

Abstract
Thousands of candidate human-specific regulatory sequences (HSRS) have been identified, supporting the hypothesis that unique to human phenotypes result from human-specific alterations of genomic regulatory networks. Collectively, a compendium of multiple diverse families of HSRS that are functionally and structurally divergent from Great Apes could be defined as the backbone of human-specific genomic regulatory networks. Here, the conservation patterns analysis of 18,364 candidate HSRS was carried out requiring that 100% of bases must remap during the alignments of human, chimpanzee, and bonobo sequences. A total of 5,535 candidate HSRS were identified that are: (i) highly conserved in Great Apes; (ii) evolved by the exaptation of highly conserved ancestral DNA; (iii) defined by either the acceleration of mutation rates on the human lineage or the functional divergence from non-human primates. The exaptation of highly conserved ancestral DNA pathway seems mechanistically distinct from the evolution of regulatory DNA segments driven by the species-specific expansion of transposable elements. Genome-wide proximity placement analysis of HSRS revealed that a small fraction of topologically associating domains (TADs) contain more than half of HSRS from four distinct families. TADs that are enriched for HSRS and termed rapidly evolving in humans TADs (revTADs) comprise 0.8–10.3% of 3,127 TADs in the hESC genome. RevTADs manifest distinct correlation patterns between placements of human accelerated regions, human-specific transcription factor-binding sites, and recombination rates. There is a significant enrichment within revTAD boundaries of hESC-enhancers, primate-specific CTCF-binding sites, human-specific RNAPII-binding sites, hCONDELs, and H3K4me3 peaks with human-specific enrichment at TSS in prefrontal cortex neurons (P < 0.0001 in all instances). Present analysis supports the idea that phenotypic divergence of Homo sapiens is driven by the evolution of human-specific genomic regulatory networks via at least two mechanistically distinct pathways of creation of divergent sequences of regulatory DNA: (i) recombination-associated exaptation of the highly conserved ancestral regulatory DNA segments; (ii) human-specific insertions of transposable elements.
Keywords: human-specific regulatory sequences, DNase I hypersensitive sites, human accelerated regions, human-specific transcription factor binding sites, exaptation of ancestral regulatory DNA
Introduction
Extensive search for human-specific genomic regulatory sequences (HSRS) revealed thousands candidate HSRS, a vast majority of which is residing within non-protein coding genomic regions (McLean et al. 2011; Konopka et al. 2012; Shulha et al. 2012; Capra et al. 2013; Marnetto et al. 2014; Glinsky 2015). Candidate HSRS comprise multiple distinct families of genomic regulatory elements, which were defined using a multitude of structural features, different statistical algorithms, as well as a broad spectrum of experimental, analytical, computational, and bioinformatics strategies. The current catalogue of candidate HSRS includes conserved in humans novel regulatory DNA sequences designated human accelerated regions, HARs (Capra et al. 2013); fixed human-specific regulatory regions, FHSRR (Marnetto et al. 2014); human-specific transcription factor-binding sites, HSTFBS (Glinsky 2015), regions of human-specific loss of conserved regulatory DNA termed hCONDEL (McLean et al. 2011); human-specific epigenetic regulatory marks consisting of H3K4me3 histone methylation signatures at transcription start sites in prefrontal neurons (Shulha et al. 2012); and human-specific transcriptional genetic networks in the frontal lobe (Konopka et al. 2012). Most recently, Gittelman et al. (2015) reported identification of 524 DNase I hypersensitive sites (DHSs) that are conserved in non-human primates but accelerated in the human lineage (haDHS) and may have contributed to human-specific phenotypes. They estimated that 70% of substitutions in haDHSs are attributable to positive selection consistent with the hypothesis that these DNA segments have been subjects to human-specific adaptive evolution resulting in creation of human-specific regulatory sequences. Finally, Prescott et al. (2015) identified thousands of enhancers associated with divergent cis-regulatory evolution of the human’s and chimpanzee’s neural crest underlying development of unique to human craniofacial features.
Definition of HARs, which is one of the most actively investigated HSRS families, is based on calculations as a baseline the evolutionary expected rate of base substitutions derived from the experimentally determined level of conservation between multiple species at the given locus. The statistical significance of differences between the observed substitution rates on a lineage of interest in relation to the evolutionary expected baseline rate of substitutions can be estimated. This method is considered particularly effective for identifying highly conserved sequences within non-coding genomic regions that have experienced a marked increase of substitution rates on a particular lineage. It has been successfully applied to humans (Pollard et al. 2006; Prabhakar et al. 2006; Bird et al. 2007), where the rapidly-evolving sequences that are highly conserved across mammals and have acquired many sequence changes in humans since divergence from chimpanzees were designated as human accelerated regions (HARs). Experimental analyses of HARs bioactivity revealed that some HARs function as non-coding RNA genes expressed during the neocortex development (Pollard et al. 2006) and human-specific developmental enhancers (Prabhakar et al. 2008). Consistent with the hypothesis that HARs function in human cells as regulatory sequences, most recent computational analyses and transgenic mouse experiments demonstrated that many HARs represent developmental enhancers (Capra et al. 2013).
In contrast to the cross-species quantitative analyses of the DNA sequence conservation and divergence, an alternative approach to discovery of candidate HSRS is based on identification of regulatory DNA segments that are functionally divergent in humans compared with our closest evolutionary relatives, chimpanzee and bonobo (Shulha et al. 2012; Prescott et al. 2015). The systematic analysis of the sequence conservation patterns of these families of candidate HSRS, which were defined based on the functional divergence from the NHP, has not been performed.
Here, the sequence conservation patterns’ analyses of 18,364 candidate HSRS was carried out using the most recent releases of reference genomes’ databases of humans and non-human primates and requiring that 100% of bases must remap during the alignments of sequences of human, chimpanzee, and bonobo genomes. This analysis identifies 5,535 regulatory DNA segments that are: (i) predominantly located within the non-coding genomic regions; (ii) highly conserved in humans and other Great Apes; (iii) do not intersect transposable elements (TE)-derived sequences; (iv) appear to acquire human-specific regulatory traits by exaptation of ancestral DNA. In contrast to the exaptation pathway of the human regulatory DNA divergence, majority of candidate HSTFBS intersect TE-derived sequences and appear seeded by TE-associated pathway of the human regulatory DNA evolution. The results of the present analyses suggest that evolution of human-specific genomic regulatory networks is driven by at least two mechanistically distinct pathways of creation of divergent regulatory DNA segments associated with either high recombination rates or species-specific expansion of TEs.
Results and Discussion
Effects of the Human Reference Database Refinements on the Validity of Molecular Definitions of 18,364 Candidate Human-Specific Regulatory Sequences
The sequence quality of reference genome databases is essential for the accurate definition of regulatory DNA segments as candidate HSRS. It was unclear how continuing database improvements would affect the validity of the HSRS’ definition. To address this problem, the most recent hg38 release of the human genome reference database (HGRD), which replaces the hg19 release as default human assembly (http://genome.ucsc.edu/cgi-bin/hgGateway, last accessed August 6, 2016), was utilized. Present analyses revealed variable effects of the human genome reference database (HGRD) refinement’s on the validity of molecular definitions of distinct families of candidate HSRS (supplementary tables S1–S10, Supplementary Material online). The large HGRD refinements’ effect was observed on the molecular definition of 583 hCONDELs (McLean et al. 2011), indicating that only 42% of the hCONDELs’ sequences, which were originally defined using the hg18 release of the HGRD, could be mapped to the most recent hg38 release of the HGRD (supplementary table S10, Supplementary Material online). A moderate HGRD refinements’ effect was observed on the molecular definition of human-specific epigenetic regulatory sequences consisting of H3K4me3 histone methylation signatures at transcription start sites (TSS) in prefrontal neurons (Shulha et al. 2012), indicating that 16 (3.9%) of 410 H3K4me3 marks defined as candidate HSRS failed to convert to the hg38 release of the HGRD at MinMatch threshold of 1.00 (supplemental table S8, Supplementary Material online). However, in most instances, the required adjustments were limited to a few sequences, thus validating the overall high sequence quality of candidate HSRS.
Sequence Conservation Analysis of Human Accelerated DNase I Hypersensitive Sites
The identified haDHSs represent relatively short DNA segments of the median size 290 bp (range from 150 to 1010 bp; average size of 323 bp), which are predominantly located within intronic and intergenic sequences (Gittelman et al. 2015). To test whether reported 524 haDHSs represent human-specific DNA sequences, the conservation analysis was carried-out using the LiftOver algorithm and Multiz Alignments of 20 mammals (17 primates) of the UCSC Genome Browser on Human Dec. 2013 (GRCh38/hg38) Assembly (http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg38&position=chr1%3A90820922-90821071&hgsid=441235989_eelAivpkubSY2AxzLhSXKL5ut7TN, last accessed August 6, 2016).
The most recent releases of the corresponding reference genome databases were utilized to ensure the use of the most precise, accurate, and reproducible genomic DNA sequences available to date. The results of these analyses are reported in the supplementary table S1, Supplementary Material online. Several thresholds of the LiftOver algorithm MinMatch function (minimum ratio of bases that must remap) were utilized to assess the sequences conservation and identify candidate human-specific regulatory sequences as previously described (Glinsky 2015). In this analysis, the candidate human-specific regulatory sequences were defined based on conversion failures to both Chimpanzee’s and Bonobo’s genomes and supported by direct visual evidence of human-specific sequence alignment differences of the Multiz Alignments of 20 mammals (17 primates). It appears that only small fractions (0.2–13.9%) of reported 524 haDHSs can be defined as candidate human-specific regulatory sequences applying these criteria at different sequence conservation thresholds (supplementary table S1, Supplementary Material online). Based on this analysis, the vast majority (86.1–99.8%) of 524 haDHSs could be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates.
Interestingly, the Multiz Alignments of 20 mammals (17 primates) revealed that 71% of candidate human-specific haDHSs defined at 0.99 MinMatch threshold (supplementary table S1, Supplementary Material online) contain small human-specific inserts of 2–15 bp, suggesting a common mutation mechanism (supplementary data set S1, Supplementary Material online). A majority (78%) of candidate human-specific haDHSs is located within the intronic (47.9%) and intergenic (30.1%) sequences (supplementary data set S2, Supplementary Material online). However, 15 of 73 (20.5%) candidate human-specific haDHSs sequences appear to intersect exons, 11 of which include intron/exon junctions (supplementary data sets S1 and Supplementary Data, Supplementary Material online). Intriguingly, this analysis identified the 18 bp. human-specific deletion within the exon 9 of the PAX8 gene, which appears to affect the structure of the PAX8-AS1 RNA as well (supplementary data set S1, Supplementary Material online).
Therefore, these analyses demonstrate that there is no detectable reference genome database refinements’ effect on the accuracy of molecular definition of haDHSs and the majority of haDHSs’ sequences are conserved in humans and non-human primates.
Sequence Conservation Analysis of Human Accelerated Regions
Strikingly similar results were observed when the sequence conservation analysis of 2,745 HARs was performed (supplementary table S2, Supplementary Material online). It appears that only small fractions (1.2–9.3%) of reported HARs can be defined as candidate human-specific regulatory sequences using different sequence conservation thresholds (supplementary table S2, Supplementary Material online). Based on this analysis, the vast majority (90.7–98.8%) of 2,745 HARs could be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates (supplementary table S2, Supplementary Material online). This conclusion remains valid when the most stringent definition of the sequence conservation threshold was used by setting the minimum sequence alignments’ match requirement (MinMatch threshold) as the 100% of bases that must remap (supplementary table S2, Supplementary Material online). Based on this analysis, it appears that there is a minor reference genome database refinements’ effect on the accuracy of molecular definition of HARs and the majority of HARs’ sequences are conserved in humans and non-human primates.
Sequence Conservation Analysis of Other Classes of Candidate HSRS
In contrast to haDHS and HARs, several other classes of candidate HSRS were defined based on the failure of alignments of human regulatory DNA segments to the reference genome databases of other species (Marnetto et al. 2014; Glinsky 2015). It appears that a majority (82.1–88.4%) of reported DNase I hypersensitive sites-derived fixed human specific regulatory regions (DHS_FHSRR) can be defined as candidate human-specific regulatory sequences using different sequence conservation thresholds (supplementary table S3, Supplementary Material online). Based on this analysis, the relatively minor fraction (11.6–17.9%) of 2,118 DHS_FHSRR may be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates (supplementary table S3, Supplementary Material online).
Similarly, a majority (79.0–86.5%) of reported HSTFBS can be defined as candidate human-specific regulatory sequences using different sequence conservation thresholds and the relatively minor fraction (13.5–21.0%) of 3,803 HSTFBS may be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates (supplementary table S4, Supplementary Material online). Strikingly similar results were documented during the analyses of other families of HSRS. A majority (70.2–79.7%) of reported hESC_FHSRR can be defined as candidate human-specific regulatory sequences using different sequence conservation thresholds and the relatively small fraction (20.3–29.8%) of 1,932 hESC_FHSRR could be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates (supplementary table S5, Supplementary Material online). A majority (84.3–89.7%) of reported other FHSRR can be defined as candidate human-specific regulatory sequences using different sequence conservation thresholds and the relatively minor fraction (10.3–15.7%) of 4,249 other_FHSRR could be classified as the candidate regulatory sequences that appear conserved in humans and non-human primates (supplementary table S6, Supplementary Material online). Based on this analysis, the conclusion has been made that there is a minor reference genome database refinements’ effect on the accuracy of molecular definition of HSTFBS and FHSRR families of candidate HSRS. The majority of HSTFBS and FHSRR sequences failed to align to both Chimpanzee and Bonobo genomes, thus meeting the criteria for definition as candidate HSRS.
Identification of Highly Conserved in Nonhuman Primates Regulatory DNA Sequences among Candidate HSRS Based on Direct and Reciprocal Alignments
To identify regulatory DNA segments that are highly conserved in non-human primates, the most stringent definition of the sequence conservation threshold was used by setting the minimum sequence alignments’ match requirement as the 100% of bases that must remap, which would require that 100% of bases must remap during the alignments. It has been noted that a direct lift over at MinMatch 1.00 from human’s genome to genomes of non-human primates may identify the aligned sequences with clearly visible base differences detectable during the visual inspections of aligned sequences, which was most often due to the losses of the ancestral DNA. To address this limitation, in the subsequent analysis a given regulatory DNA segment was defined as highly conserved only when both direct and reciprocal conversions between humans’ and non-human primates’ genomes were observed using the MinMatch threshold of 1.00, thus requiring that 100% of bases must remap during the direct and reciprocal alignments. This approach removed sequences with the ancestral DNA losses during the reciprocal alignments of the corresponding genomes of non-human primates to the human reference genome. Nevertheless, the majority of both haDHSs (404 of 524; 77.1%) and HARs (2,262 of 2,739; 82.6%) were defined as the highly conserved in humans and non-human primates regulatory sequences (table 1). In contrast, only relatively small fractions of other classes of candidate HSRS were identified as highly conserved in non-human primates regulatory sequences, scoring at 7.3% for HSTFBS; 8.3% for other_FHSRR; 9.4% for DHS_FHSRR; and 15.9% for hESC_FHSRR (table 1). Follow-up visual inspections of these highly conserved in non-human primates’ genomes candidate regulatory sequences and nucleotide BLAST analyses of selected sequences revealed examples of the overall similar sequence gap structures among the Great Apes after the divergence from the Rhesus Macaque, however, some Great Apes display the unique structure of the sequence gaps for individual species.
Table 1.
Distribution of Highly Conserved in Non-Human Primates Regulatory Sequences among 15,371 Candidate Human-Specific Regulatory Sequence
| HSRS/Genomes | haDHS | HARs | HSTFBS | DHS_FHSRR | hESC_FHSRR | Other_FHSRR |
|---|---|---|---|---|---|---|
| Human genome (hg19) | 524 | 2,745 | 3,803 | 2,118 | 1,932 | 4,249 |
| Human genome (hg38) | 524 | 2,739 | 3,714 | 2,114 | 1,928 | 4,235 |
| Mouse genome conversion (mm10) | 66 | 1,004 | 12 | 4 | 0 | 0 |
| Reciprocal conversion to human genome | 23 | 560 | 1 | 2 | 0 | 0 |
| Percent conserved in rodents' genome | 4.4 | 20.4 | 0.0 | 0.1 | 0.0 | 0.0 |
| Chimpanzee genome conversion | 439 | 2,404 | 56 | 5 | 0 | 13 |
| Reciprocal conversion to human genome | 390 | 2,146 | 40 | 0 | 0 | 1 |
| Percent conserved in Chimpanzee | 74.4 | 78.3 | 1.1 | 0 | 0 | 0 |
| Bonobo genome conversion | 425 | 2,341 | 495 | 242 | 396 | 438 |
| Reciprocal conversion to human genome | 383 | 2,123 | 262 | 199 | 306 | 350 |
| Percent conserved in Bonobo | 73.1 | 77.5 | 7.1 | 9.4 | 15.9 | 8.3 |
| Conserved in non-human primates** | 404 | 2,262 | 271 | 199 | 306 | 351 |
| Percent conserved in non-human primates | 77.1 | 82.6 | 7.3 | 9.4 | 15.9 | 8.3 |
| Bonobo and Chimp conserved | 370 | 2,004 | 31 | 0 | 0 | 0 |
| Chimp only conserved | 21 | 141 | 9 | 0 | 0 | 1 |
| Bonobo only conserved | 13 | 117 | 231 | 199 | 306 | 350 |
NOTE.—LiftOver algorithm MinMatch Minimum ratio of bases that must remap) threshold was 1.00. HSRS, human-specific regulatory sequences; HSTFBS, human-specific transcription factor-binding sites; haDHS, human accelerated DNase I hypersensitive sites; HARs, human accelerated regions; DHS, DNase I hypersensitive sites; FHSRR, fixed human-specific regulatory regions.
Chimpanzee genome PanTro4 conversion.
Conserved in non-human primates sequences were defined based on both direct and reciprocal conversions to either one or both Chimpanzee and Bonobo genomes at MinMatch threshold of 1.00.
Significantly, during the BLAST analyses of these DNA segments the consistently high levels of the sequence identities among different species of primates were observed, ranging from 91% to 100% (supplementary data sets S3 and S4, Supplementary Material online). Taken into consideration that a majority of haDHS and HARs are located within intronic and intergenic regions, it seems reasonable to conclude that these sequences manifest a high level of sequence conservation in non-human primates.
Notably, despite the setting of the MinMatch lift over threshold at 1.00 (thus, requiring that 100% of bases must remap during the alignments of the corresponding sequence), the follow-up BLAST analyses of selected sequences revealed that humans and Great Apes manifest clearly discernable species-specific patterns of single-nucleotide substitutions (supplementary data sets S3 and S4, Supplementary Material online). Specifically, this pattern was noted during the BLAST analyses of human, Chimpanzee, and Bonobo sequences. It is possible that these species-specific single-nucleotide substitutions may be of functional significance. Lastly, it has been confirmed during the present analysis that haDHS sequences display rates of mutations accelerated by 1.7- to 8.0-fold in humans compared with Bonobo and Chimpanzee genomes (supplementary data set S5, Supplementary Material online). Calculations of the increased mutation rates within human’s and primate’s lineages were made based on direct measurements of the sequence identities after the split with the Gorilla gorilla ∼17 million years ago (supplementary data set S5, Supplementary Material online). Interestingly, a sub-set of haDHS appears to remain 100% identical in both Bonobo’s and Chimpanzee’s genomes during ∼25 to 30 million years of evolution after the split with the Rhesus Macaque and undergoes single-nucleotide substitutions in the human lineage after the split with the Chimpanzee ∼13 million years ago. Examples of these haDHS sequences are shown in the supplementary data sets S4 and S5, Supplementary Material online.
Sequence Conservation Patterns’ Analyses of Candidate HSRS Defined by the Functional Divergence in Humans Compared with Chimpanzees
It was of interest to analyze the sequence conservation patterns among the candidate HSRS, which were defined based on identification of regulatory DNA segments that are functionally divergent in humans compared with our closest evolutionary relatives, chimpanzee and bonobo (Shulha et al. 2012; Prescott et al. 2015). The results of these analyses recapitulate two major patterns of sequence conservations observed for other families of candidate HSRS (supplementary tables S7–S10, Supplementary Material online). The sequence conservation patterns of both human-biased and chimp-biased CNCCs’ enhancers resemble the sequence conservation profiles of haDHSs and HARs with the majority of regulatory DNA segments (80.7 and 82.2% for human-biased and chimp-biased CNCCs enhancers, respectively) being defined as highly conserved in human, Bonobo, and Chimpanzee genomes (compare data in table 1; supplementary tables S1 and S2, Supplementary Material online; and table 2; supplementary tables S7 and S8, Supplementary Material online). In contrast, human-specific regulatory sequences consisting of H3K4me3 histone methylation signatures at transcription start sites in prefrontal neurons manifest sequence conservation patterns similar to the sequence conservation profiles of the FHSRR and HSTFBS with only the minor fraction of regulatory DNA sequences (12.7%) being identified as highly conserved in human, Bonobo, and Chimpanzee genomes (compare data in the table 2; supplementary tables S3–S6, Supplementary Material online; and table 2; supplementary table S9, Supplementary Material online).
Table 2.
Distribution of Highly Conserved in Non-Human Primates Regulatory Sequences among Candidate Human-Specific Regulatory Sequence Defined by the Functional Divergence from Chimpanzee or Deletions of Ancestral DNA in the Human Genome
| HSRS/Genomes | Human-Biased CNCC’s Enhancers | Chimp-Biased CNCC’s Enhancers | hCONDELs | H3K4me3 Signatures in Human Prefrontal Neurons | All HSRS |
|---|---|---|---|---|---|
| Human genome (hg19) | 1,000 | 1,000 | 583 | 410 | 18,364 |
| Human genome (hg38) | 996 | 998 | 245 | 394 | 17,887 |
| Mouse genome conversion (mm10) | 21 | 30 | 22 | 0 | 1,159 |
| Reciprocal conversion to human genome | 4 | 7 | 18 | 0 | 615 |
| Percent conserved in rodents' genome | 0.4 | 0.7 | 7.3 | 0 | 3.4 |
| Chimpanzee genome conversion | 871 | 884 | 17 | 86 | 4,775 |
| Reciprocal conversion to human genome | 765 | 785 | 12 | 36 | 4,175 |
| Percent conserved in Chimpanzee | 76.8 | 78.7 | 4.9 | 9.1 | 23.3 |
| Bonobo genome conversion | 844 | 847 | 71 | 74 | 6,173 |
| Reciprocal conversion to human genome | 754 | 760 | 63 | 36 | 5,236 |
| Percent conserved in Bonobo | 75.7 | 76.2 | 25.7 | 9.1 | 29.3 |
| Conserved in non-human primates** | 804 | 820 | 68 | 50 | 5,535 |
| Percent conserved in non-human primates | 80.7 | 82.2 | 27.8 | 12.7 | 30.9 |
| Bonobo and Chimp conserved | 715 | 725 | 7 | 22 | 3,874 |
| Chimp only conserved | 50 | 60 | 5 | 14 | 301 |
| Bonobo only conserved | 39 | 35 | 56 | 14 | 1,360 |
NOTE.—LiftOver algorithm MinMatch Minimum ratio of bases that must remap) threshold was 1.00. HSRS, human-specific regulatory sequences; hCONDELs, human-specific deletions of regulatory DNA; CNCCs, cranial neural crest cells; All HSRS column shows the sum of records for each categories from the corresponding entries in tables 1 and 2.
*Chimpanzee genome PanTro4 conversion.
**Conserved in non-human primates sequences were defined based on both direct and reciprocal conversions to either one of both Chimpanzee and Bonobo genomes at MinMatch threshold of 1.00.
In total, 5,535 candidate HSRS, which were defined by either the acceleration of mutation rates on the human lineage or the functional divergence from chimpanzee, appear highly conserved in humans and NHP. Nonetheless, these sequences manifest clearly discernable species-specific patterns of single-nucleotide substitutions in humans, chimpanzee, and bonobo genomes suggesting that they evolved by the exaptation of ancestral regulatory DNA.
Identification of Topologically-Associating Domains Rapidly-Evolving in the hESC Genome
Two important experimentally testable predictions can be derived from the proposed model of evolution of HSRS (fig. 1):
Fig. 1.—
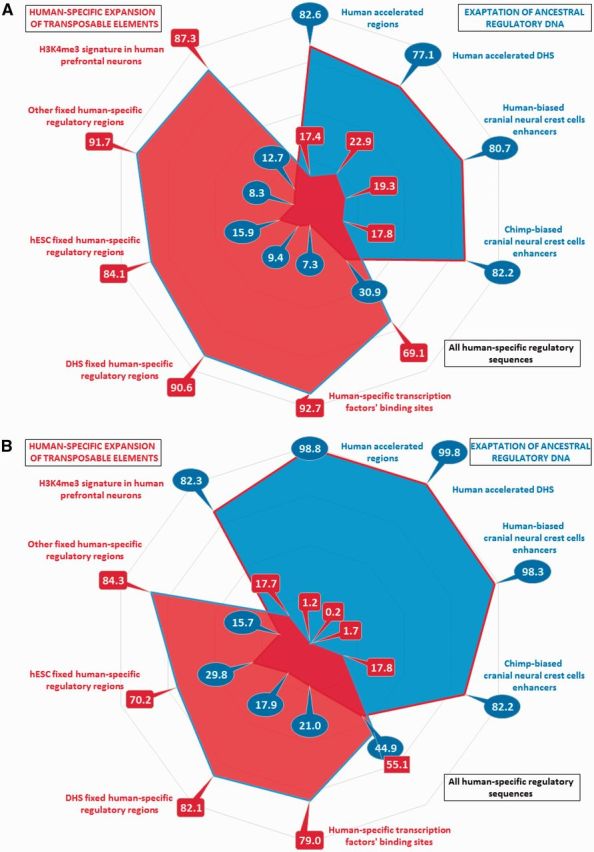
Two distinct pathways of human regulatory DNA divergence during evolution of human-specific genomic regulatory networks. (A) Sequence conservation analyses of 18,364 candidate human-specific regulatory sequences (HSRS) revealed two distinct patterns of regulatory DNA alignments to genomes of non-human primates (NHP): (i) an alignment pattern with a significant majority (from 77.1% to 82.6%) of candidate HSRS being highly conserved in genomes of Bonobo and Chimpanzee (blue colored features in the figure); (ii) an alignment pattern with only a minority (from 7.3% to 15.9%) of candidate HSRS being highly conserved in genomes of Bonobo and Chimpanzee (red colored features in the figure). It is proposed that these two distinct sequence conservation patterns reflect two mechanistically distinct pathways of human regulatory DNA divergence during evolution (see text for details). For each family of HSRS the percentage of highly conserved in NHP (blue) and human-specific (red) regulatory DNA segments are shown. The results in the (A) represent the graphical summary of the primary data reported in the tables 1 and 2 based on definition of the sequence conservation threshold of 1.00 during both direct and reciprocal conversions thus requiring that 100% bases must remap during the alignments. The results in the (B) illustrate the sequence conservation analyses based on definition of the sequence conservation threshold of 0.95 during direct conversion from human to NHP genomes without reciprocal conversion corrections.
1. Genomic locations of HSRS must reflect the apparently non-random patterns of HSRS placement and/or retention in the human genome;
2. Different HSRS families that are created via two mechanistically distinct pathways of divergent regulatory DNA evolution should manifest distinct location patterns within chromosomal domains.
To test these predictions, genome-wide proximity placement analyses were carried out integrating data on DNA sequences of individual regulatory elements comprising four distinct families of candidate HSRS within the context of the principal regulatory components of the interphase chromosome domain structures defined by recent studies of interphase chromatin interactions and chromosome folding patterns in human and mouse cells. Pioneering work on the interphase chromosome structures revealed specific and highly reproducible folding patterns of the chromosome fibers into spatially segregated domain-like segments (Dixon et al. 2012; Gorkin et al. 2014). In the mammalian nucleus, beads on a string linear strands of interphase chromatin fibers are folded into continuous megabase-sized topologically associating domains (TADs) that are readily detectable by the high-throughput analysis of interactions of chemically cross-linked chromatin (Dixon et al. 2012; Hou et al. 2012; Nora et al. 2012; Sexton et al. 2012). It has been hypothesized that TADs represent spatially segregated neighborhoods of high local frequency of intrachromosomal contacts reflecting individual physical interactions between long-range enhancers and promoters of target genes in live cells (Dixon et al. 2012; Gorkin et al. 2014). Definition of TADs implies that neighboring TADs are separated by the sharp boundaries, across which the intrachromosomal contacts are relatively infrequent (Dixon et al. 2012; Gorkin et al. 2014), indicating that TADs constitute relatively autonomous transcription regulatory domains of mammalian interphase chromosomes.
Using genomic coordinates of 3,127 topologically-associating domains (TADs) in hESC (Dixon et al. 2012), a proximity placement analysis of 10,598 DNA sequences representing four distinct families of candidate HSRS was performed (supplementary table S11, Supplementary Material online). The primary criterion for selection of this set of regulatory DNA sequences was the fact that they were identified in human cells lines and primary human tissues whose karyotype were defined as “normal.” Based on the origin and definition of corresponding HSRS, the four HSRS families were assigned the following designations:
Human accelerated regions (HARs; Capra et al. 2013);
Human-specific transcription factor-binding sites (HSTFBS; Glinsky 2015);
hESC-derived fixed human-specific regulatory regions (hESC-FHSRR; Marnetto et al. 2014);
DNase hypersensitive sites-derived fixed human-specific regulatory regions (DHS-FHSRR; Marnetto et al. 2014).
The number of HSRS placed within a given TAD was computed for every TAD in the hESC genome and the HSRS placement enrichment was calculated as the ratio of observed values to expected values estimated from a random distribution model at the various cut-off thresholds (supplementary table S11, Supplementary Material online). Regardless of the chosen cut-off thresholds, placement of most HSRS appears markedly restricted to the small fraction of TADs in the hESC genome (supplementary table S11, Supplementary Material online). Notably, a majority of individual sequences of each HSRS family is placed within 0.8–10.3% of TADs in the human genome (supplementary table S11, Supplementary Material online). Of the 3,127 TADs in the hESC genome, 24 (0.8%); 53 (1.7%); 259 (8.3%); and 322 (10.3%) TADs are populated by 1,110 (52.4%); 1,936 (50.9%); 1,151 (59.6%); and 1,601 (58.3%) individual sequences assigned to DHS-FHSRR, HSTFBS, hESC-FHSRR, and HAR families of HSRS, respectively (supplementary table S11, Supplementary Material online). The genome-wide enrichment factors varied for different HSRS families ranging from 6- to 16-fold for HARs; 7- to 17-fold for hESC-FHSRR; 30- to 45-fold for HSTFBS; and 43- to 88-fold for DHS-FHSRR (P < 0.0001 in all instances; supplementary table S11, Supplementary Material online). Based on these observations, TADs manifesting a statistically significant accumulation of HSRS compared to the random distribution model were defined as the rapidly evolving in humans TADs (revTADs; supplementary data set S7, Supplementary Material online). In agreement with the model prediction, results of these analyses demonstrate the apparent non-random patterns of placement and/or retention of HSRS in the human genome.
Follow-Up Analyses of the Sixty revTADs Enriched for Placement of HARs and HSTFBS
Subsequent analyses were focused on the revTAD set harboring at least 10 individual regulatory DNA sequences assigned to either or both of two distinct HSRS families: 2,745 HARs and 3,803 HSTFBS. According to the model prediction (fig. 1), the emergence of these two HSRS families is most likely a result of mechanistically distinct processes, because a vast majority of HSTFBS (99%) is represented by human-specific sequences of regulatory DNA which are located within transposable elements (TE)-derived DNA segments (Glinsky 2015), whereas HARs represent evolutionary highly conserved sequences that have experienced a marked increase of base substitution rates on a human lineage (Capra et al. 2013). A threshold of ten HSRS per TAD was chosen for the revTAD selection based on a consideration that it would exceed ∼10-fold the expected placement number of individual HSRS per TAD based on a random distribution model estimates.
In the hESC genome, there are 60 TADs (1.9% of all TADs) meeting these criteria (table 3), 60% of which (36 revTADs) harbor both HARs and HSTFBS. Notably, 50 of 60 revTADs (83%) assigned to this revTAD set harbor at least one HAR. Fourteen revTADs contain within their boundaries at least ten HARs and no HSTFBS, while ten revTADs harbor at least twelve HSTFBS and no HARs. Placement of both HARs and HSTFBS is markedly enriched in this set of revTADs, significantly exceeding the expected numbers for HARs (7.4-fold; P < 0.0001) and HSTFBS (18.8-fold; P < 0.0001). Among HSTFBS, human-specific CTCF-binding sites manifest the most pronounced placement enrichment (28.4-fold; P < 0.0001).
Table 3.
Genomic Features Associated with 60 Rapidly Evolving in Humans Topologically Associating Domains
| Genomic features | Genome | revTADs | Expected | Enrichment | P-value |
|---|---|---|---|---|---|
| Human Accelerated Regions (HARs) | 2,745 | 378 | 53 | 7.4 | <0.0001 |
| Human-specific TFBS | 3,803 | 1,370 | 73 | 18.8 | <0.0001 |
| Lamina-associated domains (LADs) | 1,344 | 54 | 26 | 2.1 | 0.0019 |
| Human-specific CTCF-binding sites | 591 | 312 | 11 | 28.4 | <0.0001 |
| Human-specific NANOG-binding sites | 826 | 192 | 16 | 12 | <0.0001 |
| Human-specific RNAPII-binding sites | 290 | 181 | 6 | 30.2 | <0.0001 |
| Human-specific regulatory regions identified in H1-hESC | 1,932 | 109 | 37 | 2.9 | <0.0001 |
| Human-specific regulatory regions identified in multiple cells | 4,249 | 417 | 82 | 5.1 | <0.0001 |
| DHS-defined human-specific regulatory regions | 2,118 | 558 | 41 | 13.6 | <0.0001 |
| Human-specific conservative deletions (CONDELs) | 583 | 29 | 11 | 2.6 | <0.0001 |
| Human ESC enhancers | 6,823 | 240 | 131 | 1.8 | <0.0001 |
| Human-specific transcriptional network in the brain | 6,622 | 147 | 127 | 1.2 | 0.3856 |
| Primate-specific CTCF-binding sites | 29,081 | 1,269 | 558 | 2.3 | <0.0001 |
| H3K27ac peaks with human-specific enrichment in embryonic limb at E33 stage | 780 | 31 | 15 | 2.1 | 0.0238 |
| H3K4me3 peaks with human-specific enrichment in prefrontal cortex (PFC) neurons | 410 | 29 | 8 | 3.6 | <0.0001 |
NOTE.—hESC, human embryonic stem cells; TFBS, transcription factor-binding site; HARs, human accelerated region; LAD, lamina-associated domain; TAD, topologically-associating domain; RNAPII, RNA polymerase II; PFC, prefrontal cortex; DHS, DNase hypersensitive sites; CONDELs, conservative deletions; E33, embryonic day 33; Expected number of genomic features was estimated based on the ratio of the number of human rapidly-evolving TADs (n = 60) to the total number of TADs in hESC (n = 3,127).
Next, the placement enrichment estimates were computed for multiple other genomic regulatory elements that were previously implicated as candidate regulatory loci with putative impact on human-specific phenotypes and were not considered during the revTAD selection process. Remarkably, placement of hESC enhancers, primate-specific CTCF-binding sites, human-specific RNAPII-binding sites, regions of human-specific conserved deletions (hCONDELs), and H3K4me3 peaks with human-specific enrichment at transcription start sites (TSS) in prefrontal cortex neurons appears significantly enriched within the revTAD boundaries (P < 0.0001 in all instances; table 3). Placement of H3K27ac peaks with human-specific enrichment in embryonic limb at E33 stage of human embryogenesis (Cotney et al. 2013) is significantly higher in the revTADs than expected by chance alone (table 3). However, no increase of placements was observed for H3K27ac peaks with human-specific enrichment in embryonic limb at the later stages of embryogenesis, including E37; E41; and E44 stages (data not shown). These results seem to implicate the enhancers and promoters that are engaged during the first five weeks of human embryogenesis in limb development as putative targets for human-specific regulatory elements residing within the revTADs.
One notable exception was the lack of significant placement enrichment for genes comprising human-specific transcriptional genetic networks in the frontal lobe (table 3), which were defined based on the analyses of adult brain tissues (Konopka et al. 2012). However, the FOXP2 gene encoding one of the principal transcription factors presumably contributing to the human-specific transcriptional control of these networks (Konopka et al. 2012) and previously implicated in evolution of human language and cognition, is residing within the revTAD harboring 12 HAR sequences, one human-specific NANOG-binding site, and 22 primate-specific TFBS, including ten primate-specific CTCF-binding sites. Interestingly, the promoter of the FOXP1 gene, which can form heterodimers with FOXP2 to regulate transcription and has been implicated in language impairment, intellectual disability, and autism, is also located within another revTAD harboring 10 HAR sequences and 17 primate-specific TFBS, including eight primate-specific CTCF-binding sites. One of the well-known FOXP2 target genes, LMO4, is also located within yet another revTAD harboring 10 HAR sequences and 17 primate-specific TFBS, including seven primate-specific CTCF-binding sites. Several lines of experimental evidence strongly argue that LMO4 plays an important role in regulation of asymmetrically developed cognitive processes in humans such as language (Konopka et al. 2012). Nevertheless, the proximity placement analysis does not support the hypothesis that a majority of genes comprising human-specific transcriptional networks in adult brain are located within revTAD regions of human genome. This conclusion is consistent with the previous observations that HSRS are placed in close proximity to genes having important regulatory functions during the early embryogenesis (Glinsky 2015).
Correlation Screens Revealed Distinct Patterns of Associations between Individual Members of HSRS Families Residing within the revTADs
The highly complex patterns of the genomic architecture of individual revTADs harboring hundreds regulatory elements create a significant analytical challenge (fig. 2). The UCSC Genome Browser view of the revTAD on human chr6 is shown in figure 2A to illustrate this problem. This particular revTAD harbors 10 Human Accelerated Regions, HARs (red bars), 10 hESC-enriched enhancers (black bars), 52 primate-specific TFBS for NANOG (26 sites), POU5F1 (10 sites), CTCF (26 sites), and 72 recombination hotspots with recombination rates at least 10 cM/Mb (blue bars). It was reasonable to expect that deconvolution of this exceedingly high complexity may provide a clue regarding the underlying mechanisms of creation of such regulatory structures.
Fig. 2.—

High-complexity patterns of the genomic architecture of individual rapidly evolving in humans Topologically Associating Domains (revTADs) harboring hundreds regulatory elements and reflecting distinct association profile between placements of HARs and TFBS residing within the revTADs. UCSC Genome Browser view of the revTAD on human chr6 harboring 10 Human Accelerated Regions, HARs (red bars), 10 hESC-enriched enhancers (black bars), 52 primate-specific TFBS for NANOG (26 sites), POU5F1 (10 sites), CTCF (26 sites), and 72 recombination hotspots with recombination rates at least 10 cM/Mb (blue bars). Genomic coordinates of POU3F2 super-enhancer domain in the hESC genome is depicted by the horizontal arrow. Supplementary figure S1, Supplementary Material online reports multiple correlation screens revealing distinct patterns of associations between placements of HARs and TFBS residing within the revTADs.
At the next stage of the revTAD analysis, a series of correlation screens was performed to determine the relationships between the individual HSRS residing within the revTAD boundaries (fig. 2 and supplementary fig. S1, Supplementary Material online). To this end, the numbers of individual members of each HSRS family and primate-specific TFBS located within the boundaries of each revTAD were calculated and corresponding correlation coefficients were computed. Notably, the placement patterns of HSTFBS and individual members of HSTFBS family manifested highly significant positive correlations with the number of primate-specific CTCF-binding sites located within the revTAD boundaries (supplementary fig. S1, Supplementary Material online). The most significant positive correlation coefficients were observed for human-specific TFBS and the weakest correlation was recorded for HSTFBS and non-human primate-specific CTCF-binding sites.
In striking contrast, the significant inverse correlations were documented between the placement patterns of HARs and primate-specific CTCF-binding sites residing within the revTADs (supplementary fig. S1, Supplementary Material online). The most significant negative correlation coefficients were observed between the placement numbers of HARs and human-specific TFBS and the weakest inverse correlation was recorded between HARs and non-human primate-specific CTCF-binding sites.
The results of these analyses are highly consistent with the idea that placement and/or retention patterns of HARs and HSTFBS within the revTADs are guided and governed by distinct mechanisms. Placement and/or retention of HSTFBS appear to follow the CTCF-binding sites’ patterns, whereas locations of HARs seem to favor the revTAD regions harboring relatively fewer CTCF-binding sites resulting in highly significant inverse correlation between placement patterns of HARs and HSTFBS within the revTADs (supplementary fig. S1, Supplementary Material online).
Distinct Correlation Profiles of HSRS and Recombination Rates within the revTADs Distinguish Placement Patterns of HARs and HSTFBS
It has been reported that a prevalent mode of mutations in HARs is base substitutions that change a weak (A, T) bond into a strong (G, C) bond, which may occur during meiotic recombination as a result of a biochemical bias towards strong G/C alleles during the mismatch repair of heteroduplex DNA molecules (Kostka et al. 2012). Consistent with this notion, the enrichment of GC-biased substitutions of DNA sequences near recombination hotspots and a significant correlation between GC bias and recombination rate in the human genome have been reported (Katzman et al. 2011). Direct measurements of fine-scale recombination rates in genomic regions surrounding hominid accelerated conserved regions demonstrated significantly higher mean recombination rate estimates for 30 Kb DNA segments around HARs (Freudenberg et al. 2007). These observations prompted detailed examination of recombination rates within the revTADs. Recombination rates were downloaded from the HapMap Project (The International Hapmap Consortium 2007) and the number of DNA segments with the recombination rates of 10 cM/Mb or greater were identified for each revTAD. The results were plotted for visualization of spatial distributions (fig. 3A and B) and corresponding correlation coefficients were computed (supplementary fig. S2, Supplementary Material online). In these analyses, the total numbers of recombination hotspots having recombination rates of 10 cM/Mb or greater within boundaries of a given revTAD were determined and designated as the recombination scores (see “Materials and Methods” section for details).
Fig. 3.—
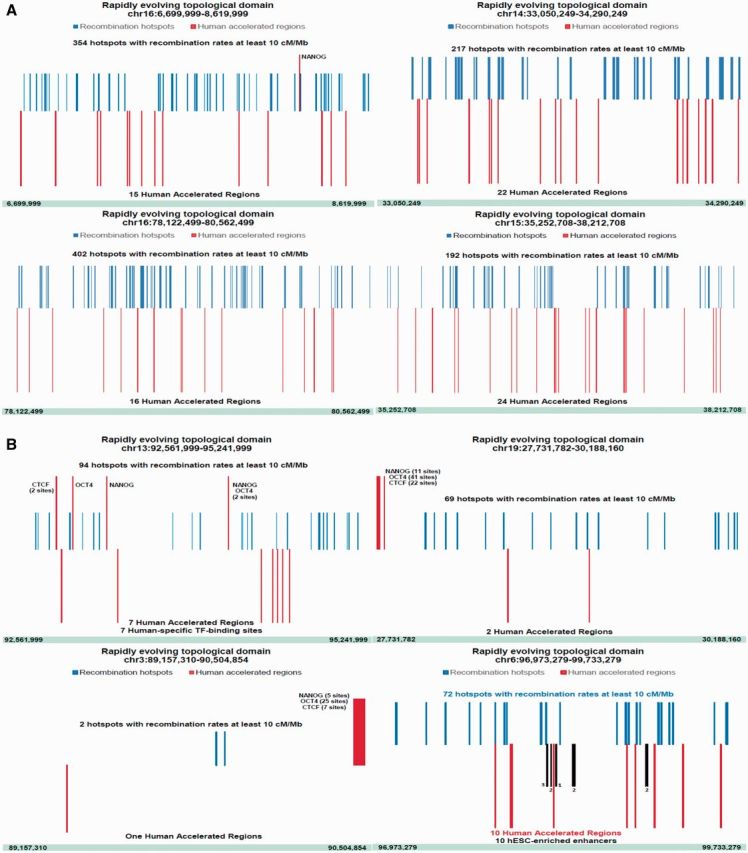

Distinct correlation profiles of HSRS and recombination rates within the revTADs distinguish placement patterns of HARs and HSTFBS. (A, B) Visualization of the placement distribution patterns of HARs (low positioned red bars), recombination hotspots, RHs (blue bars), and HSTFBS (high positioned red bars with designations of TF names) within the revTADs. Note that revTADs containing high numbers of RHs (192–402 RHs) tend to harbor higher numbers of HARs (15–24 HARs) and no HSTFBS (figures in the panel A). In contrast, revTADs containing intermediate (69–94 RHs) or low (2 RHs) numbers of RHs tend to harbor intermediate and low numbers of HARs and multiple HSTFBS (figures in the panel B). (C) A model of genome evolution driven by the increasing complexity of genomic regulatory networks (GRNs). It is proposed that mechanistically distinct processes creating HSRS occur within the context of the intrinsic division of mammalian genomes into regions of high and low recombination rates. Genomic regions of high and low recombination rates are associated with the low and high probabilities of TE insertion and/or retention as well as C/G and A/T alleles’ bias, respectively. According to this model, the continuing emergence of new enhancer elements constitutes a critical creative event driving the increasing complexity of GRNs in the hESC genome. Potential mechanisms of HSRS-mediated effects on principal regulatory structures of interphase chromatin involve: (i) creation of new TFBS and novel enhancer elements; (ii) increasing density of conventional enhancers which would facilitate a transition to super-enhancer structures; (iii) emergence of overlapping CTCF/cohesin-binding sites and LMNB1-binding sites; (iv) continuing insertion of clusters of Alu elements near the putative DNA bending sites. Collectively, the ensemble of these structural changes facilitated by the targeted placements and retention of HSRS at defined genomic locations would enable the emergence of new super-enhancer domains and facilitate the remodeling of existing TADs to drive evolution of GRNs. MADE, cytosine methylation associated DNA editing.
Significant inverse correlations were observed between recombination scores and the numbers of HSTFBS residing within the revTADs (supplementary fig. S2, Supplementary Material online), whereas no significant correlation was recorded between recombination scores and non-human primate-specific CTCF-binding sites. In striking contrast with HSTFBS, a highly significant positive correlation was observed between recombination scores and the numbers HARs located within the revTADs (supplementary fig. S2, Supplementary Material online).
Interactions of DNA strands are required for recombination process. The interphase chromosome contact counts obtained from Hi-C experiments reflect the likelihood of placement of DNA strands in close proximity, which should correlate with the probability of direct physical interactions of DNA strands. Based on these considerations, it was reasonable to expect that genomic regions of high chromatin contact counts may display a tendency for increased recombination rates. Consistent with this notion, a significant positive correlation was recorded between the numbers of intrachromosomal contacts observed within a given revTAD region and the corresponding recombination scores (supplementary fig. S2, Supplementary Material online). This observation offers an opportunity to analyze the relationships between recombination rates and placement of distinct HSRS in sub-groups of revTADs that were segregated solely based on the mean values of cumulative numbers of intrachromosomal contacts. Intrachromosomal contacts represent analytically and technically distinct set of genomic variables, which has been measured genome-wide in entirely independent set of experiments by design, execution, and technical protocols (Jin et al. 2013). To carry-out these analyses, the mean values of corresponding variables were computed and analyzed for the distinct revTAD sub-sets. The strikingly distinct association profiles between recombination rates and placement patterns of either HARs or HSTFBS were documented. In one of these analyses, the revTADs were segregated into quartiles based on the cumulative values of numbers of intrachromosomal contacts observed within the regions without prior knowledge of their recombination scores (supplementary fig. S2, Supplementary Material online). Strikingly, the median recombination scores for the revTADs placed in the top quartile based on the quantity of observed intrachromosomal interactions was 40-fold greater compared to the median recombination score of the revTADs assigned to the bottom quartile (supplementary fig. S2, Supplementary Material online).
Collectively, the results of these analyses are highly consistent with the hypothesis that placements of HARs and HSTFBS within the revTADs are associated with distinct molecular processes and support the idea connecting the biogenesis of HARs with high recombination rates. It has been demonstrated that HSTFBS are located almost exclusively within TE-derived DNA sequences (Glinsky 2015), strongly implicating activity of TE in biogenesis of HSTFBS. Present observations of significant inverse correlations between the HSTFBS placement numbers and recombination rates within the revTADs are highly congruent with this hypothesis, because TE insertions are known to evade the genomic regions with high recombination rates (Rizzon et al. 2002). The results of these analyses are consistent with the model prediction that different HSRS families that are created via two mechanistically distinct pathways of divergent regulatory DNA evolution should manifest distinct patterns of placements in the human genome.
According to this model of genome evolution, one of the key elements of the evolution of genomic regulatory networks is the creation of new enhancer elements (fig. 3). Conventional enhancers comprise discrete DNA segments occupying a few hundred base pairs of the linear DNA sequence and harboring multiple TFBS. Super-enhancers consist of clusters of conventional enhancers that are densely occupied by the master transcription factors and Mediator (Whyte et al. 2013). Therefore, it is logical to expect that creation of new TFBS and increasing density of TFBS would increase the probability of the emergence of new enhancer elements at specific genomic locations. In turn, creation of new enhancers and increasing their density would facilitate the emergence of new super-enhancer domain structures. In this context, creation of human-specific CTCF-binding sites seems particularly important, because CTCF-binding sites play a crucial role in defining the TAD boundaries (Dixon et al. 2012; Li et al. 2013) and in establishing the genomic architecture of super-enhancer domains (Dowen et al. 2014).
Conclusions
The results of the present analyses have important implications for our understanding of mechanisms of biogenesis and evolution of the majority of candidate HSRS, in particular, HARs and haDHS. Based on the sequence conservation analyses using the most recent releases of the reference genome databases, it is proposed to define these predominantly intronic and intergenic DNA segments manifesting more than 90% sequence identities among the Great Apes as the candidate HSRS that are highly conserved in both human and NHP lineages. Using this approach, a total of 5,535 regulatory DNA segments (supplementary data set S6, Supplementary Material online) are classified as the highly conserved in humans and NHP regulatory DNA sequences. This sub-set of candidate HSRS appears to evolve by the exaptation pathway of ancestral regulatory DNA segments, which is mechanistically distinct from the evolution of regulatory DNA driven by the species-specific expansion of transposable elements. Consistent with this notion, it has been demonstrated that transposable element-derived sequences, most notably LTR7/HERVH, LTR5_HS/HERVK, and L1HS, harbor 99.8% of the candidate human-specific regulatory loci with putative transcription factor-binding sites in the genome of hESC (Glinsky 2015). Intriguingly, transcriptional activation of these endogenous human stem cell-associated retroviral sequences has been documented in pre-implantation human embryos [reviewed in Robbez-Masson and Rowe (2015) and Glinsky (2015)] and associated with development of clinically intractable malignancies (Glinsky 2015, 2016).
Present analysis revealed a variable reference database refinement’s effect on the validity of molecular definitions of different families of candidate HSRS. It identifies limitations of the current computational cross-species sequence alignment algorithm and underscores the requirement of the careful follow-up analyses of each individual candidate HSRS using the most recent releases of the reference genome databases of Great Apes and other non-human primates. A large fraction of regulatory DNA segments representing candidate HSRS appears highly conserved in humans and other Great Apes. Reported herein sequence conservation analysis reveals that a significant majority of haDHSs, HARs, and CNCCs’ enhancers appears to represent highly conserved in humans and non-human primates candidate regulatory sequences that are consistently manifest species-specific patterns of single-nucleotide substitutions and accelerated mutation rates on the human lineage. Collectively, these observations imply that human-specific phenotypes may evolve as a result of combinatorial interplay of both conserved in non-human primates and human-specific (unique to humans) regulatory sequences.
Based on the present analyses, it seems reasonable to propose that at least two mechanistically distinct pathways of creation of divergent sequences of regulatory DNA drive the evolution of human-specific regulatory networks (fig. 1). In agreement with the proposed model, genome-wide proximity placement analyses of HSRS within TADs revealed the apparent non-random patterns of placement and/or retention of HSRS in the human genome. These analyses facilitated discovery of the relatively small number of chromosomal domains termed revTADs, which are significantly enriched for multiple, structurally and functionally diverse families of HSRS. Diverse families of candidate HSRS, which were defined by either the acceleration of mutation rates on the human lineage or the functional divergence from chimpanzee, appear highly conserved in humans and non-human primates, strongly arguing that they evolved via the exaptation of ancestral regulatory DNA. This conclusion is in agreement with recent reports describing exaptation of ancestral DNA as a mechanism of creation of human-specific enhancers active in embryonic limb (Cotney et al. 2013) and as a prevalent mechanism of recently evolved enhancers’ creation during the mammalian genome evolution (Villar et al. 2015). Despite the exceedingly high interspecies sequence identities for non-coding genomic regions and only minor differences of DNA sequences estimated in the range of ∼3 to 6 substitutions per 500 bp of the regulatory sequence (Prescott et al. 2015), it appears that the acquisition of a small number of mutations was sufficient to confer biologically discernable divergence of regulatory activities.
Methods
Data Source
Candidate Human-Specific Regulatory Sequences
A total of 18,364 candidate HSRS were analyzed in this study, including 2,745 human accelerated regions (Capra et al. 2013); 524 human accelerated DNase I hypersensitive sites (Gittelman et al. 2015); 3,083 human-specific transcription factor binding sites (Glinsky 2015); 8,229 fixed human-specific regulatory regions, FHSRR (Marnetto et al. 2014), which were divided into 2,118 DHS_FHSRR; 1,932 hESC_FHSRR; and 4,249 FHSRR identified in different human cell lines, excluding hESC (Other_FHSRR); 583 regions of human-specific loss of conserved regulatory DNA termed hCONDELs (McLean et al. 2011); 410 human-specific epigenetic regulatory marks consisting of H3K4me3 histone methylation signatures at transcription start sites in prefrontal neurons (Shulha et al. 2012); 1,000 human-biased and 1,000 chimp-biased cranial neural crest cells (CNCC) enhancers, which are associated with divergent cis-regulatory evolution of the human’s and chimpanzee’s neural crest and development of unique to human craniofacial features (Prescott et al. 2015).
Additional Data Sources and Analytical Protocols
Solely publicly available datasets and resources were used in this contribution as well as methodological approaches and a computational pipeline validated for discovery of primate-specific gene and human-specific regulatory loci (Kent et al. 2002; Schwartz et al. 2003; Tay et al. 2009; Capra et al. 2013; Marnetto et al. 2014; Glinsky 2015). The analysis is based on the University of California Santa Cruz (UCSC) LiftOver conversion of the coordinates of human blocks to corresponding non-human genomes using chain files of pre-computed whole-genome BLASTZ alignments with a minMatch of 0.95 and other search parameters in default setting (http://genome.ucsc.edu/cgi-bin/hgLiftOver, last accessed August 6, 2016). Extraction of BLASTZ alignments by the LiftOver algorithm for a human query generates a LiftOver output “Deleted in new”, which indicates that a human sequence does not intersect with any chains in a given non-human genome. This indicates the absence of the query sequence in the subject genome and was used to infer the presence or absence of the human sequence in the non-human reference genome. Human-specific regulatory sequences were manually curated to validate their identities and genomic features using a BLAST algorithm and the latest releases of the corresponding reference genome databases for time periods between April, 2013 and December, 2015.
Genomic coordinates of 3,127 topologically-associating domains (TADs) in hESC; 6,823 hESC-enriched enhancers; 6,322 conventional and 684 super-enhancers (SEs) in hESC; 231 SEs and 197 SEDs in mESC were reported in the previously published contributions (Dixon et al. 2012; Hnisz et al. 2013; Whyte et al. 2013; Xie et al. 2013; Dowen et al. 2014). The primary inclusion criterion for selection of the human-specific regulatory sequences (HSRS) analyzed in this contribution was the fact that they were identified in human cells lines and primary human tissues whose karyotype were defined as “normal”. The following four HSRS families comprising of 10,598 individual regulatory DNA sequences were subjected to the proximity placement analyses in this study: (1) Human accelerated regions (HARs; Capra et al. 2013); (2) Human-specific transcription factor-binding sites (HSTFBS; Glinsky 2015); (3) hESC-derived fixed human-specific regulatory regions (hESC-FHSRR; Marnetto et al. 2014); (4) DNase hypersensitive sites-derived fixed human-specific regulatory regions (DHS-FHSRR; Marnetto et al. 2014). Individual TADs were regarded as autonomous transcription regulatory units of human interphase chromosomes (Dixon et al. 2012; Gorkin et al. 2014). The number of HSRS placed within a given TAD was computed for every TAD in the hESC genome and the HSRS placement enrichment was calculated for each individual HSRS family as the ratio of observed values to expected values estimated from a random genome-wide distribution model at the various cut-off thresholds. Expected number of genomic features was estimated based on the ratio of the number of human revTADs to the total number of TADs in hESC (n = 3,127). The significance of the differences in the expected and observed numbers of events was calculated using two-tailed Fisher’s exact test. Additional placement enrichment tests were performed for individual revTADs and sub-sets of revTADs taking into account the size in bp of corresponding genomic regions. Datasets of NANOG-, POU5F1-, and CTCF-binding sites and human-specific TFBS in hESCs were reported previously (Kunarso et al. 2010; Glinsky 2015) and are publicly available. Recombination rates were downloaded from the HapMap Project (The International Hapmap Consortium 2007) and the numbers of DNA segments with the recombination rates of 10 cM/Mb or greater were counted. This threshold exceeds ∼10-fold the mean intensity of recombination rates in telomeric regions, which were identified as the regions with the higher recombination rates in the human genome. It is well known that over large genomic scales, recombination rates tend to be higher in telomeric as compared to centromeric chromosomal regions. In telomeric regions, the mean detected hotspot spacing is 90 kb and the mean intensity (total rate across the hotspot) per hotspot is 0.115 cM, whereas for centromeric regions the mean spacing is 123 kb and the mean intensity is 0.070 cM (The International Hapmap Consortium 2007).
Data Analysis
To determine the conservation patterns of reported 18,364 candidate human-specific regulatory DNA sequences, the conservation analysis was carried-out using the LiftOver algorithm and Multiz Alignments of 20 mammals (17 primates) of the UCSC Genome Browser (Kent et al. 2002) on Human Dec. 2013 Assembly (GRCh38/hg38) (http://genome.ucsc.edu/cgi-bin/hgTracks?db=hg38&position=chr1%3A90820922-90821071&hgsid=441235989_eelAivpkubSY2AxzLhSXKL5ut7TN, last accessed August 6, 2016).
The most recent releases of the corresponding reference genome databases were utilized to ensure the use of the most precise, accurate, and reproducible genomic DNA sequences available to date. A candidate HSRS was considered conserved if it could be aligned to either one or both Chimpanzee or Bonobo genomes using defined sequence conservation thresholds of the LiftOver algorithm MinMatch function. LiftOver conversion of the coordinates of human blocks to non-human genomes using chain files of pre-computed whole-genome BLASTZ alignments with a specified MinMatch levels and other search parameters in default setting (http://genome.ucsc.edu/cgi-bin/hgLiftOver, last accessed August 6, 2016). Several thresholds of the LiftOver algorithm MinMatch function (minimum ratio of bases that must remap) were utilized to assess the sequences conservation and identify candidate human-specific (MinMatch of 0.95; 0.99; and 1.00) and conserved in non-human primates (MinMatch of 1.00) regulatory sequences as previously described (Glinsky 2015). The Net alignments provided by the UCSC Genome Browser were utilized to compare the sequences in the human genome (hg38) with the mouse (mm10), Chimpanzee (PanTro4), and Bonobo genomes. A given regulatory DNA segment was defined as the highly conserved regulatory sequence when both direct and reciprocal conversions between humans’ and non-human primates’ genomes were observed using the MinMatch sequence alignment threshold of 1.00 requiring that 100% of bases must remap during the alignments of the corresponding sequences (tables 1 and 2). A given regulatory DNA segment was defined as the candidate human-specific regulatory sequence when sequence alignments failed to both Chimpanzee and Bonobo genomes using the specified MinMatch sequence alignment thresholds (supplementary tables S1–S10, Supplementary Material online).
Statistical Analyses of the Publicly Available Datasets
All statistical analyses of the publicly available genomic datasets, including error rate estimates, background and technical noise measurements and filtering, feature peak calling, feature selection, assignments of genomic coordinates to the corresponding builds of the reference human genome, and data visualization, were performed exactly as reported in the original publications and associated references linked to the corresponding data visualization tracks (http://genome.ucsc.edu/). Any modifications or new elements of statistical analyses are described in the corresponding sections of the Results. Statistical significance of the Pearson correlation coefficients was determined using GraphPad Prism version 6.00 software. The significance of the differences in the numbers of events between the groups was calculated using two-sided Fisher’s exact and Chi-square test, and the significance of the overlap between the events was determined using the hypergeometric distribution test (Tavazoie et al. 1999).
Supplementary Material
Acknowledgments
This work was made possible by the open public access policies of major grant funding agencies and international genomic databases and the willingness of many investigators worldwide to share their primary research data. I thank you Dr Joshua Akey for the insightful comments during the preparation of the final versions of the manuscript. I would like to thank my anonymous colleagues for their valuable critical contributions during the peer review process of this work. This work was supported by the personal, private, and institutional funds. This is a single author contribution. All elements of this work, including the conception of ideas, formulation, and development of concepts, execution of experiments, analysis of data, and writing of the paper, were performed by the author.
Literature Cited
- Bird C, et al. 2007. Fast-evolving noncoding sequences in the human genome. Genome Biol. 8:R118.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capra JA, Erwin GD, McKinsey G, Rubenstein JL, Pollard KS. 2013. Many human accelerated regions are developmental enhancers. Philos Trans R Soc Lond B Biol Sci. 368:20130025.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotney J, et al. 2013. The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154:185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, et al. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowen JM, et al. 2014. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenberg J, Fu YH, Ptácek LJ. 2007. Human recombination rates are increased around accelerated conserved regions—evidence for continued selection? Bioinformatics 23:1441–1443. [DOI] [PubMed] [Google Scholar]
- Gittelman RM, et al. 2015. Comprehensive identification and analysis of human accelerated regulatory DNA. Genome Res. 25:1245–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. 2015. Transposable elements and DNA methylation create in embryonic stem cells human-specific regulatory sequences associated with distal enhancers and non-coding RNAs. Genome Biol Evol. 7:1432–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. 2015. Viruses, stemness, embryogenesis, and cancer: a miracle leap toward molecular definition of novel oncotargets for therapy-resistant malignant tumors? Oncoscience 2:751–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinsky GV. 2016. Activation of endogenous human stem cell-associated retroviruses (SCARs) and therapy-resistant phenotypes of malignant tumors. Cancer Lett. 376:347–359. [DOI] [PubMed] [Google Scholar]
- Gorkin DU, Leung D, Ren B. 2014. The 3D genome in transcriptional regulation and pluripotency. Cell Stem Cell 14:762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, et al. 2013. Super-enhancers in the control of cell identity and disease. Cell 155:934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Li L, Qin ZS, Corces VG. 2012. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol Cell 48:471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, et al. 2013. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman S, Capra JA, Haussler D, Pollard KS. 2011. Ongoing GC-biased evolution is widespread in the human genome and enriched near recombination hot spots. Genome Biol Evol. 3:614–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, et al. 2002. The Human Genome Browser at UCSC. Genome Res. 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G, et al. 2012. Human-specific transcriptional networks in the brain. Neuron 75:601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostka D, Hubisz MJ, Siepel A, Pollard KS. 2012. The role of GC-biased gene conversion in shaping the fastest evolving regions of the human genome. Mol Biol Evol. 29:1047–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunarso G, et al. 2010. Transposable elements have rewired the core regulatory network of human embryonic stem cells. Nat Genet. 2:631–634. [DOI] [PubMed] [Google Scholar]
- Li Y, et al. 2013. Characterization of constitutive CTCF/cohesin loci: a possible role in establishing topological domains in mammalian genomes. BMC Genomics 14:553.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnetto D, Molineris I, Grassi E, Provero P. 2014. Genome-wide identification and characterization of fixed human-specific regulatory regions. Am J Hum Genet. 95:39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, et al. 2011. Human-specific loss of regulatory DNA and the evolution of human-specific traits. Nature 471:216–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, et al. 2012. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, et al. 2006. An RNA gene expressed during cortical development evolved rapidly in humans. Nature 443:167–172. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Noonan JP, Paabo S, Rubin EM. 2006. Accelerated evolution of conserved noncoding sequences in humans. Science 314:786.. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, et al. 2008. Human specific gain of function in a developmental enhancer. Science 321:1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott SL, et al. 2015. Enhancer divergence and cis-regulatory evolution in the human and chimp neural crest. Cell 163:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzon C, Marais G., Gouy M, Biémont C. 2002. Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res. 12:400– 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbez-Masson L, Rowe HM. 2015. Retrotransposons shape species-specific embryonic stem cell gene expression. Retrovirology 12:45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, et al. 2003. Human-mouse alignments with BLASTZ. Genome Res. 13:103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulha HP, et al. 2012. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 10:e1001427.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, et al. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148:458–472. [DOI] [PubMed] [Google Scholar]
- Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. 1999. Systematic determination of genetic network architecture. Nat Genet. 22:281–285. [DOI] [PubMed] [Google Scholar]
- Tay SK, Blythe J, Lipovich L. 2009. Global discovery of primate-specific genes in the human genome. Proc Natl Acad Sci U S A. 106:12019–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Hapmap Consortium. 2007. A second generation human haplotype map of over 3.1 million SNPs. Nature 449:851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar D, et al. 2015. Enhancer evolution across 20 mammalian species. Cell 160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, et al. 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153:307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, et al. 2013. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153:1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.