work has shown that EF-P enhances translation of particular codon combinations encoding prolines in both Bacteria and Eukaryotes (Doerfel, 2013, Ude, 2013, and Gutierrez, 2013). The mechanism by which EF-P potentiates translation is poorly understood but EF-P structurally resembles a tRNA molecule, has been shown to bind between the E and P sites of the ribosome, and may function catalytically by increasing entropy (Blaha, 2009, Hanawa-Suetsugu, 2004, Doerfel. 2015 and Schmidt, 2016). In nearly all systems studied to date, depletion of EF-P is either lethal or results in severe growth limitation, presumably due to the impaired ability to translate target sequences in essential genes (Balibar, 2013, Patel, 2009, Schnier, 1991, and Yanagisawa, 2016). The one exception thus far is the Gram positive firmicute bacterium Bacillus subtilis where EF-P is not required for growth and is instead specifically required for swarming motility, a flagellar-mediated multicellular behavior in which cells rapidly move across a semi-solid surface (Kearns, 2004; Rajkovic, 2016).
In all organisms tested to date, EF-P is post-translationally modified on a conserved lysine/arginine residue at a position analogous to the amino acid acceptor site of tRNAs (Katz, 2013). The chemical nature of the EF-P post-translational modification varies between species. Within the proteobacteria, Escherichia coli and Salmonella enterica modify EF-P lysine 34 with a β-lysine group (Blaha, 2010 and Navarre, 2010), whereas Neisseria meningitidis, Pseudomonas aeruginosa and Shewanella oneidensis modify EF-P arginine 32 with a rhamnose moiety (Lassak, 2015, Rajkovic, 2015, and Yanagisawa, 2016). The EF-P ortholog in Eukaryotes, eIF5A, is modified by the addition of hypusine (Cooper, 1983). In each case mentioned thus far the function of the modification is thought to be essential for EF-P activity because mutations in the modification system result in growth defects similar to efp deletion mutants and reduce the translation rate of consecutive prolines (Patel, 2009, Sasaki, 1996, Yanagisawa, 2009, Doerfel et al., 2013, and Yanagisawa, 2015). The mechanism by which the various modification groups enhance EF-P activity is unknown.
B. subtilis modifies EF-P lysine 32 with yet another chemical variant, 5-aminopentanol. Substitution of the conserved modified lysine to alanine (EF-PK32A) was shown to impair surface motility but the enzymes required for 5-aminopentanolylation were unknown (Rajkovic, 2016). Here, we revisit the swarming mutants from the same screen that first discovered EF-P as a swarming motility activator and we identify YmfI as a protein required for EF-P 5-aminopentanolylation. YmfI is paralogous to the fatty acid biosynthesis gene FabG and catalyzes the reduction of 5-aminopentanone to 5-aminopentanol as, in the absence of YmfI, EF-P was modified with a 5-aminopentanone group and cells exhibit attenuated swarming motility. Finally, in a forward genetic screen, we identify mutations in efp that bypass the requirement for YmfI in swarming motility and in one of the mutants, EF-PK32R, post-translational modification was abolished. Thus, YmfI is the first example of an enzyme involved in EF-P modification in B. subtilis, and the B. subtilis EF-P is the first example that retains activity in the absence of modification.
RESULTS
The absence of YmfI impairs swarming motility
EF-P proteins are thought to be activated by post-translational modification and B. subtilis EF-P is post-translationally modified by a 5-aminopentanol group attached to lysine 32 (Rajkovic, 2016). B. subtilis EF-P resolved as two bands in semi-native gel electrophoresis and Western blot analysis, and we hypothesized that the two bands could represent different states of EF-P modification and/or activity (Fig 1A left, lane T0). To determine whether the two species of EF-P were differentiated by a post-translational mechanism, translation was inhibited by the addition of spectinomycin and EF-P electrophoretic mobility in semi-native gel electrophoresis was monitored over time. Within 15 minutes of inhibiting translation, the lower band species of EF-P diminished in abundance (Fig 1A left, lanes T0-T240). We hypothesize that the two bands of EF-P represent different post-translational modification states.
Figure 1. EF-P resolves as two species on a semi-native gel in a YmfI-dependent manner.
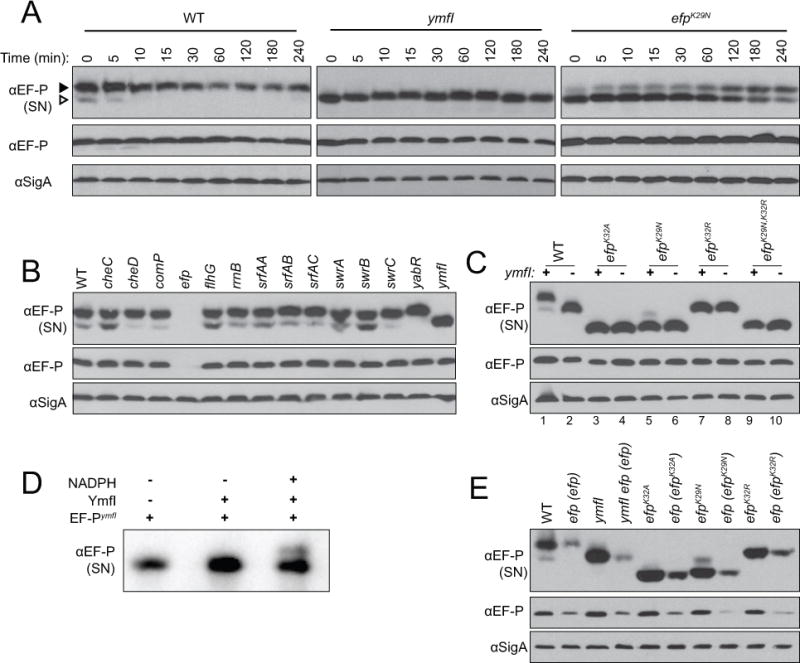
(A) Translation was inhibited in mid-log phase cultures by the addition of spectinomycin, lysates were subsequently harvested at the indicated time points, resolved by semi-native (top panel, “SN”) or denaturing (middle and bottom panels) polyacrylamide gel electrophoresis, electroblotted, and probed with anti-EF-P or anti-SigA polyclonal antisera (used as a loading control) as indicated. The following strains were used to generate the samples: wild type (DK1042), ymfI (DK3621), and efpK29N (DK4282). For denaturing gels, EF-P resolved at approximately 22 kDa and SigA resolved at approximately 46 kDa. The black arrow indicates the upper band and the white arrow indicates the lower band. (B) Lysates of mid-log phase cultures were resolved by semi-native (top panel, “SN”) or denaturing (middle and bottom panels) polyacrylamide gel electrophoresis, electroblotted, and probed with anti-EF-P or anti-SigA polyclonal antisera as indicated. The following strains were used to generate the samples: wild type (3610), cheC (DS1045), cheD (DS1064), comP (DS1028), efp (DS1124), flhG (DS1164), rrnB-16S (DS1146), srfAA (DS1102), srfAB (DS1044), srfAC (DS1122), swrA (DS1026), swrB (DS1107), swrC (DS1113), yabR (DS1078), and ymfI (DS1029) (C) Lysates of mid-log phase cultures were resolved by semi-native (top panel, “SN”) or denaturing (middle and bottom panels) polyacrylamide gel electrophoresis, electroblotted, and probed with anti-EF-P or anti-SigA polyclonal antisera as indicated. Lanes are numbered at the bottom of the panels for clarity in text. The presence and absence of a wild type copy of the ymfI gene is indicated by (+) and (-) respectively at the top of the panels. The following strains were used to generate samples: wild type (DK1042), ymfI (DK3621), efpK32A (DK3235), efpK32A ymfI (DK3712), efpK29N (DK4282), efpK29N ymfI (DK4396), efpK32R (DK4359), efpK32R ymfI (DK4397), efpK29N,K32R (DK4420), and efpK32A,K32R ymfI (DK4436). (D) EF-P-FLAG purified from a ymfI mutant (EF-P-FLAGymfI) was incubated alone, with YmfI, or with YmfI and 150 mM NADPH for 30 min at 37C. Reactions were subsequently resolved by semi-native gel electrophoresis, electroblotted, and probed with anti-EF-P antisera. (E) Lysates of mid-log phase cultures were resolved by semi-native (top panel, “SN”) or denaturing (middle and bottom panels) polyacrylamide gel electrophoresis, electroblotted, and probed with anti-EF-P or anti-SigA polyclonal antisera as indicated. The following strains were used to generate the samples: wild type (DK1042), efp (efp) (DK3780), ymfI (DK3621), ymfI efp (efp) (DK3789), efpK32A (DK3235), efp (efpK32A) (DK2248), efpK29N (DK4282), efp (efpK29N) (DK4043), efpK32R (DK4359), efp (efpK32R) (DK4072).
To find proteins involved in EF-P post-translational modification, whole cell lysates of transposon mutants taken from the same screen that identified the swarming motility defect of an efp mutant were resolved by semi-native gel electrophoresis and Western blot analysis (Kearns and Losick, 2004). Mutation of one gene, ymfI, resulted in a single species of EF-P (Fig 1B). When resolved side-by-side, EF-P of the ymfI mutant exhibited an electrophoretic mobility similar to the lower band found in the wild type (Fig 1C, compare lanes 1 and 2). Moreover, the band intensity of ymfI EF-P did not change over time after spectinomycin treatment (Fig 1A middle). We conclude that the protein encoded by the ymfI gene, YmfI, alters EF-P mobility in semi-native gels and we hypothesize that YmfI might control EF-P post-translational modification.
Whereas mutation of efp results in a severe defect in swarming motility, mutation of ymfI resulted in periodic cessation and reinitiation of swarming motility to create a terraced colony appearance on swarm plates (Fig 2A,B) (Kearns and Losick, 2004). The terracing pattern of swarming was reminiscent of an EF-P mutant defective in post-translation modification in which the modification site lysine 32 was mutated to an alanine (Fig 2A,C) (Rajkovic et al., 2016). In both qualitative and quantitative analyses, the swarming defect of the efpK32A mutant was more severe than that of a ymfI mutant (Fig 2A,C). Furthermore, the swarming motility of a ymfI efpK32A double mutant resembled that of the efpK32A mutant alone, suggesting that the efpK32A mutation was epistatic (Fig 2A,C). As further evidence of epistasis, Western blots of EF-P resolved by semi-native gel electrophoresis indicated an altered mobility of the mutant EF-PK32A protein that was not further altered by the absence of YmfI (Fig 1C, compare lanes 3 and 4). We conclude that YmfI requires EF-P lysine residue 32 to promote swarming motility and alter EF-P electrophoretic mobility.
Figure 2. Cells mutated for ymfI are defective in swarming motility and swarming can be restored by mutations in efp.
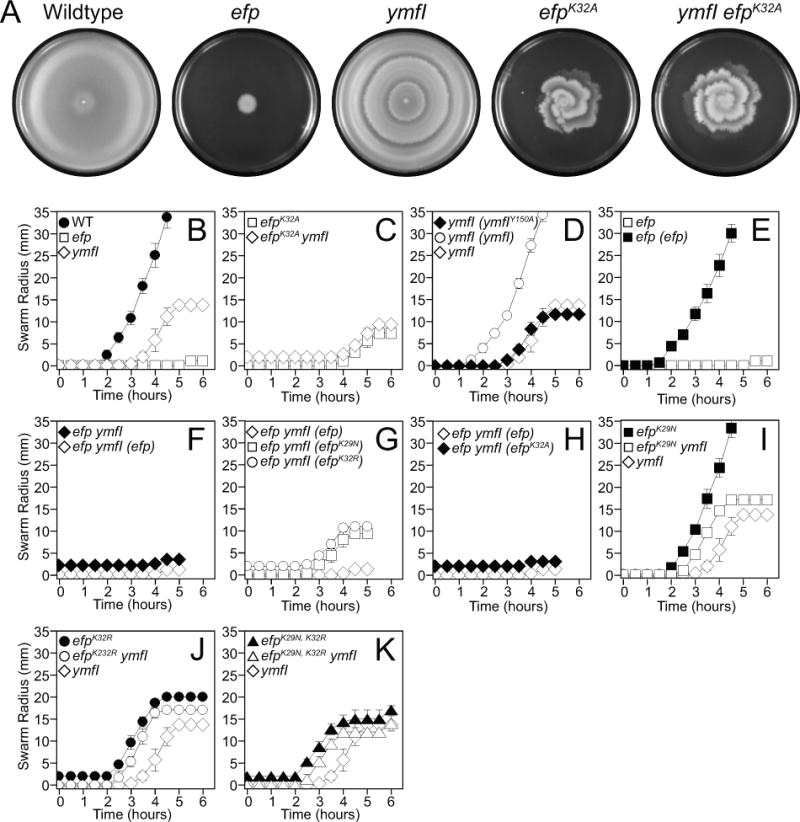
(A) Top views of centrally-inoculated swarm plates incubated overnight at 37°C were imaged against a black background. Zones of colonization appear light grey. The plate inoculated with the ymfI mutant has internal rings that mark the locations at which the population stopped moving and restarted at a later time point. Note: a comparable cessation can be seen at hour 5 in the quantitative swarm expansion assay in panel 2B. The following strains were used to generate the panels: wild type (DK1042), efp (DK2050), ymfI (DK3621), efpK32A (DK3235), and ymfI efpK32A (DK3712).(B-K) Quantitative swarm expansion assays in which mid-log phase cultures were concentrated and used to inoculate swarm plates. Swarm expansion was monitored along the same axis every 30 min for 5–6 hours. Each data point represents the average of three replicates and error bars represent the standard deviation. The following strains were used as the inoculum (B) wild type (DK1042), efp (DK2050), and ymfI (DK3621). (C) efpK32A (DK3235) and efpK32A ymfI (DK3712). (D) ymfI (ymfIY150A) (DK4233), ymfI (ymfI) (DK3969), and ymfI (DK3621). (E) efp (DK2050) and efp (efp) (DK3780). (F) efp ymfI (DK2886), and efp ymfI (efp) (DK3789). (G) efp ymfI (efp) DK3789, efp ymfI (efpK29N) (DK4043), and, efp ymfI (efpK32R) (DK4072). (H) efp ymfI (efp) (DK3789) and efp ymfI (efpK32A) (DK2889). (I) efpK29N (DK4282), efpK29N ymfI (DK4396), and ymfI (DK3621). (J) efpK32R (DK4359), efpK32R ymfI (DK4397), and ymfI (DK3621). (K) efpK29N,K32R (DK4420), efpK29N,K32R ymfI (DK4436), and ymfI (DK3621).
YmfI is homologous to FabG, a 3-keto-acyl ACP reductase essential for fatty acid biosynthesis in E. coli, and contains conserved active site residues (Fig S1A) (Price, 2001). Whereas FabG is essential for growth, cells mutated for ymfI exhibited only a 5% decrease in growth rate resembling the slight growth rate reduction found in cells mutated for efp (Fig S2). To determine whether the YmfI active site residues are required for promoting swarming motility and altering EF-P semi-native gel mobility, we first complemented the ymfI mutation in trans. The ymfI gene appeared to be the third gene in a putative four gene operon (ymfFHIJ) (Fig 3A), and swarming motility was restored to the ymfI mutant when ymfI was cloned downstream of the PymfF promoter and inserted at an ectopic site in the chromosome (amyE∷PymfF-ymfI) (Fig 2D). Thus, the ymfI mutant defect was due to the absence of ymfI, not due to a polar effect on other genes in the operon which, when mutated, had no effect on swarming motility or EF-P native gel electrophoretic mobility (Fig S3A–E). The ectopic ymfI complementation construct did not restore swarming motility to the ymfI mutant, however, when the ymfI putative active site residue tyrosine 150 was mutated to an alanine (amyE∷PymfF-ymfIY150A) (Fig 2D, Fig S1A). We conclude that the active site residue of YmfI is required for either YmfI activity or stability and we infer that YmfI may activate EF-P enzymatically.
Figure 3. Genetic architecture and phylogenetic distribution of the ymfI locus.
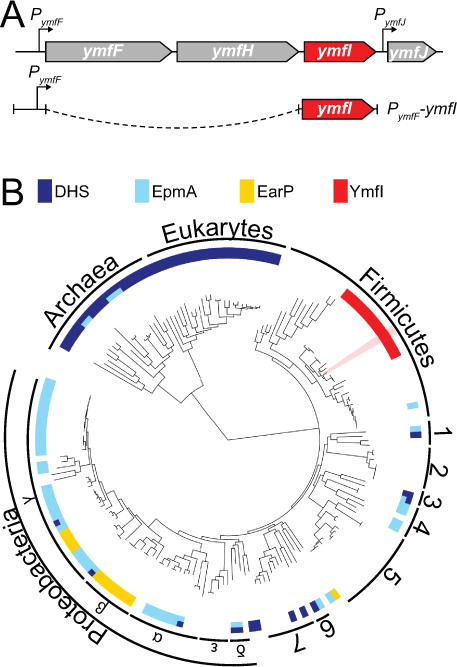
(A) Predicted genetic architecture of the ymfI locus and the design of the PymfF-ymfI complementation construct. Both ymfF and ymfH are predicted to encode peptidases and ymfJ encodes a DUF3235-domain containing protein. (B) The distribution of homologs of the EF-P modification enzymes deoxyhypusine synthase, DHS (dark blue), EpmA (light blue), EarP (yellow), and YmfI (red) across the three domains of life. YmfI is a member of the large family of alcohol dehydrogenases and the proximity to neighboring genes as indicated in Fig 3A was used to aid in the identification of YmfI homologs. Where multiple enzymes are encoded in the same genome, the bar is split accordingly. White space indicates the absence of a homolog to any EF-P modification enzyme. Numbers indicate the following clades (1) Flavobacterium-Cytophaga-Bacteroides group, (2) Chlamydiales, (3) Planctomycetes, (4) Spirochaetes, (5) Actinobacteria, (6) Deinococcus-Thermus group, and (7) Cyanobacteria. Bacillus subtilis is highlighted in pink. See Fig S5 enlarged map with species names. Table S3 contains the accession numbers for each species and Table S4 contains all sequences called to be YmfI in all completed bacterial genomes to date.
To determine if YmfI directly participates in the alteration of EF-P electrophoretic mobility, recombinant YmfI was purified from E. coli and EF-P-FLAG was purified from the B. subtilis ymfI mutant (EF-P-FLAGymfI). EF-P-FLAGymfI resolved as a single band on semi-native gel electrophoresis in the absence of YmfI or in the presence of YmfI but the absence of NADPH (Fig 1D, left and middle lanes). Upon incubation of EF-P-FLAGymfI in the presence of both purified YmfI and NADPH, however, a faint upper band resolved on semi-native gel electrophoresis (Fig 1D, right lane). We conclude that YmfI is an enzyme that requires the NADPH cofactor and is sufficient in vitro to alter EF-P electrophoretic mobility on a semi-native gel.
YmfI is required for 5-aminopentanolylation of EF-P
One way in which YmfI might alter semi-native gel electrophoretic mobility in a manner dependent on its enzymatic activity is if YmfI is involved in 5-aminopentanolylation of EF-P. In order to assess the modification status of EF-P, EF-P was affinity tagged and ectopically overexpressed from an IPTG-inducible promoter in efp and efp ymfI mutant backgrounds and subjected to chymotrypsin in-gel digestion. The digested protein was then resolved on an orbitrap elite mass spectrometer, and subjected to electron transfer dissociation fragmentation to generate a complete MS2 spectrum of the peptide containing Lys32 (QHVKPGKGAAF). In the wild type, both unmodified (Fig 4A, 1138.624 Da) and 5-aminopentanolylated (Fig 4B, 1239.709 Da) EF-P peptides were detected. The additional mass of 101.085 Da is consistent with previous reports for 5-aminopentanol C5H12NO (Rajkovic et al., 2016). In the absence of YmfI, unmodified peptides were detected but instead of a 101.085 Da mass, a mass of 99.068 Da was identified on Lys32 (Fig 5). From the mass difference of 2 Da we determined that in the absence of YmfI, the post-translational modification on Lys32 is C5H10NO. Although C5H10NO could represent multiple different structures, based on previous MS3 data of 5-aminopentanol and the proposed reductase activity of ymfI, we suggest it likely corresponds to 5-aminopentanone (Fig 4F). Further, while the precise position of the carbonyl can only be unequivocally determined with the use NMR or other high resolution structural methods, we suggest it likely corresponds to the position of the hydroxyl group in 5-aminopentanol at either the C3 or C4 position (Rajkovic 2016).
Figure 4. Deletion of ymfI results in 5-aminopentanonation of EF-P and can be suppressed by abolishing EF-P post-translational modification.
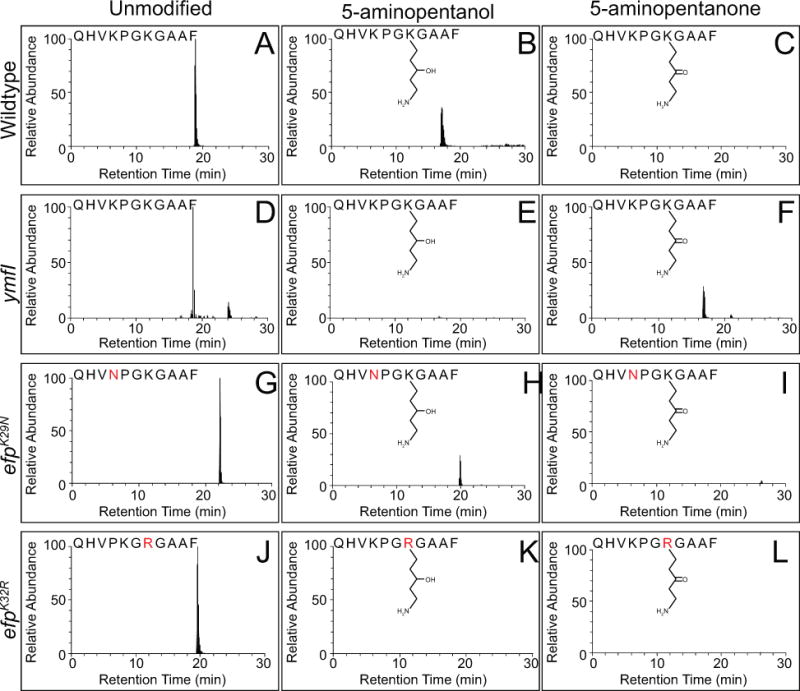
Extracted ion chromatograms of chymotrypsin-digested EF-P peptide from wild type (A–C), ymfI mutant (D–F), efpK29N mutant (G–I), and efpK32R mutant (J–L). Three different species were detected for the peptide corresponding to the wild type sequence QHVKPGKGAAF containing including unmodified (A,D,G,J), 5-aminopentanolylated (B,E,H,K), and 5-aminopentanonated (C,F,I,L) lysine residue 32. All chromatograms for ymfI represent the 2+ ion for the indicated peptide. All other chromatograms represent the 3+ ion for the indicated peptide. Point mutations are indicated in bold red lettering. Predicted chemical structures are indicated. The cartoon indicates that the hydroxyl/carbonyl group is at the C3 position but it is possible that the hydroxyl group is at the C4 position instead. The precise position of the carbonyl can only be unequivocally determined with the use NMR or other high resolution structural methods.
Figure 5. EF-P is 5-aminopentanonated in the absence of YmfI.
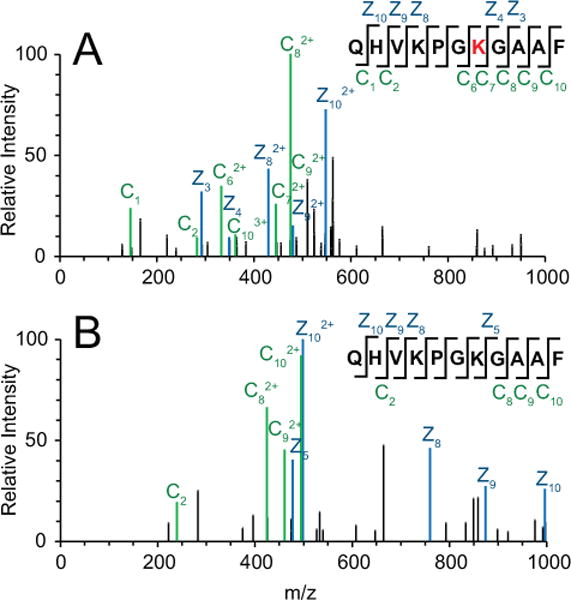
MS2 spectrum generated from ETD fragmentation of the QHVKPGKGAAF peptide. Both (A) Unmodified (precursor m/z of 570.320) and (B) 5-aminopentanonated (precursor m/z of 413.752) peptide were detected in the absence of ymfI. z ions are indicated in blue, and c ions are indicated in green. Site of 5-aminopentanonation (Lys32) is marked in red.
Very low levels of 5-aminopentanolylated EF-P also appeared to be detected in the absence of ymfI (Fig 4E). As the reduced and oxidized forms differ by two hydrogen atoms, however, it is possible that the parent ion predicted to be the 5-aminopentanolylated peptide is in reality a heavy isotope of the 5-aminopentanonated peptide. In order to address this possibility, a predicted isotope distribution was calculated for QHVKPGKGAAF with either modification, and compared to the measured isotope distribution (Fig S4). Actual measured abundances for each isotope of the 5-aminopentanonated peptide in the ymfI background corresponded with predicted abundances precisely, but not for the 5-aminopentanolylated peptide. Thus, the apparent 5-aminopentanolylated peptide in the ymfI mutant is likely an artifact of heavy isotope incorporation. Although the possibility that extremely low levels of 5-aminopentanol exist in the absence of ymfI cannot be ruled out, the differences between the predicted and actual isotope distribution for 5-aminopentanolylated EF-P makes this possibility remote. We conclude that YmfI is an enzyme that directly catalyzes the reduction of 5-aminopentanone to 5-aminopentanol. We further conclude, based on YmfI’s ability to alter EF-P electrophoretic mobility in vitro, that YmfI alone is capable of catalyzing the reduction reaction when the substrate is ligated to EF-P.
During the mass spectrometry analyses, we noted that there appeared to be a relative excess of unmodified EF-P that did not seem consistent with the relative band intensity in the wild type (Compare Fig 1A left and Fig 4A,B). As the EF-P-FLAG construct was expressed from an IPTG-inducible promoter, we wondered whether 1 mM IPTG induction was causing over-representation of the unmodified form. Thus, an IPTG-titration was conducted for both the swarming motility phenotype and the semi-native gel Western blots. The Physpank-efp-FLAG construct restored swarming motility to the efp mutant even in the absence of IPTG, suggesting that leakiness of the promoter was sufficient to rescue the phenotype (Fig 6A, upper) though EF-P protein was not detectable by Western blot analysis until 0.01 mM IPTG was added (Fig 6A, lower). Moreover, the EF-P upper band saturated in intensity while the lower band continued to accumulate (Fig 6A, lower). We conclude that the affinity-tagged version of EF-P is functional at very low levels in the efp mutant and that IPTG-induction resulted in hyper-accumulation of the unmodified form. In the efp ymfI double mutant, however, 0.03 mM IPTG induction of EF-P-FLAG was required to restore partial swarming (Compare first four lanes of Fig 6A,B upper). Further, 0.05 mM IPTG and greater allowed the efp ymfI double mutant to swarm at a faster rate than the ymfI mutant alone (Fig 6B, upper). Regardless, in the absence of YmfI, only one EF-P band was detected in Western blot analysis at all IPTG concentrations tested (Fig 6B, lower). We conclude that the efp ymfI double mutant requires more EF-P for swarming motility and that EF-P overexpression could partially suppress the absence of YmfI.
Figure 6. Overexpression of EF-P results in hyper-accumulation of the unmodified form and partially bypasses the need for YmfI.
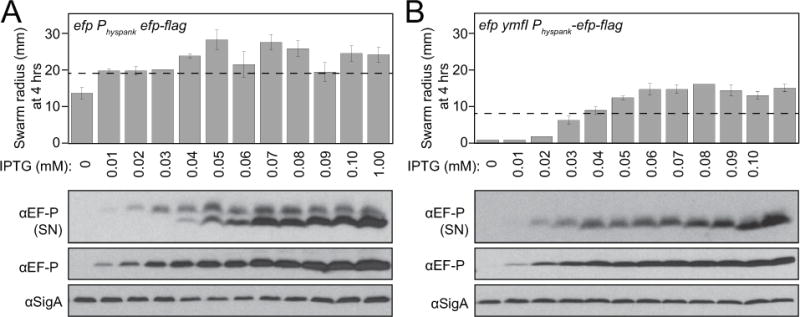
A) An efp mutant and B) a efp ymfI double mutant each with an ectopically integrated Physpank-EF-P-FLAG construct was induced with the indicated concentrations of IPTG and measured for swarming motility radius after 4 hours of incubation (upper panel) and Western blot analysis following semi-native gel (SN) (top row) and denaturing (middle and bottom rows) electrophoresis of cell lystates (lower panel). Note, dashed line on 4 hr swarm expansion graph indicates the extent of expansion of either wild type (A) or a ymfI mutant (B). The following strains were used to generate samples: efp Physpank-efp-flag (DK2448) and efp ymfI Physpank-efp-flag (DK3828). Wild type (DK1042) and ymfI (DK3621) were used to generate the dashed lines.
Modification independent alleles of EF-P bypass the need for YmfI
To explain the swarming defect of the ymfI mutant, we hypothesized that either the reduced 5-aminopentanol modification was essential for EF-P activity, or the oxidized 5-aminopentanone modification was inhibitory. To distinguish the two possibilities, we randomly mutagenized the efp gene and screened for mutations that bypass the need for YmfI. If the 5-aminopentanol group was essential for activity, we predicted that there would be no mutation in efp sufficient to bypass the absence of YmfI. To set up the screen, we first complemented the efp mutation in trans. The efp gene appears to be the third gene in a putative three gene operon (yqhS-papA-efp) (Fig S3F), and swarming motility was restored to the efp mutant when efp was cloned downstream of the PyqhS promoter region and inserted at an ectopic site in the chromosome (amyE∷PyqhS-efp) (Fig 2E). Thus the efp mutant defect was due to the absence of EF-P, not due to a polar effect on other genes in the region which, when mutated had no effect on swarming motility or on EF-P semi-native gel electrophoretic mobility (Fig S3E,G,H). The PyqhS-efp complementation construct, however, did not restore swarming to the efp ymfI double mutant (Fig 2F). The failure to complement the swarming defect of efp ymfI similar to that of ymfI alone appeared to be due a reduced level of ectopic EF-P expression relative to that of the native locus (Fig 1E), consistent with the observation that ymfI rescue depends on EF-P levels (Fig 6B). We conclude that transcription of efp from PyqhS is sufficient under most conditions but additional promoters may be needed to amplify expression when ymfI is mutated. We note that the upstream PyqhR may contribute to EF-P expression as transcriptomics indicates that there is no transcriptional terminator between the yqhR and yqhS genes (Fig S3F) (Nicolas et al., 2012). We conclude that, in the absence of YmfI, a threshold of EF-P protein level and thus activity is required to initiate swarming motility.
Next, the PyqhS-efp complementation construct was randomly mutagenized using an error prone polymerase in seven separate tubes and each PCR product was separately transformed into an efp ymfI double mutant background to generate seven parallel pools of transformants. Each pool of transformants was collected and spotted as a mixed population onto swarming motility agar. Cells initially grew as a tight colony in the center of the plate but, in each pool, motile cells emerged with a prolonged lag period and swarmed outward from the origin. One colony was isolated from each motile swarm and was confirmed to have restored swarming motility. Sequencing of the PyqhS-efp construct indicated that each isolate contained either a mutation of Lys29 to an asparagine (EF-PK29N) or a mutation of Lys32 to an arginine (EF-PK32R) (Table 1). In three swarming proficient isolates, K29N or K32R was the only mutation present (Fig 2G). Mutation of K32R was allele-specific for the rescue of swarming motility in an efp ymfI background as ectopic introduction of K32A was not sufficient (Fig 2H). Furthermore, neither ectopic integrant of K29N nor K32R exhibited increased protein levels relative to ectopic integrant of the wild type allele in otherwise wild type cells indicating that suppression was not simply due to increased stability or expression of the mutant proteins (Fig 1E). We conclude that particular substitutions of Lys29 and Lys32 yielded EF-P at least partially YmfI-independent and sufficient to restore swarming in the absence of YmfI.
Table 1.
Mutations in efp that improve swarming in the absence of YmfI
| Strain Number | Genotype | Nucleotide substitution(s) |
|---|---|---|
| DK4043 | efpK29N | AAA > AAT (residue 29) |
| DK4978 | efpK29N | AAA > AAT (residue 29) |
| DK4072 | efpK32R | AAA > AGG (residue 32) |
| DK4075 | efpK32R,E101V | AAA > AGA (residue 32) GAA > GTA (residue 101) |
| DK4076 | efpK32R,V111M | AAA > AGA (residue 32) GTG > ATG (residue 111) |
| DK4073 | efpK32R,E124G | AAA > AGA (residue 32) GAA > GGA (residue 124) |
To further characterize the YmfI-independent alleles of EF-P, each single mutant was introduced by allelic replacement at the native locus. Cells expressing EF-PK29N at the native site swarmed like the wild type and migrated as two bands on a semi-native gel (Fig 2I, Fig 1C lane 5). The majority of the EF-PK29N resolved as the lower species, unlike wild type EF-P which resolved mostly as the upper species (Fig 1C, compare lane 1 to lane 5). One way in which the ratio of upper/lower bands could be altered in this way is if EF-PK29N affected the rate of post-translational modification. To test this hypothesis, translation was inhibited in an efpK29N mutant and EF-P electrophoretic mobility on a semi-native gel was monitored over time. The dominant EF-PK29N lower band appeared to decrease slowly with a concomitant increase in the intensity of the upper band indicative of a slow conversion of the oxidized to reduced modification (Fig 1A, right). Further, EF-PK29N expressed from the native site was not fully independent of YmfI as mutation of ymfI diminished swarming migration and abolished the faint upper band in semi-native gels (Fig 1C, compare lanes 5 and 6; Fig 2I). We conclude that EF-PK29N has a decreased rate of post-translational modification thereby preventing accumulation of 5-aminopentanonated EF-P.
Cells expressing EF-PK32R at the native site arrested swarming prematurely and resolved as a single band on native gels (Fig 1C lane 7; Fig 2J). Unlike efpK29N, mutation of ymfI did not diminish swarming migration of efpK32R or alter mobility on semi-native gel electrophoresis of the EF-PK32R protein (Fig 1C compare lanes 7 and 8; Fig 2J). Finally, wild type cells expressing the combined double mutant allele EF-PK29N,K32R exhibited a premature swarming arrest and resolved as a single band on native gels regardless of the presence of YmfI, similar to the EF-PK32R single mutant alone (Fig 1C compare lanes 9 and 10; Fig 2K). We conclude that both lysine residues contribute to the same function as the phenotype of the double mutant was not additive, and that mutation of K32R yields EF-P YmfI-independent and is epistatic toK29N.
To determine the consequence of the suppressor mutations on post-translational EF-P modification, the EF-P alleles were fused to a FLAG tag, overexpressed, purified from B. subtilis, and analyzed by mass spectrometry. For the wild type and EF-PK29N protein, both unmodified and 5-aminopentanolylated EF-P were detected (Fig 4G–I). By contrast, only the unmodified fragment was detected for the EF-PK32R protein and no additional masses could be detected on the peptide (Fig 4J–L). We conclude that one way to bypass the need for YmfI and restore EF-P activity is to abolish EF-P modification on Lys32. We also conclude that cells mutated for ymfI exhibit a swarming defect because they accumulate an inhibitory 5-aminopentanone group on EF-P. Swarming inhibition may be relieved either by converting the group to 5-aminopentanol or by the accumulation of unmodified EF-P. Unmodified EF-P can accumulate by mutation of K32R, by decreasing the rate of post-translation modification as seen in K29N mutants, or by simply overexpressing EF-P protein. Finally, we conclude that, to the best of our knowledge, B. subtilis EF-P is the only member of its family that functions in the absence of post-translational modification.
DISCUSSION
Elongation factor P is a highly conserved protein found in all domains of life that structurally resembles a tRNA, associates with the ribosome, and assists in translating particular primary sequences such as polyprolines (Blaha, 2009, Hanawa-Suetsugu, 2004, Schmidt, 2016, Doerfel, 2013, Ude, 2013, and Gutierrez, 2013). Furthermore, the EF-P position analogous to the site of amino acid attachment on tRNA is post-translationally modified by species-specific, structurally-divergent molecules (Lassak, 2016). Here we identify YmfI of Bacillus subtilis as the first protein shown to be involved in the post-translational modification of EF-P by 5-aminopentanol. YmfI is a protein of previously unknown function that is a paralog of the fatty acid biosynthesis protein FabG and requires a conserved active site residue for its function (Fig S1). Furthermore, YmfI catalyzes the NADPH-dependent alteration of EF-P electrophoretic mobility in vitro and the reduction of EF-P 5-aminopentanone to 5-aminopentanol in vivo (Fig 7A). To date, B. subtilis is the only species known to modify EF-P with 5-aminopentanol but YmfI homologs were found by sequence analysis and synteny with upstream genes in the Firmicutes order of Bacilliales (Fig 3A,B). Based on our analyses, we predict that the genera of Listeria and Staphylococcus also modify EF-P with 5-aminopentanol.
Figure 7. Model of EF-P activation by YmfI.
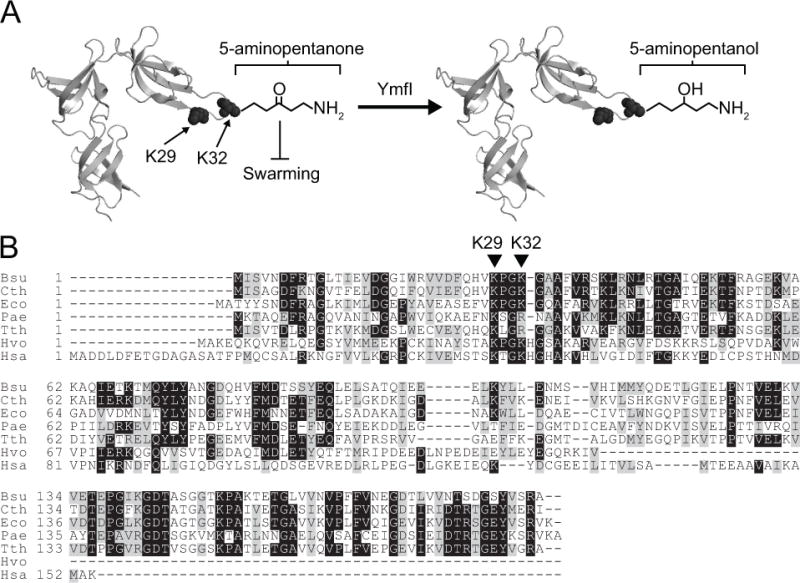
(A) The crystal structure of EF-P isolated from the close relative of B. subtilis, Clostridium thermocellum (PDB ID 1YBY) with the location of Lys 29 (K29) and Lys 32 (K32) indicated. YmfI catalyzes the converstion of 5-aminopentanone (left) to 5-aminopentanol (right) groups on Lys32. In the absence of YmfI, 5-aminopentanone accumulates on EF-P and inhibits swarming motility (T-bar). (B) A multiple sequence alignment of EF-P orthologs from B. subtilis (Bsu), C. thermocellum (Cth), E. coli (Eco), Pseudomonas aeruginosa (Pae), Thermus thermophilus (Tth), the archaeon Haloferax volcanii (Hvo), and the eukaryote Homo sapiens (Hsa). The location of B. subtilis EF-P conserved residues K29 and K32 are indicated by carets.
In Bacillus subtilis, EF-P is required for a flagellar-mediated form of surface motility called swarming and mutation of YmfI conferred a swarming defect. The swarming motility defect was not due to the inability to synthesize the mature 5-aminopentanol group and was rather due to the accumulation of the inhibitory oxidized 5-aminopentanone group as indicated by suppressor mutations that altered the EF-P primary sequence. One suppressor mutation changed the modified EF-P residue Lys32 to an arginine, and while the allele impaired swarming in a wild type cell, it bypassed the requirement for YmfI by abolishing modification altogether. Both a swarming impairment and lack of modification were previously reported in a site-directed mutation that changed Lys 32 to Ala (Rajkovic, 2016), but unlike the EF-PK32R allele, the EF-PK32A allele was unable to bypass the swarming requirement for YmfI. We infer that a positive charge at position 32 assists in the function of EF-P and that a lysine residue in particular is ideal in B. subtilis. The partial phenotypes of the EF-P K32R and K32A mutations however, indicate that 5-aminopentanolylation per se is not explicitly required for EF-P activity. To the best of our knowledge, the EF-P protein of B. subtilis is the first of its kind to retain substantial biological activity in the absence of post-translational modification possibly due to the below-expected number of genomic polyprolines encoded in B. sutilis (Rajkovic, 2016).
Another suppressor mutation changed the highly conserved EF-P residue Lys29 to an asparagine. EF-PK29N appears to reduce the rate of 5-aminopentanolylation on Lys32 as indicated by a dramatic reduction in upper EF-P band on semi-native gels. The lower band accumulated in the EF-PK29N mutant but could represent at least two different molecular species. 5-aminopentanonated EF-P resolves as the lower band as it was the only band observed in the absence of YmfI. The lower band also accumulated when EF-P was overexpressed, and unmodified EF-P predominated in mass spectrometry analyses. Thus, the lower band on semi-native gels may represent two molecular species, one in which Lys32 is unmodified and one in which Lys32 is 5-aminopentanonated. We infer that EF-PK29N restores swarming in the absence of YmfI by reducing the rate of post-translational modification thereby accumulating EF-P unmodified at Lys32. EF-P Lys29 could play a role in the catalysis of 5-aminopentanoylation as a binding site for modification proteins or as a residue for allosteric regulation. While we cannot distinguish whether Lys29 is an active or allosteric site, we note that Lys29 appears to be as highly conserved as Lys32 in EF-P proteins (Fig 7B, Fig S5).
Post-translational modification is thought to be essential for EF-P activity for the organisms in which it has been studied and is generally assumed to directly participate in the synthesis of certain primary sequences, but how the modification participates in potentiating translation is unknown. If the modification does directly participate in synthesis, it seems unusual that a conserved EF-P function can tolerate a wide variety of different chemical modifications including but likely not limited to lysine, hypusine, rhamnose, and 5-aminopentanol groups depending on the organism. Furthermore, some modifications such as deoxyhypusine in the fungus Fusarium alter EF-P function and here we show in B. subtilis that 5-aminopentanonation has similar consequences, perhaps for regulatory reasons (Martinez-Rocha, 2016). Consistent with regulation, we show that in B. subtilis mutation of Lys32 to an Arg (a residue naturally found in other EF-P proteins; Fig 7B, Fig S5) partially preserves EF-P function in the absence of the 5-aminopentanol group. Moreover, overexpression of EF-P and mutation of Lys29 to an asparagine results in the hyper-accumulation of unmodified EF-P and bypasses the swarming defect caused by the absence of YmfI. Thus, we hypothesize an unmodified Lys32 may be fully functional in Bacillus subtilis. We speculate that post-translational modification of EF-P may serve a regulatory function in B. subtilis and perhaps other organisms.
Experimental Procedures
Strains and growth conditions
B. subtilis and E. coli strains were grown in lysogeny broth (LB) (10 g tryptone, 5 g yeast extract, 5 g NaCl per L) or on LB plates fortified with 1.5% Bacto agar at 37°C. When appropriate, antibiotics were included at the following concentrations: 10 μg/ml tetracycline, 100 μg/ml spectinomycin, 5 μg/ml chloramphenicol, 5 μg/ml kanamycin, and 1 μg/ml erythromycin plus 25 μg/ml lincomycin (mls). Isopropyl β-D-thiogalactopyranoside (IPTG, Sigma) was added to the medium at the indicated concentration when appropriate.
For quantitative swarm assays, strains were grown to mid-log phase (OD600 0.3–1.0) concentrated to an OD600 of 10 in PBS pH 7.4 (0.8% NaCl, 0.02% KCl, 100 mM Na2HPO4, and 17.5 mM KH2PO4) plus 0.5% India ink. LB plates fortified with 0.7% agar were dried for 10 min open-faced in a laminar flow hood and subsequently inoculated by spotting 10 uL cell resuspensions onto the center of the plate. Plates were dried an additional 10 min open-faced in a laminar flow hood and then incubated at 37°C in a humid chamber. Swarm radius was measured along the same axis every 30 minutes.
Images of swarm plates were obtained by toothpick-inoculating a colony into the center of an LB plate fortified with 0.7% agar. Plates were dried open-faced in a laminar flow hood for 12 min and incubated at 37°C in a humid chamber for 20 hrs. Images were taking using a BioRad Gel Doc.
Western Blotting
Strains were grown to mid-log phase, concentrated to an OD600 of 10 in lysis buffer (17.2 mM Tris pH 7.0, 8.6 mM EDTA pH 8.0, 1 mg/mL Lysozyme, 0.1 mg/mL RNaseA, 20 μg/mL DNase I, and 50 μg/mL phenylmethane sulfonyl fluoride) and incubated at 37°C for 30 min. SDS sample buffer (500 mM Tris pH 6.8, 22% glycerol, 10% SDS, and 0.12% bromophenol blue) was added, and samples were boiled for 5 min. 12 μl boiled samples were loaded onto 10% polyacrylamide native (with no added SDS) or 15% polyacrylamide denaturing (with 0.1% SDS) gels. Lysates were resolved at 150 V for 1.25 hours, transferred onto nitrocellulose membranes, and subsequently probed with a 1:40,000 dilution of anti-EF-P or a 1:80,000 dilution anti-SigA polyclonal antiserum. Following incubation with the primary antibodies, nitrocellulose membranes were probed with horseradish peroxidase conjugated goat anti-rabbit immunoglobulin G. Blots were developed using Pierce ECL substrate (Thermo Fisher Scientific). To inhibit translation, 200 ug/mL spectinomycin was added to mid-log phase cultures.
Strain construction
All constructs were introduced into DK1042, a competent derivative of ancestral strain 3610 (Konkol et al., 2013) or laboratory strain PY79 and then transferred to the 3610 background using SPP1-mediated generalized phage transduction (Yasbin and Young, 1974). All strains used in this study are listed in Table 2. All plasmids used in this study are listed in Supplemental Table S1. All primers used in this study are listed in Supplemental Table S2.
Table 2.
Strains
| Strain | Genotype | Reference |
|---|---|---|
| 3610 | Wild type | |
| DK1042 | comIQ12L | Konkol, 2013 |
| DK1962 | comIQ12L ΔyqhS | |
| DK2050 | comIQ12L Δefp | Rajkovic, 2016 |
| DK2248 | comIQ12L Δefp amyE∷PyqhS-efpK32A cat | |
| DK2448 | comIQ12L Δefp amyE∷Physpank-efp-flag spec | Rajkovic, 2016 |
| DK2886 | comIQ12L Δefp ymfI∷tet | |
| DK2889 | comIQ12L Δefp ymfI∷tet amyE∷PyqhS-efpK32A cat | |
| DK3159 | comIQ12L ΔpapA | |
| DK3235 | comIQ12L efpK32A | Rajkovic, 2016 |
| DK3621 | comIQ12L ymfI∷tet | |
| DK3712 | comIQ12L efpK32A ymfI∷tet | |
| DK3726 | comIQ12L ymfF∷tet | |
| DK3727 | comIQ12L ymfH∷tet | |
| DK3728 | comIQ12L ymfFH∷tet | |
| DK3780 | comIQ12L Δefp amyE∷PyqhS-efp cat | |
| DK3789 | comIQ12L Δefp ymfI∷tet amyE∷PyqhS-efp cat | |
| DK3828 | comIQ12L Δefp ymfI∷tet amyE∷Physpank-efp-flag spec | |
| DK3969 | comIQ12L ymfI∷tet amyE∷PymfF-ymfI spec | |
| DK4043 | comIQ12L Δefp ymfI∷tet amyE∷PyqhS-efpK29N cat | |
| DK4072 | comIQ12L Δefp ymfI∷tet amyE∷PyqhS-efpK32R cat | |
| DK4073 | comIQ12 L Δefp ymfI∷tet amyE∷PyqhS-efpK32R,E124G cat | |
| DK4075 | comIQ12 L Δefp ymfI∷tet amyE∷PyqhS-efpK32R,E101V cat | |
| DK4076 | comIQ12L Δefp ymfI∷tet amyE∷PyqhS-efpK32R,V111M cat | |
| DK4233 | comIQ12L ymfI∷tet amyE∷PymfF-ymfIY150A spec | |
| DK4246 | comIQ12L Δefp amyE∷Physpank-efpK29N-flag spec | |
| DK4247 | comIQ12L Δefp amyE∷Physpank-efpK32R-flag spec | |
| DK4282 | comIQ12L efpK29N | |
| DK4359 | comIQ12L efpK32R | |
| DK4396 | comIQ12L efpK29N ymfI∷tet | |
| DK4397 | comIQ12L efpK32R ymfI∷tet | |
| DK4420 | comIQ12L efpK29N,K32R | |
| DK4436 | comIQ12L efpK29N,K32R ymfI∷tet | |
| DS235 | ymfI∷tet | Kearns, 2004 |
| DS236 | ymfJ∷tet | |
| DS1026 | swrA∷Tn10 spec | Kearns, 2004 |
| DS1028 | comP∷Tn10spec | Kearns, 2004 |
| DS1029 | ymfI∷Tn10spec | Kearns, 2004 |
| DS1044 | srfAB∷Tn10spec | Kearns, 2004 |
| DS1045 | cheC∷Tn10spec | Kearns, 2004 |
| DS1064 | cheD∷Tn10spec | Kearns, 2004 |
| DS1078 | yabR∷Tn10spec | Kearns, 2004 |
| DS1102 | srfAA∷Tn10spec | Kearns, 2004 |
| DS1107 | swrB∷Tn10spec | Kearns, 2004 |
| DS1113 | swrC∷Tn10spec | Kearns, 2004 |
| DS1122 | srfAC∷Tn10spec | Kearns, 2004 |
| DS1124 | efp∷Tn10spec | Kearns, 2004 |
| DS1146 | rrnB∷Tn10spec | Kearns, 2004 |
| DS1164 | flhG∷Tn10spec | Kearns, 2004 |
| PY79 | sfp0 swrA |
Complementation constructs
To create the efp complementation construct, the PyqhS promoter was amplified using primers 477/478 and the efp gene was amplified using primers 483/486 using B. subtilis 3610 genomic DNA as a template. Fragments were digested with EcoRI/XhoI and XhoI/BamHI respectively and subsequently ligated into the EcoRI and BamHI restriction sites of the plasmid pDG364, containing a polylinker and chloramphenicol resistance cassette between two arms of the amyE gene (Guérout-Fleury et al., 1996), to produce pDP85. pDP85 was transformed into DK2050 to produce DK3780. Site directed mutation of efp to change lysine 32 to an alanine was performed by amplifying DK3780 genomic DNA with primer pairs 3177/4039 and 3180/4038 (primers 4038 and 4039 encode the efpK32A mutation). The resulting fragments were subjected to Gibson assembly (Gibson, 2009), and transformed into DK2050 followed by selection for chloramphenicol resistance to produce DK2248.
To create the ymfI complementation construct, the PymfF promoter was amplified using primers 4724/4884 and the ymfI gene was amplified using primers 4721/4722 using B. subtilis 3610 chromosomal DNA as a template. Fragments were digested with EcoRI/HinDIII and HinDIII/NheI respectively were subsequently ligated into the EcoRI and NheI restriction sites of the plasmid pAH25, containing a polylinker and spectinomycin resistance cassette between two arms of the amyE gene, to produce pKRH50. pKRH50 was transformed into DK3621 followed by selection for spectinomycin resistance to produce DK3969. Site directed mutation of ymfI to change tyrosine 150 to an alanine was performed by amplifying DK3969 genomic DNA with primer pairs 4884/4971 and 4722/4970. The resulting fragments were subjected to Gibson assembly (Gibson, 2009) followed by PCR amplification by primers 4722/4884. The resulting DNA fragment was subsequently ligated into the EcoRI and NheI restriction sites of the plasmid pAH25 to produce pKRH67. Plasmid pKRH67 was transformed into DK3621 followed by selection on spec to produce DK4233.
Site directed mutation of the Physpank-efp-flag construct was performed by amplifying DK2448 genomic DNA (Rajkovic, 2015) with primer pairs 861/5133 and 862/5132 for efpK29N and 861/5143 861/5142 for efpK32R. Cognate fragments were subjected to Gibson assembly (Gibson, 2009) and transformed into DK2050 followed by selection for spectinomycin resistance to yield DK4246 and DK4247.
YmfI expression construct
To create the His-Sumo-YmfI expression construct, ymfI was amplified using primers 5439/5440 using B. subtilis 3610 chromosomal DNA as a template. The resulting fragment was digested with XhoI/SapI and subsequently ligated into the XhoI and SapI restriction sites of the plasmid pTB146 to produce pKRH76.
Native site mutants
Site directed mutation of efp at the native site was performed by allelic replacement. 3610 genomic DNA was amplified using primer pairs 4031/5133 4034/5132 for efpK29N, 4031/5143 4034/5042 for efpK32R, and 4031/5145 4034/5144 for efpK29N,K32R. Cognate fragments were introduced into the SmaI restriction site of pMiniMAD using Gibson assembly (Patrick and Kearns, 2008; Gibson, 2009) to produce pKRH59, pKRH60, and pKRH61 which were subsequently passaged through the recA+ Escherichia coli strain TG1 and transformed into DK1042. Plasmid pMiniMAD (and its derivatives pKRH59, pKRH60, and pKRH61) encodes an mls resistance cassette and a temperature sensitive origin that is active at room temperature but not at 37°C. Thus, mls-resistant colonies were isolated at 37°C (ensuring integration of the plasmid into the chromosome) and subsequently grown in overnight at room temperature, thereby activating the plasmid-encoded temperature sensitive origin and inciting plasmid excision. Mls-sensitive colonies were isolated and the efp locus was sequenced to determine the allele present at that site.
Random mutagenesis of efp
Mutagenesis of the efp complementation construct was achieved by amplification of DK3789 genomic DNA with primer pair 4891/3180 using the error prone Taq polymerase (New England BioLabs Inc.). To decrease the frequency of obtaining mutations that increase expression of EF-P through altering its promoter, the portion of the complementation construct upstream of efp (including PyqhS) was amplified with primers 3177/4892 using the high fidelity Phusion polymerase (New England BioLabs Inc.). The two fragments were ligated together using Gibson assembly (Gibson, 2009) and the products subsequently PCR amplified with primers 954/3180. The resulting 7 pools of mutagenized amyE∷PyqhS-efp were transformed into DK2886 and chloramphenicol-resistant colonies were selected.
Approximately 1700 colonies for each of 7 pools were scraped off of selection plates as a mixture, diluted into LB, and grown to mid-log phase at 37°C. Cell mixtures were then subjected to swarm assays as described above. Following a 9–10 hour incubation on swarm plates, swarming-competent cells began to emerge from the site of inoculation and one colony per pool was isolated from the edge of the swarm front. The amyE∷PyqhS-efp complementation construct from each of the ymfI suppressors was backcrossed into DK2886 by SPP1-mediated transduction and the retention of swarming motility was confirmed. For each ymfI suppressor, the efp complementation construct was sequenced to determine the mutations present.
Insertion/deletion mutants
ymfF∷tet
Mutation of ymfF was performed by amplifying the regions upstream (with primers 4788/4789) and downstream (with primers 4790/4791) of ymfF using 3610 chromosomal DNA as a template, and amplifying the tetracycline resistance cassette from pDG1515 with primers 3250/3251 (Guérout-Fleury, 1995). The three fragments were ligated by Gibson assembly and transformed into DK1042 to produce DK3726. The mutation was confirmed by PCR length polymorphism analysis.
ymfH∷tet
Mutation of ymfH was performed by amplifying the regions upstream (with primers 4792/4793) and downstream (with primers 4832/4795) of ymfH using 3610 chromosomal DNA as a template, and amplifying the tetracycline resistance cassette from pDG1515 with primers 3250/3251. The three fragments were ligated using Gibson assembly and transformed into DK1042 to produce DK3727. The mutation was confirmed by PCR length polymorphism analysis.
ymfFH∷tet
Simultaneous mutation of ymfF and ymfH was performed by amplifying the regions upstream of ymfF (with primers 4788/4789) and downstream of ymfH (with primers 4832/4795) using 3610 chromosomal DNA as a template, and amplifying the tetracycline resistance cassette from pDG1515 with primers 3250/3251. The three fragments were ligated using Gibson assembly and transformed into DK1042 to produce DK3727. The mutation was confirmed by PCR length polymorphism analysis.
ymfJ∷tet
Mutation of ymfJ was performed by amplifying the regions upstream (with primers 105/106) and downstream (with primers 107/108) of ymfJ using 3610 chromosomal DNA as a template, the fragments were purified and used to amplify the tetracycline resistance cassette from HaeII-digested pDG1515. The amplification product was used as a template to cement the fragment using primers 105/108. The final long-flanking homology disruption cassette (Wach, 1996) was transformed into laboratory strain PY79 and transduced to 3610 by SPP1-mediated phage transduction to produce DS236. The mutation was confirmed by PCR length polymorphism analysis.
ΔyqhS
Deletion of yqhS was achieved through allelic replacement. 3610 genomic DNA was amplified using primer pairs 4023/4024 and 4025/4026 and the resulting fragments were introduced into the SmaI restriction site of pMiniMAD using Gibson assembly (Gibson, 2009) to produce pKRH1. Plasmid pKRH1 was passaged through the recA+ Escherichia coli strain TG1, transformed into DK1042, integrated by selecting mls resistant colonies at 37°C and evicted by passage at room temperature. Mls-sensitive colonies were isolated and confirmed to encode the deletion by PCR length polymorphism analysis.
ΔpapA
Deletion of papA was achieved through allelic replacement. 3610 genomic DNA was amplified using primer pairs 4027/4028 and 4029/4030 and the resulting fragments were introduced into the SmaI restriction site of pMiniMAD using Gibson assembly (Gibson, 2009) to produce pKRH2. Plasmid pKRH2 was passaged through the recA+ Escherichia coli strain TG1, transformed into DK1042, integrated by selecting mls resistant colonies at 37°C and evicted by passage at room temperature. Mls-sensitive colonies were isolated and confirmed to encode the deletion by PCR length polymorphism analysis.
SPP1-mediated transduction
SPP1-mediated transductions were performed as described previously (Yasbin, 2004). In short, lysates were created on B. subtilis strains grown in TY (1% Tryptone, 0.5% yeast extract, 0.5% NaCl, 10mM MgSO4, and 1mM MnSO4). Recipient strains were grown in TY to stationary phase, 1 mL diluted into 9 mL TY, and 10 μL (for tetracycline selection) or 25 μL (for spectinomycin and chloramphenicol selection) lysates were added, followed by incubation at room temperature for 30 min and selection for the respective antibiotic resistance at 37°C overnight. For transductions in which spectinomycin or chloramphenicol-resistance was selected for, 10 mM sodium citrate was added to the selection plates.
Mass Spectrometry
Flag-tagged EF-P was overexpressed and purified from each mutant strain (DK2448, DK3828, DK4246, and DK4247) as previously described (Rajkovic, 2016). Following purification, the eluate was concentrated and resolved on a 13% SDS-PAGE gel. Protein was visualized with colloidal Coomassie, excised from the gel, and in-gel digested with chymotrypsin. The generated peptide samples were brought up in 2% acetonitrile in 0.1% formic acid (20 μL) and analyzed (4 μL) by LC/ESI MS/MS with a Thermo Scientific Easy-nLC II (Thermo Scientific, Waltham, MA) coupled to a hybrid Orbitrap Elite ETD (Thermo Scientific, Waltham, MA) mass spectrometer using an instrument configuration as described (Yi et al). In-line de-salting was accomplished using a reversed-phase trap column (100 μm × 20 mm) packed with Magic C18AQ (5-μm 200Å resin; Michrom Bioresources, Auburn, CA) followed by peptide separations on a reversed-phase column (75 μm × 250 mm) packed with Magic C18AQ (5-μm 100Å resin; Michrom Bioresources, Auburn, CA) directly mounted on the electrospray ion source. A 40-minute gradient from 2% to 40% acetonitrile in 0.1% formic acid at a flow rate of 400 nL/minute was used for chromatographic separations. A spray voltage of 2750 V was applied to the electrospray tip and the Orbitrap Elite instrument was operated in the data-dependent mode, switching automatically between MS survey scans in the Orbitrap (AGC target value 1,000,000, resolution 120,000, and injection time 250 milliseconds) with MS/MS spectra detected in the Orbitrap (AGC target value of 50,000, 15,000 resolution and injection time 250 milliseconds). The 3 most intense ions from the Fourier-transform (FT) full scan were selected for fragmentation in the Orbitrap using ETD 100 ms activation time with supplemental CID activation with normalized collision energy of 35%. Selected ions were dynamically excluded for 10 seconds.
Isotope Distribution Analysis
Predicted isotope distributions were calculated using the Scientific Instrument Services, Inc. Isotope Distribution Calculator and Mass Spec Plotter. Actual abundances of each ion were quantified using Thermo Xcalibur software with a Genesis peak picking algorithm and an m/z range of 0.02. To compare the predicted and actual distributions, the measured abundances were set relative to the most abundant ion for the peptide with the indicated modification.
Phylogenetic Analyses
All phylogenetic trees are presented using the Interactive Tree of Life visualization software (Letunic, 2016). To identify the conservation of the different EF-P post-translational modification systems and B. subtilis EF-P residues 29 and 32 (analogous to residues 31 and 34 respectively in E. coli) across all domains of life, the genomes of 191 organisms identified by Ciccarelli, et al. were annotated with the Pfam 29 library using the software hmmer v 3.1b2 and an E value threshold of 1e-5 (Ciccarelli, 2006; Finn, 2016; Eddy, 1998). The presence of EarP was established by the annotation of a DUF2331 domain and the presence of DHS was established by the annotation of a DS domain. The presence of EpmA was established by the annotation of a protein that contained a tRNA-synt_2 domain with homology beginning at residue 15–30 of the profile hidden Markov model (HMM) and without a tRNA_anti-codon domain. The presence of YmfI was established by the presence of 3 co-oriented open reading frames separated by no more than 500 bp that encode proteins with domains found in the YmfFHI proteins. Specifically, YmfF and YmfH were identified by the presence of Peptidase_M16 or Peptidase_M16_C domains, and YmfI homologs were identified by the presences of adh_short or adh_shortC2 domains.
To identify EF-P and eIF5a homologs, proteins that most closely aligned to the eIF-5a, EFP_N, EFP, or Elongation-fact-P_C were identified and subsequently aligned to the EFP_N domain. The identity of EF-P residue 32 was determined by the residue aligning to position 30 of the EFP_N profile HMM and the identity of EF-P residue 29 was determined by the residue aligning to position 27 of the EFP_N profile HMM.
To gain further clarity on which bacteria are likely to encode a YmfI homolog and thereby 5-aminopentanolylated EF-P, all species taxIDs with a completed bacterial genome available on the NCBI reference sequence database were analyzed for the presence of a YmfFHI operon structure as described above.
In vitro Reactions
The His-Sumo-YmfI expression construct, pKRH76, was transformed into E. coli Rosetta gami cells and grown in Terrific broth (12 g tryptone, 24 g yeast extract, 4 mL glycerol, 2.31 g monobasic potassium phosphate and 12.54 g dibasic potassium phoasphate) to mid-log phase. 1 mM IPTG was then added and the culture was grown overnight at 16°C. Cells were pelleted, resuspended in lysis buffer (50 mM Na2HPO4, 300 mM NaCl, and 10 mM imidazole) and lysed by sonication. Cell debris was pelleted by centrifugation at 31,000 × g for 30 min and Ni-nitrolotriacetic acid resin (Novagen) was added to the clarified supernatant. The bead-lysate mixture was incubated at 4°C overnight. Beads were sedimented, the supernatant was removed, and the beads were washed 3 times with wash buffer (50 mM Na2HPO4, 300 mM NaCl, and 30 mM imidazole). Beads were resuspended in wash buffer, applied to a 1-cm separation column (Bio-Rad), and His-SUMO-YmfI was eluted with wash buffer containing 250 mM imidazole. To cleave the His-SUMO tag from the purified YmfI, ubiquitin ligase/protease was added and the reaction was incubated at 4°C overnight. To remove the free His-SUMO and any remaining His-SUMO-YmfI from the cleavage reaction, Ni-nitrolotriacetic acid resin (Novagen) was added and incubated at 4°C for 1 hr. Beads were pelleted by centrifugation and the supernatant, containing untagged YmfI, was dialyzed into PBS pH 7.4 plus 50% glycerol and stored at 4°C. EF-PymfI-FLAG was purified from DK3828 as described previously (RajKovic, 2016).
10 μL reactions containing 5 μg EF-PymfI-FLAG was incubated in 100 mM sodium phosphate buffer at 37°C for 30 min. 4 μg YmfI with or without 150 μM NADPH was added to the reaction, where appropriate. Reactions were stopped by the addition of 50% glycerol, resolved by native gel electrophoresis, electroblotted, and probed with anti-EF-P polyclonal antisera.
Supplementary Material
SUMMARY.
Elongation factor P (EF-P) is found in all living things and helps ribosomes translate proline rich sequences. Moreover, EF-P is modified with different chemical groups depending on the organism and modification is thought to be essential for activity. Here we find the first protein, YmfI, involved in modification of B. subtilis EF-P with 5-aminopentanol and show that, in the absence of YmfI, an inhibitory intermediate accumulates. Further, we show that EF-P biological activity can be restored to cells lacking YmfI by by specific mutations that reduce or abolish modification, suggesting that the presence of the modification may be regulatory. Phylogenetic analysis suggests that EF-P may be similarly modified in Listeria and Staphylococcus.
Acknowledgments
This work was supported by National Science Foundation Graduate Research Fellowship Grant 1342962 to KRH, National Institutes of Health Training Grant T32 GM086252 and an OSU Center for RNA Biology Fellowship to AW, National Institutes of Health Grant GM065183 to MI, and National Institutes of Health Grant GM093030 to DBK. We thank Masaya Fujita and Winston Winkler for strains and reagents. The Fred Hutchinson Cancer Research Center Proteomics Facility is funded by Cancer Center Support Grant P30 CA015704 from the National Institutes of Health.
References
- Balibar CJ, Iwanowicz D, Dean CR. Elongation Factor P is dispensable in Escherichia coli and Pseudomonas aeruginosa. Curr Microbiol. 2013;67:293–299. doi: 10.1007/s00284-013-0363-0. [DOI] [PubMed] [Google Scholar]
- Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. RodZ (YfgA) is required for proper assembly of MreB actin cytoskeleton and cell shape in E. coli. EMBO J. 2009;28:581–590. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaha G, Stanley RE, Steitz TA. Formation of the first peptide bond: the structure of EF-P bound to the 70S ribosome. Science. 2009;353:966–969. doi: 10.1126/science.1175800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: acrchitecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper HL, Park MH, Folk JE, Safer B, Braverman R. Identification of the hypusine-containing protein Hy+ as translation initiation factor eIF-4D. Proc Natl Acad Sci. 1983;80:1854–1857. doi: 10.1073/pnas.80.7.1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, Doerks T, von Mering C, Creevey CJ, Snel B, Bork P. Toward automatic reconstruction of a highly resolved tree of life. Science. 2006;311:1283–1288. doi: 10.1126/science.1123061. [DOI] [PubMed] [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. EF-P is essential for rapid synthesis of proteins containing conservative proline residues. Science. 2013;339:85–88. doi: 10.1126/science.1229017. [DOI] [PubMed] [Google Scholar]
- Doerfel LK, Wohlgemuth I, Kubyshkin V, Starosta AL, Wilson DN, Budisa N, Rodnina MV. Entropic contribution of Elongation Factor P to proline positioning at the catalytic center of the ribosome. J Am Chem Soc. 2015;137:12997–13006. doi: 10.1021/jacs.5b07427. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14(9):755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- Finn RD, Coggil P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2015;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganoza MC, Kiel MC, Aoki H. Evolutionary conservation of reactions in translation. Microbiol Mol Biol Rev. 2002;66(3):460–485. doi: 10.1128/MMBR.66.3.460-485.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchinson CA, III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods. 2009;6(5):343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- Guérout-Fleury AM, Shazand K, Frandsen N, Stragier P. Antibiotic-resistance cassettes for Bacillus subtilis. Gene. 1995;167:335–336. doi: 10.1016/0378-1119(95)00652-4. [DOI] [PubMed] [Google Scholar]
- Guérout-Fleury AM, Frandsen N, Stragier P. Plasmids for ectopic integration in Bacillus subtilis. Gene. 1996;180:57–61. doi: 10.1016/s0378-1119(96)00404-0. [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, Buskirk AR, Dever TE. eIF5A promotes translation of polyproline motifs. Molec Cell. 2013 doi: 10.1016/j.molcel.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawa-Suetsugu K, Sekine S, Sakai H, Hori-Takemoto C, Tereda T, Unzai S, et al. Crystal structure of elongation factor P from Thermus thermophiles HB8. Proc Nat Acad Sci. 2004;101(26):9595–9600. doi: 10.1073/pnas.0308667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaoka T, Ochi K. Glucose uptake pathway-specific regulation of synthesis of neotrehalosadiamine, a novel autoinducer produced in Bacillus subtilis. J Bacteriol. 2007;189(1):65–75. doi: 10.1128/JB.01478-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz A, Solden L, Zou SB, Navarre WW, Ibba M. Molecular evolution of protein-RNA mimicry as a mechanism for translation control. Nuc Acid Res. 2013;42:3261–3271. doi: 10.1093/nar/gkt1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns DB, Chu F, Rudner R, Losick R. Genes governing swarming in Bacillus subtilis and evidence for a phase variation mechanism controlling surface motility. Mol Microbiol. 2004;52:357–369. doi: 10.1111/j.1365-2958.2004.03996.x. [DOI] [PubMed] [Google Scholar]
- Lassak J, Keilhauer EC, Fürst M, Wuichet K, Gödeke J, Starosta AL, et al. Arginine-rhamnosylation as new strategy to activate translation elongation factor P. Nat Chem Biol. 2015;11:266–270. doi: 10.1038/nchembio.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassak J, Wilson DN, Jung K. Stall no more at polyproline stretches with the translation elongation factors EF-P and IF-5A. Mol Microbiol. 2016;99(2):219–235. doi: 10.1111/mmi.13233. [DOI] [PubMed] [Google Scholar]
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–W245. doi: 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Rocha AL, Woriedh M, Chemnitz J, Willingmann P, Kröger C, Hadeler B, et al. Posttranslational hypusination of the eukaryotic translation initiation factor-5A regulates Fusarium graminearum virulence. Sci Rep. 2016;6:24698. doi: 10.1038/srep24698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Zou BS, Roy H, Xie JL, Savchenko A, Singer A, et al. PoxA, YjeK, and Elongation Factor P coordinately modulate virulence and drug resistance in Salmonella enterica. Mol Cell. 2010;39:209–221. doi: 10.1016/j.molcel.2010.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas P, Mäder U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, et al. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science. 2012;335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- Patel HP, Costa-Mattioli M, Schulze KL, Bellen HG. The Drosophila deoxyhupusine hydroxylase homologue nero and its target eIF5A are required for cell growth and the regulation of autophagy. J Cell BIol. 2009;185(7):1181–1194. doi: 10.1083/jcb.200904161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrick JE, Kearns DB. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol Microbiol. 2008;70:1166–1179. doi: 10.1111/j.1365-2958.2008.06469.x. [DOI] [PubMed] [Google Scholar]
- Price AC, Zhang YM, Rock CO, White SW. Structure of β-Ketoacyl-[acyl carrier protein] reductase from Escherichia coli: negative cooperativity and its structural bias. Biochemistry. 2001;40:12772–12781. doi: 10.1021/bi010737g. [DOI] [PubMed] [Google Scholar]
- Rajkovic A, Erickson S, Witzky A, Branson OE, Seo J, Gafken PR, et al. Cyclic rhamnosylated Elongation factor P extablishes antibiotic resistance in Pseudomonas aeruginosa. MBio. 2015;6(3):e00823–15. doi: 10.1128/mBio.00823-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkovic A, Hummels KR, Witzky A, Erickson S, Gafken PR, Whitelegge JP, et al. Translation Control of Swarming Proficiency in Bacillus subtilis by 5-Amino-pentanolylated Elongation Factor P. J Biol Chem. 2016;291(21):10976–10985. doi: 10.1074/jbc.M115.712091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkovic A, Ibba M. Elongation Factor P and the Control of Translation Elongation. Annual Review of Microbiology. 2017 doi: 10.1146/annurev-micro-090816-093629. in press. [DOI] [PubMed] [Google Scholar]
- Sasaki K, Abid MR, Miyazaki M. Deoxyhypusine synthase gene is essential for cell viability in the yeast Saccharomyces cerevisiae. FEBS Lett. 1996;384:151–154. doi: 10.1016/0014-5793(96)00310-9. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Becker T, Heuer A, Braunger K, Shanmuganathan V, Pech M, et al. Structure of the hypusinylated eukaryotic translation factor eIF-5A bound to the ribosome. Nucleic Acids Res. 2016;44(4):1944–1951. doi: 10.1093/nar/gkv1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnier J, Schwelberger HG, Smit-McBride Z, Kang HA, Hershey JWB. Translation initiation factor 5A and its hypusine modification are essential for cell viability in the yeast Saccharomyces cerevisiae. Mol Cell Biol. 1991;11(6):3105–3144. doi: 10.1128/mcb.11.6.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K. Translation elongation factor EF-P alleviates ribosome stalling at polyproline stretches. Science. 2013;339:82–85. doi: 10.1126/science.1228985. [DOI] [PubMed] [Google Scholar]
- Wach A. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast. 1996;12:259–265. doi: 10.1002/(SICI)1097-0061(19960315)12:3%3C259::AID-YEA901%3E3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Sumida T, Ishii R, Takemoto C, Yokoyama S. A paralog of lysyl-tRNA synthetase aminoacylates a conserved lysine residue in translation elongation factor P. Nat Struct Mol Bio. 2010;17(9):1136–1143. doi: 10.1038/nsmb.1889. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T, Takahashi H, Suzuki T, Masuda A, Dohmae N, Yokoyama S. Neisseria meningitidis translation elongation factor P and its active-site arginine residue are essential for cell viability. PLoS One. 2016;11(2):e0147907. doi: 10.1371/journal.pone.0147907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasbin RE, Young FE. Transduction in Bacillus subtilis by bacteriophage SPP1. J Virol. 1974;14:1343–1348. doi: 10.1128/jvi.14.6.1343-1348.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi EC, Lee H, Aebersold R, Goodlett DR. A microcapillary trap cartridge-microcapillary high-performance liquid chromatography electrospray ionization emitter device capable of peptide tandem mass spectrometry at the attomole level on an ion trap mass spectrometer with automated routine operation. Rapid Commun Mass Spectrom. 2003;17:2093–8. doi: 10.1002/rcm.1150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.