

Abstract
Fusobacterium nucleatum is considered to be a key oral bacterium in recruiting periodontal pathogens into subgingival dental plaque. Currently F. nucleatum can be subdivided into five subspecies. Our previous genome analysis of F. nucleatum W1481 (referred to hereafter as W1481), isolated from an 8-mm periodontal pocket in a patient with chronic periodontitis, suggested the possibility of a new subspecies. To further investigate the biology and relationships of this possible subspecies with other known subspecies, we performed comparative analysis between W1481 and 35 genome sequences represented by the five known Fusobacterium subspecies. Our analyses suggest that W1481 is most likely a new F. nucleatum subspecies, supported by evidence from phylogenetic analyses and maximal unique match indices (MUMi). Interestingly, we found a horizontally transferred W1481-specific genomic island harboring the tripartite ATP-independent (TRAP)-like transporter genes, suggesting this bacterium might have a high-affinity transport system for the C4-dicarboxylates malate, succinate, and fumarate. Moreover, we found virulence genes in the W1481 genome that may provide a strong defense mechanism which might enable it to colonize and survive within the host by evading immune surveillance. This comparative study provides better understanding of F. nucleatum and the basis for future functional work on this important pathogen.
Keywords: Fusobacterium nucleatum, comparative genomics, F. nucleatum W1481
Introduction
The genus Fusobacterium can be distinguished by its Gram-negative, nonmotile and nonsporulating features, as described in the studies conducted by Bachmann and Gregor (1936). Cultures of Fusobacterium grow only under strictly anaerobic conditions and they are extremely susceptible to atmospheric oxygen (Spaulding and Rettger 1937b). Fusobacterium are normally present in the human oral cavity and throat (Spaulding and Rettger 1937a), however, recent studies have shown that the oropharynx, as well as the gastrointestinal and genitourinary tracts may also be populated with Fusobacterium spp. (Castellarin et al. 2012). Fusobacterium spp. can occasionally cause localized infections such as tonsillitis, para-tonsillar abscess or dental sepsis, invasive disease including septicaemia, abscesses of the brain, liver and lung, or Lemierre’s syndrome, a septic thrombophlebitis of the internal jugular vein that is often associated with oropharyngeal infection (Huggan and Murdoch 2008; Kuppalli et al. 2012). The majority of research has focused on the role of Fusobacterium spp. in gingivitis and periodontitis, where Fusobacterium nucleatum (F. nucleatum) is the most frequently isolated species from dental plaque, where it is believed to play an important role in recruiting periodontal pathogens into subgingival dental plaque (Socransky et al. 1998). Based on the polyacrylamide gel electrophoresis pattern of whole-cell proteins and DNA homology, glutamate dehydrogenase and 2-oxoglutarate reductase electrophoretic patterns, DNA–DNA hybridization comparisons and phenotypic characteristics, F. nucleatum was further classified into five subspecies (nucleatum, polymorphum, animalis, vincentii and fusiforme) (Gharbia and Shah 1990; Morris et al. 1996).
In this study, we present a detailed genomic and comparative analysis of F. nucleatum W1481 (referred to hereafter as W1481), which was isolated from an 8-mm periodontal pocket in a patient with chronic periodontitis and was subsequently sequenced and reported by our group (Ang et al. 2014). An in-depth phylogenetic and comparative genomic analysis of this genome reconfirmed our initial assumption that W1481 belongs to F. nucleatum, but it is quite distinct from the other existing subspecies of F. nucleatum and is possibly a new subspecies. The distinct taxonomic status of F. nucleatum W1481 prompted us to investigate the genome more closely, especially its virulence profile and compare it with the other subspecies of F. nucleatum to provide a better insight into the biology of this unique strain. This genomic study is likely to form the basis for future functional work on this important pathogen.
Materials and Methods
Genome Annotation
To ensure the uniformity in the annotations of the genomes which is important for comparative analysis, all the genome sequences of Fusobacterium spp. used in the study were annotated by uploading their sequence files to the Rapid Annotation using Subsystem Technology (RAST) web server (Aziz et al. 2008) for genomic annotations. The RAST pipeline produced putative protein sequences and functions based on the subsystems in FIGfams (Meyer et al. 2009), a new collection of over 100,000 protein families that were the product of manual curation and close strain comparison across hundreds of bacteria and archaea genomes.
Phylogenetic Analyses
16S rRNA sequences for 25 identified Fusobacterium strains (comprising of F. nucleatum, F. periodonticum, F. ulcerans, F. mortiferum, F. varium, F. russii, F. gonidiaformans and F. necrophorum) were obtained from National Centre for Biotechnology Information (NCBI) RefSeq (Pruitt et al. 2007) and were aligned with that of W1481 using Clustal Omega (Sievers et al. 2011). To construct a core genome SNP-based tree, all genome sequences were uploaded to PanSeq (Laing et al. 2010) for alignment and the Single Nucleotide Polymorphism (SNPs) of the core/common genome regions were extracted. MEGA version 5.2 (Tamura et al. 2011) was used for DNA substitution model testing and also to generate the phylogenetic trees. The model selection was based on Bayesian information criterion (BIC). All the neighbor-joining phylogenetic trees were based on Kimura’s two parameter model, inferred with 1,000 bootstrap replications.
Genomic Distance Calculation
Calculation of genomic distance was performed based on the number of maximal unique and exact matches (MUMs) of a minimal length shared by two genomes when being compared. For each genome pairs, MUMs were generated using MUMmer 3 software package (Delcher et al. 2003) using the parameters: -mum, -b, -c and -119. MUMmer package automatically detects matches that may not be unique while a Perl script developed by Marc and his colleague (Deloger et al. 2009) was used to trim overlapping MUMs and to calculate the MUM index (MUMi) values. MUMi varies between 0 for very similar and 1 for very distant genomes.
Whole-Genome Average Nucleotide Identity Analysis
To evaluate the genetic relatedness between W1481 and the other Fusobacterium genomes the average nucleotide identity (ANI) (Konstantinidis and Tiedje 2005) was calculated based on the method proposed by Goris et al. (2007). Two-way BLAST was chosen and only forward and reversed-matched orthologs were used in the calculations. For the robustness, the BLAST match have been set at least 50% identity at the nucleotide and amino acid level and a sequence coverage of at least 70%.
Amino Acid Identity Analysis
The calculation of average amino acid identity (AAI) was performed by the method as described by Konstantinidis and Tiedje (2005). The RAST annotated protein-coding sequences of the W1481 genome was used as the reference for comparison against other Fusobacterium genomes (also annotated using RAST), using the BLAST search to determine the conserved genes. The cut-off was set at ≥30% sequence identity and ≥70% sequence coverage at the amino acid level for the BLAST search. The average of the amino acid identity of all conserved genes between a pair of genomes was computed to measure the genetic relatedness between them.
Comparative Genomic Island Analysis
All genomes sequences were submitted to IslandViewer (Langille and Brinkman 2009) for genomic island (GI) prediction. IslandViewer implements a sequence composition based approaches derived from other validated GI prediction software packages, which were SIGI-HMM (Waack et al. 2006) and IslandPath-DIMOB (Hsiao et al. 2003). Both the composition based approach were shown to have a higher than 86% overall accuracy compared with other prediction software. IslandViewer also integrated IslandPick, which was developed based on comparative genomics approach (Langille et al. 2008). To cluster these GIs for comparison across different F. nucleatum strains, the GI nucleotide sequences were clustered using BLASTClust (Altschul et al. 1990) with the thresholds of at least 50% sequence identity and 50% sequence coverage.
Comparative Pathogenomic Analyses
Homology searches were performed on the protein sequences of 36 Fusobacterium strains against the Virulence Factors Database (VFDB) (Chen et al. 2005, 2012), by applying BLASTP of the BLAST software package (Altschul et al. 1997). The BLAST hits were processed using in-house Python script filtering and accepting only orthologs at the threshold of at least 50% sequence identity and 50% sequence completeness between the query and subject sequences. Instead of using the tabular comparison for pathogenomic composition provided in VFDB release of 2012, the data collected from VFDB was used to construct a graphical heat map representation using our in-house R Scripts, allowing for the comparison of the virulence gene profiles across all Fusobacterium strains.
Pan-Genome Analyses
Functional ortholog clustering was performed using PGAP (Pan-genome Analysis Pipeline) (Zhao et al. 2012). The protein sequences of 21 F. nucleatum strains were used as the input file. The orthologs among these 21 strains were searched using the Gene Family (GF) method included in the PGAP pipeline. Protein sequences of each strain were clustered together and denoted with strain identifiers. BLASTALL program (Altschul et al. 1990) was performed among the protein sequences with the minimum score value of 50 and E-value at 1e-8. The filtered BLAST results were clustered by MCL algorithm (Enright et al. 2002). In order to group the same genes into the same cluster, the global match region must have at least 70% of the longer gene protein sequence (coverage) and 40% sequence identity.
Results and Discussion
Genome Overview and Annotation
The size of the sequenced genome of F. nucleatum W1481 was 2,477,971 bp, with a GC composition of 27.6%. About 2,163 coding sequences (CDSs) and 56 RNAs were predicted by RAST in W1481 (Ang et al. 2014). An illustration of genomic contents in the genome of W1481 is shown in figure 1A. The subsystem distribution statistic of W1481 based on the genome annotation performed by RAST pipeline is presented in figure 1B.
Fig. 1.—
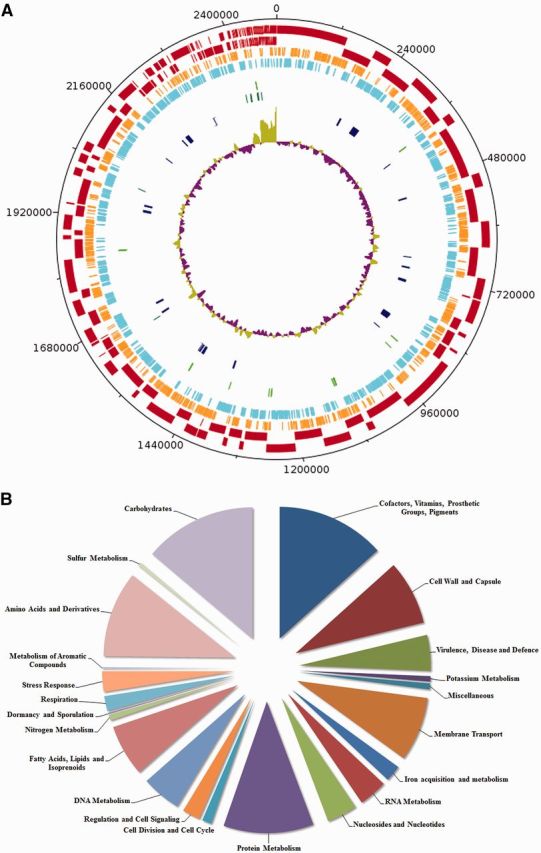
(A) Circular representation of the W1481 genome. The indication for each feature from the outermost layer: (a) Contigs (Dark Red); (b) Forward protein-coding genes (Orange); (c) Reverse protein-coding genes (Light Blue); (d) tRNAs (Light Green); (e) rRNAs (Dark Green); (f) Predicted Genomic Islands (Dark Blue); and (g) GC plot (Purple and Green). (B) Subcellular distribution as predicted by RAST.
Phylogenetic Analysis Using Different Biomarker
Twenty-six (26) published Fusobacterium strains (including F. nucleatum strain W1481) were obtained from the National Center for Biotechnology Information (NCBI) database (Pruitt et al. 2007). Phylogenetic trees were reconstructed by maximum likelihood method using the Kimura 2-parameter (K2P) distance model. The 16S rRNA phylogenetic tree clearly clustered all the subspecies together, as previously shown (Manson McGuire et al. 2014). Based on the 16S rRNA phylogenetic tree (fig. 2A), W1481 was clustered together with F. nucleatum strains, indicating that it was indeed a F. nucleatum species. To further confirm the taxonomic position of W1481, 271,947 Single Nucleotide Polymorphisms (SNPs) in the core genomes were concatenated and aligned. We reconstructed a more robust phylogenetic tree using the core-genome SNPs by using K2P distance model (fig. 2B). The core-genome SNP-based tree showed W1481 was clearly clustered together with F. nucleatum strains, further supporting that W1481 is F. nucleatum.
Fig. 2.—

Phylogenetic classification of W1481. (A) Phylogenetic tree constructed with 16S rRNA gene sequences of 26 strains of Fusobacterium strains. (B) Phylogenetic tree constructed with the core-genome SNPs of 23 strains of Fusobacterium.
Interestingly, W1481 did not cluster with any of the known subspecies, pointing to the possibility that W1481 might be a novel subspecies of F. nucleatum.
Genomic Distance Based on Maximum Unique Matches Index (MUMi)
To further study the genetic relationship between W1481 and other subspecies of F. nucleatum at the subspecies level, we performed genomic distance-type index calculations. Genomic distance based on MUMi analysis was then performed by comparing two sets of data, in which the first set consisted of all the 36 genomes of Fusobacterium spp. and the second set consisted only of F. nucleatum genomes. The results of the MUMi analysis of the first set as summarized in figure 3A showed that W1481 was quite distant compared to other Fusobacterium strains/species with the range of values between 0.64 and 0.98. The closest strains to W1481 were all of F. nucleatum, reinforcing our view that this isolate is indeed F. nucleatum.
Fig. 3.—
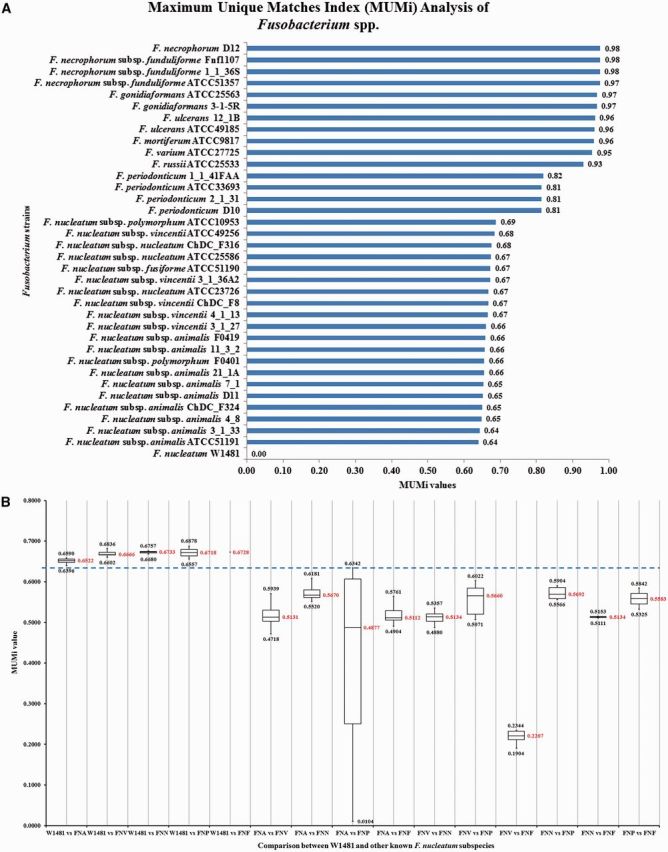
(A) MUMi analysis between W1481 and other Fusobacterium spp. The initial result showed that W1481 has the closest value (0.64) when compared to F. nucleatum subsp. animalis. (B) Comparison between the MUMi values among W1481 and the groups of five existing subspecies of F. nucleatum. The MUMi results of each subspecies are summarized and compared. The upper value of the box and whisker box showed the highest MUMi value, whereas the lower value showed the lowest MUMi value while the value in middle showed the average MUMi value when we performed comparison. A clear separation of W1481 from the rest of the known F. nucleatum subspecies is able to be performed at the threshold of 0.6342. (Note: FNA denotes subsp. animalis; FNV denotes subsp. vincentii; FNN denotes subsp. nucleatum; FNP denotes subsp. polymorphum and FNF denotes subsp. fusiforme).
Although W1481 had the closest value (0.64) with F. nucleatum subsp. animalis, it was still unclear if this value was distant enough to support W1481 as a novel subspecies since we did not have a specific cut-off to define a subspecies. To define a cut-off value, we calculated the MUMi distance between all pairs of known F. nucleatum subspecies. To use a very stringent criterion, we chose the highest value (most distant) to separate a pair of two known subspecies as a cut-off to discriminate subspecies. Our MUMi analysis showed that the highest value between the subsp. animalis (F. nucleatum subsp. animalis 11_3_2) and subsp. polymorphum (F. nucleatum subsp. polymorphum ATCC10953), which was 0.6342 (fig. 3B). Therefore, we selected 0.6342 as a cut-off value to discriminate subspecies.
Interestingly, the MUMi distances between W1481 with any strain of known subspecies were all above the defined cut-off, supporting our view that W1481 might be a novel subspecies. W1481 was distantly separated from all known subspecies compared to the distances between any pair of known subspecies (fig. 3A). Supplementary table S1, Supplementary Material online, shows the full sets of MUMi values generated by comparing all 36 strains of Fusobacterium species.
The MUMi results were further supported by the ANI and AAI analysis (using the same set of genomes with W1481 as the reference genome) which also showed that the closest strain to W1481, belonged to F. nucleatum subsp. animalis as shown in supplementary figure S1, Supplementary Material online.
Pan-Genome and Genome Composition Analysis
To further investigate the genomic structure and gene content of F. nucleatum W1481, we performed pan-genome analysis using the protein sequences of 21 F. nucleatum strains. All the genome sequences of Fusobacterium used in the study were also annotated using the RAST pipeline to ensure uniformity. The genome size of the strains analyzed ranged from 1.71 to 3.68 Mb with GC composition varying between 25.2% and 35.2%. An overview of all the 21 Fusobacterium genomes used in the present study is shown in table 1. The results showed that F. nucleatum pan-genome contained 6,666 gene clusters which were composed of 880 (13%) core gene clusters, 2,850 (43%) dispensable gene clusters and 2,936 (44%) strain-specific gene clusters. The accessory genes (dispensable and strain-specific genes) which cover 87% of the total F. nucleatum pan-genome suggested that this species contains a very diverse genomic structure.
Table 1.
Overview of the genome statistics
| # | Strain name | Strain status | Genome size (Mb) | GC content (%) | Number of contigs | Contig N50 | Number of ORFs | Number of tRNAs | Number of rRNAs |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Fusobacterium nucleatum W1481 | I | 2.48 | 27.5 | 186 | 7,109 | 2,219 | 46 | 10 |
| 2 | F. nucleatum subsp. animalis 11_3_2 | I | 2.7 | 26.9 | 81 | 101,807 | 2,725 | 47 | 14 |
| 3 | F. nucleatum subsp. animalis 21_1A | I | 2.15 | 27 | 2 | 1,755,614 | 2,083 | 46 | 7 |
| 4 | F. nucleatum subsp. animalis 3_1_33 | I | 2.29 | 27 | 18 | 266,619 | 2,135 | 44 | 3 |
| 5 | F. nucleatum subsp. animalis 4_8 | C | 2.26 | 27.1 | 1 | – | 2,202 | 56 | 15 |
| 6 | F. nucleatum subsp. animalis 7_1 | I | 2.47 | 26.8 | 95 | 45,444 | 2,477 | 42 | 2 |
| 7 | F. nucleatum subsp. animalis ATCC 51191 | I | 2.48 | 25.2 | 465 | 4,679 | 2,443 | 42 | 5 |
| 8 | F. nucleatum subsp. animalis ChDC F324 | I | 2.27 | 26.9 | 123 | 39,283 | 2,210 | 35 | 0 |
| 9 | F. nucleatum subsp. animalis D11 | I | 2.34 | 26.9 | 355 | 11,476 | 2,387 | 44 | 4 |
| 10 | F. nucleatum subsp. animalis F0419 | I | 2.44 | 27 | 5 | 2,257,469 | 2,345 | 39 | 13 |
| 11 | F. nucleatum subsp. nucleatum ATCC 23726 | I | 2.24 | 27 | 67 | 69,905 | 2,143 | 43 | 3 |
| 12 | F. nucleatum subsp. nucleatum ATCC 25586 | C | 2.17 | 27.2 | 1 | – | 2,102 | 47 | 15 |
| 13 | F. nucleatum subsp. nucleatum ChDC F316 | I | 2.16 | 27 | 83 | 68,085 | 2,112 | 32 | 3 |
| 14 | F. nucleatum subsp. polymorphum ATCC 10953 | C | 2.43 | 26.8 | 1 | – | 2,390 | 45 | 10 |
| 15 | F. nucleatum subsp. polymorphum F0401 | I | 2.51 | 27 | 2 | 2,507,721 | 2,533 | 51 | 19 |
| 16 | F. nucleatum subsp. vincentii 3_1_27 | I | 2.2 | 27 | 77 | 111,961 | 2,115 | 46 | 13 |
| 17 | F. nucleatum subsp. vincentii 3_1_36A2 | C | 2.27 | 27.1 | 1 | – | 2,189 | 43 | 3 |
| 18 | F. nucleatum subsp. vincentii 4_1_13 | I | 2.25 | 26.9 | 50 | 106,163 | 2,207 | 43 | 5 |
| 19 | F. nucleatum subsp. vincentii ATCC 49256 | I | 2.12 | 27.3 | 302 | 14,248 | 2,321 | 44 | 12 |
| 20 | F. nucleatum subsp. vincentii ChDC F8 | I | 1.99 | 26.9 | 186 | 23,738 | 1,833 | 28 | 1 |
| 21 | F. nucleatum subsp. fusiforme ATCC 51190 | I | 1.84 | 27.2 | 198 | 23,781 | 1,778 | 45 | 11 |
Note.—C, complete genome; I, incomplete genome.
In order to know more about the genomic structure of F. nucleatum, we further predicted the pan-genome size by extrapolation through calculation of the gene clusters and core clusters for N genomes, where N is the number of F. nucleatum genomes (N = 1, 2, 3 ….19, 20, 21). To avoid bias, the pan-genome size and core genome for each of the permutations of genome comparisons was predicted. Results show that the pan-genome size of F. nucleatum increased with the increase in the number of genomes. Our mathematical functional model (Y = 1260.48511544134 *X**0.5 + 854.557552658844; where Y = pan-genome size; X = number of genomes added) (Laing et al. 2010) represents the pan-genome of F. nucleatum.
From the curve (fig. 4) generated from this model, we can observe that F. nucleatum contains an open pan-genome, which suggested that F. nucleatum was able to acquire novel genes during the evolution of its genome. From the pan-genome results it was observed that 44% of the genes were strain-specific gene clusters. Further investigation revealed that, among the 21 F. nucleatum genomes, W1481 was the fourth highest in term of the number of strain-specific genes (281) (fig. 5).
Fig. 4.—

Pan-genome and core genome size prediction for 21 F. nucleatum genomes. The blue diamonds, represent the functional pan-genome size for each combination and the blue dotted line represents the relationship between the number of genomes and functional pan-genome size. The green dots represent the core functional clusters for each combination and the green dotted line represents the relationship between the number of genomes and the number of core functional cluster.
Fig. 5.—

Strain-specific genes for the 21 Fusobacterium nucleatum genomes.
Interestingly, except for one bacteriophage-type DNA polymerase and one phage-related protein, W1481 did not show the presence of any other phage-related proteins. This might be attributed to the presence of abortive infection proteins AbiGI and AbiGII which were present only in W1481. The abortive infection (Abi) system limits phage replication within a bacterial population by promoting cell death (O’Connor et al. 1999; Dy et al. 2014). W1481 also showed the presence of two distinct restriction modification system (RM) Type I and III (Murray 2000; Rao et al. 2014), as well as CRISPR-Cas system (Horvath and Barrangou 2010; Marraffini and Sontheimer 2010; Jore et al. 2012), conferring resistance against foreign genetic elements such as plasmids and bacteriophages. This was further supported by the fact that no prophage was detected in the W1481 genome by PHAST (PHAge Search Tool) (Zhou et al. 2011). Additionally, W1481 is equipped with a number of efflux systems. W1481 contains the macrolide efflux system (MacA and MacB) along with TolC. In Escherichia coli MacA and MacB, along with TolC, have been reported to confer resistance to erythromycin (Kobayashi et al. 2001). W1481 also carries genes encoding beta-lactamase, beta-lactamase domain protein, beta-lactamase-like protein and Zn-dependent hydrolase, thus, constituting a beta-lactamase superfamily protein in the genome, indicating that W1481 might be resistant against beta-lactam antibiotics. It also showed the presence of Spectinomycin 9-O-adenylyltransferase and a number of multi-drug resistance proteins and efflux pumps such as multidrug resistance protein 2, permease of the drug/metabolite transporter (DMT) superfamily, multi-antimicrobial extrusion protein (Na(+)/drug antiporter), MATE family of MDR efflux pumps and Na+ driven multidrug efflux pump. We also found the presence of hemolysin and putative large exoprotein involved in heme utilization or adhesion of ShlA/HecA/FhaA family in the W1481 genome.
W1481 showed the presence of ATP-dependent oligopeptide transporter OppABCDF, a member of the ATP-Binding Cassette (ABC) Superfamily of transporters (Pearce et al. 1992). OppABCDF have been reported to transport oligopeptides up to five amino acids in length but not free amino acids as shown by studies based on binding affinity and competition assays (Guyer et al. 1986). The system is involved in the uptake of oligopeptides, as well as recycling of cell wall peptides (Hiles et al. 1987). OppA is the periplasmic substrate-binding component that binds oligopeptides (Tame et al. 1994), OppB and OppC are the membrane components of the ABC transporter, whereas OppD and OppF are the ATP-binding components of the ABC transporter (Pearce et al. 1992). Another ABC transport system present in W1481 is the nik operon which is responsible for uptake of nickel ions, an essential nutrient participating in various cellular processes. The first five genes of the nik operon (nikABCDE) were identified in W1481, however, nikR the DNA-binding protein repressing the transcription of nikABCDE (Navarro et al. 1993) was not detected in W1481.
We also found two putative genes, ltrA (encoding low temperature requirement protein A). These ltrA genes were not present in the other F. nucleatum strains. ltrA genes are essential for bacterial growth at low temperature (Zheng and Kathariou 1995). We suggest that the presence of these genes in W1481 might allow it to survive in lower temperatures as compared to other F. nucleatum strains thus enabling it to survive in more diverse environments in term of temperature.
Genomic Island Prediction
Genomic islands were predicted for the genome of W1481 together with the other 20 F. nucleatum genomes by the IslandViewer 3 software. A total of 91 nonredundant GIs were predicted in all F. nucleatum genomes used in this study (supplementary table S2, Supplementary Material online). Of the 91 putative GIs, two GIs (GI41 and GI73) were conserved across all F. nucleatum genomes. Inside GI43, there are two genes which encode for pyruvate formate-lyase enzyme. Insertion of these GI to the F. nucleatum genomes allow catalyzation of the reversible conversion of pyruvate and coenzyme-A into formate and acetyl-CoA (Becker et al. 1999). For the W1481 genome, we found 23 GIs, of which three GIs (GI1, GI7, and GI10) were only present in W1481, and not in other F. nucleatum strains (supplementary table S2, Supplementary Material online). We found the presence of putative genes code for adenosylcobinamide amidohydrolase, which is required in the salvage pathway of cobinamide (Woodson and Escalante-Semerena 2004), and the components of cobalt chelatase (CobN, ChlI/ChlD) involved in B12 (cobalamin) biosynthesis (Rodionov et al. 2003) in the GI1. We have also found L(+)-tartrate dehydratase alpha subunit gene and L(+)-tartrate dehydratase beta subunit gene in another W1481 specific GI (GI7). These genes are involved in the catabolism of lactate and succinate (Do et al. 2015). Thus, GI1 and GI7 may allow W1481 to synthesize or catabolize cobinamide, lactate and succinate.
In addition, we also observed some interesting genes and features in the GIs shared between W1481 and some of the F. nucleatum strains. For example, we found that the horizontally transferred GI3, which was shared by W1481 with the strains of subsp. animalis (11_3_2), subsp. fusiforme (ATCC51190) and subsp. nucleatum (ATCC23726), carries the tripartite ATP-independent (TRAP)-like transporter genes, encoding for TRAP-type C4-dicarboxylate transport system-periplasmic component, TRAP-type C4-dicarboxylate transport system-small permease component and TRAP-type C4-dicarboxylate transport system-large permease component. These TRAP transporters are a high-affinity transport system for the C4-dicarboxylates malate, succinate, and fumarate (Forward et al. 1997). Strikingly, the GI4 which was shared by W1481 with the strains of subsp. polymorphum (ATCC10953 & F401) and three strains of subsp. animalis (F0419, D11 & 7_1), contain a cluster of eight putative CRISPRs genes which may provide the host with acquired and heritable resistance to foreign DNA (Horvath and Barrangou 2010; Marraffini and Sontheimer 2010; Jore et al. 2012). The distribution of the GIs among the strains indicates that W1481 share a number of GIs with the strains of subsp. animalis, vincentii and polymorphum.
Comparative Pathogenomic Analysis
To better understand the pathogenic potential of W1481, we performed a comparative pathogenomic analysis of W1481 along with the strains of the five subspecies of F. nucleatum (fig. 6). W1481 showed the presence of a number of virulence genes which were shared by all or most of the other strains under study mostly related to lipopolysaccharide layer of the bacterial cell surface, such as galE (UDP-glucose 4-epimerase), galU (glucose-1-phosphate uridylyltransferase), gtaB (UTP–glucose-1-phosphate uridylyltransferase), acpXL (acyl carrier protein), kdsA (2-dehydro-3-deoxyphosphooctonate aldolase), lpxA (UDP-N-acetylglucosamine acyltransferase), gmhA/lpcA (phosphoheptose isomerase) and eno (phosphopyruvate hydratase). W1481 also shared the presence of groEL a 60-kDa chaperonin, htpB (Hsp60, 60K heat shock protein), hemL (glutamate-1-semialdehyde aminotransferase), tuf (elongation factor Tu) and plr/gapA (glyceraldehyde-3-phosphate dehydrogenase) with the other strains.
Fig. 6.—
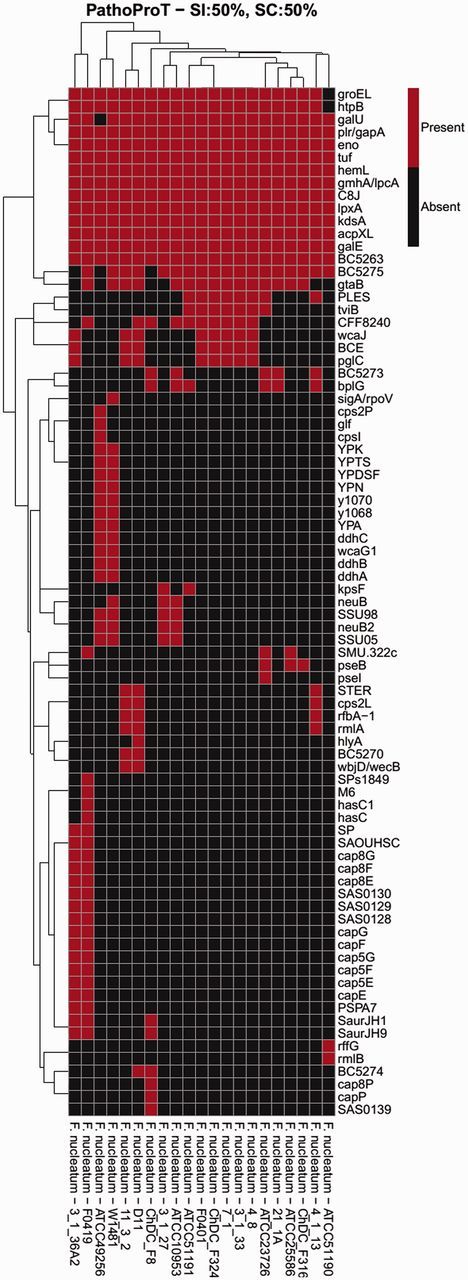
Comparative pathogenomic analysis of W1481. The threshold used in this analysis was 50% sequence identity and 50% sequence completeness.
One interesting observation was the presence of ddhABC genes which are related to CDP-glucose pathways. These genes were present in W1481 and only one other strain of F. nucleatum subsp. vincentii ATCC49256. ddhABC genes are involved in lipopolysaccharide O-antigen biosynthesis which have been reported to play a crucial role in effective colonization of host tissues, and evasion of the host immune system by gram negative bacteria (Lerouge and Vanderleyden 2002; Skurnik and Bengoechea 2003). None of the other strains under study showed the presence of these genes. W1481 also showed the presence of Putative O-antigen ligase WaaL which is responsible for the ligation of O-antigen onto the core region of the lipid A-core block a crucial step in the lipopolysaccharide biosynthetic pathway (Han et al. 2012).
Another interesting set of genes were that of neuramic acid synthetase (neuB and neuB2) reported to be catalyzing the last step of the de novo sialic acid biosynthetic pathway, which is a major element of bacterial surface structure (Feng et al. 2012). Again these were present in only W1481 and two strains of F. nucleatum subsp. vincentii (ATCC49256 and 3_1_27) and one other strain, F. nucleatum subsp. polymorphum ATCC10953. It has been suggested that bacterial pathogens employ the strategy of “molecular mimicry” by incorporating sialic acid on their cell-surface to disguise themselves as host cells to circumvent and/or counteract the host’s immune responses (Severi et al. 2007). W1481 also possess Alpha-2-macroglobulin (A2M) which are located in the periplasm and are believed to trap external proteases through a covalent interaction with an activated thioester thus providing protection to the cell. Studies in Salmonella enterica ser. typhimurium have shown that A2M mimics the innate immune system of eukaryotes (Wong and Dessen 2014). The comparative analysis results suggest that W1481 might be slightly more effective in colonization and resisting the host immune system.
Conclusion
The results of phylogenetic and MUMi distance analyses all suggest the possibility that W1481 is likely a novel subspecies of F. nucletum. W1481 has several unique horizontally transferred GIs, indicating the HGT may play an important role in the evolution of this bacterium. Comparative analysis suggests that W1481 is well equipped to defend itself, colonize and survive within the host evading its immune system, although experimental verification is required to confirm these observations. This comparative in silico study provides novel insights into the genome of W1481 and lays the foundation for future experimental studies.
Supplementary Material
Acknowledgments
Fusobacterium nucleatum W1481 was originally isolated by William Wade (currently at Barts and the London School of Medicine and Dentistry, Queen Mary, University of London). We would like to thank all members of Genome Informatics Research Group (GIRG) in contributing to this research. The work was supported by University of Malaya and Ministry of Education, Malaysia under the High Impact Research (HIR) grant (grant number: UM.C/625/HIR/MOHE/CHAN-08) and UMRG grant from University of Malaya and Ministry of Education, Malaysia (grant number: RG541-13HTM).
Literature Cited
- Altschul SF, et al. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Ang MY, et al. 2014. Genome sequence of Fusobacterium nucleatum strain W1481, a possible new subspecies isolated from a periodontal pocket. Genome Announc. 2: 1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz RK, et al. 2008. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9:75.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann W, Gregor uH. 1936. Kulturelle und immunbiologische Differenzierung von Stämmen der Gruppe “Fusobakterium”. Z Immun Forsch. 8 238–251. [Google Scholar]
- Becker A, et al. 1999. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat Struct Biol. 6:969–975. [DOI] [PubMed] [Google Scholar]
- Castellarin M, et al. 2012. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, et al. 2005. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33:D325–D328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Xiong Z, Sun L, Yang J, Jin Q. 2012. VFDB 2012 update: toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 40:D641–D645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Salzberg SL, Phillippy AM. 2003. Using MUMmer to identify similar regions in large sequence sets. Curr Protoc Bioinformatics Chapter 10:Unit 10 13. [DOI] [PubMed] [Google Scholar]
- Deloger M, El Karoui M, Petit MA. 2009. A genomic distance based on MUM indicates discontinuity between most bacterial species and genera. J Bacteriol. 191:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T, Sheehy EC, Mulli T, Hughes F, Beighton D. 2015. Transcriptomic analysis of three Veillonella spp. present in carious dentine and in the saliva of caries-free individuals. Front Cell Infect Microbiol. 5:25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy RL, Przybilski R, Semeijn K, Salmond GP, Fineran PC. 2014. A widespread bacteriophage abortive infection system functions through a type IV toxin-antitoxin mechanism. Nucleic Acids Res. 42:4590–4605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright AJ, Van Dongen S, Ouzounis CA. 2002. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 30:1575–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, et al. 2012. Attenuation of Streptococcus suis virulence by the alteration of bacterial surface architecture. Sci Rep. 2:710.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forward JA, Behrendt MC, Wyborn NR, Cross R, Kelly DJ. 1997. TRAP transporters: a new family of periplasmic solute transport systems encoded by the dctPQM genes of Rhodobacter capsulatus and by homologs in diverse gram-negative bacteria. J Bacteriol. 179:5482–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharbia SE, Shah HN. 1990. Identification of Fusobacterium species by the electrophoretic migration of glutamate dehydrogenase and 2-oxoglutarate reductase in relation to their DNA base composition and peptidoglycan dibasic amino acids. J Med Microbiol. 33:183–188. [DOI] [PubMed] [Google Scholar]
- Goris J, et al. 2007. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 57:81–91. [DOI] [PubMed] [Google Scholar]
- Guyer CA, Morgan DG, Staros JV. 1986. Binding specificity of the periplasmic oligopeptide-binding protein from Escherichia coli. J Bacteriol. 168:775–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, et al. 2012. Defining function of lipopolysaccharide O-antigen ligase WaaL using chemoenzymatically synthesized substrates. J Biol Chem. 287:5357–5365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiles ID, Gallagher MP, Jamieson DJ, Higgins CF. 1987. Molecular characterization of the oligopeptide permease of Salmonella typhimurium. J Mol Biol. 195:125–142. [DOI] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. 2010. CRISPR/Cas, the immune system of bacteria and archaea. Science 327:167–170. [DOI] [PubMed] [Google Scholar]
- Hsiao W, Wan I, Jones SJ, Brinkman FS. 2003. IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19:418–420. [DOI] [PubMed] [Google Scholar]
- Huggan PJ, Murdoch DR. 2008. Fusobacterial infections: clinical spectrum and incidence of invasive disease. J Infect. 57:283–289. [DOI] [PubMed] [Google Scholar]
- Jore MM, Brouns SJ, van der Oost J. 2012. RNA in defense: CRISPRs protect prokaryotes against mobile genetic elements. Cold Spring Harb Perspect Biol. 4: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi N, Nishino K, Yamaguchi A. 2001. Novel macrolide-specific ABC-type efflux transporter in Escherichia coli. J Bacteriol. 183:5639–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. 2005. Genomic insights that advance the species definition for prokaryotes. Proc Natl Acad Sci U S A. 102:2567–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppalli K, Livorsi D, Talati NJ, Osborn M. 2012. Lemierre’s syndrome due to Fusobacterium necrophorum. Lancet Infect Dis. 12:808–815. [DOI] [PubMed] [Google Scholar]
- Laing C, et al. 2010. Pan-genome sequence analysis using Panseq: an online tool for the rapid analysis of core and accessory genomic regions. BMC Bioinformatics 11:461.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Brinkman FS. 2009. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics 25:664–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MG, Hsiao WW, Brinkman FS. 2008. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 9:329.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerouge I, Vanderleyden J. 2002. O-antigen structural variation: mechanisms and possible roles in animal/plant-microbe interactions. FEMS Microbiol Rev. 26:17–47. [DOI] [PubMed] [Google Scholar]
- Manson McGuire A, et al. 2014. Evolution of invasion in a diverse set of Fusobacterium species. MBio 5:e01864.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. 2010. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 11:181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer F, Overbeek R, Rodriguez A. 2009. FIGfams: yet another set of protein families. Nucleic Acids Res. 37:6643–6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris ML, Andrews RH, Rogers AH. 1996. The use of allozyme electrophoresis to assess genetic heterogeneity among previously subspeciated isolates of Fusobacterium nucleatum. Oral Microbiol Immunol. 11:15–21. [DOI] [PubMed] [Google Scholar]
- Murray NE. 2000. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol Mol Biol Rev. 64:412–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro C, Wu LF, Mandrand-Berthelot MA. 1993. The nik operon of Escherichia coli encodes a periplasmic binding-protein-dependent transport system for nickel. Mol Microbiol. 9:1181–1191. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Tangney M, Fitzgerald GF. 1999. Expression, regulation, and mode of action of the AbiG abortive infection system of Lactococcus lactis subsp. cremoris UC653. Appl Environ Microbiol. 65:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce SR, et al. 1992. Membrane topology of the integral membrane components, OppB and OppC, of the oligopeptide permease of Salmonella typhimurium. Mol Microbiol. 6:47–57. [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. 2007. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35:D61–D65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DN, Dryden DT, Bheemanaik S. 2014. Type III restriction-modification enzymes: a historical perspective. Nucleic Acids Res. 42:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodionov DA, Vitreschak AG, Mironov AA, Gelfand MS. 2003. Comparative genomics of the vitamin B12 metabolism and regulation in prokaryotes. J Biol Chem. 278:41148–41159. [DOI] [PubMed] [Google Scholar]
- Severi E, Hood DW, Thomas GH. 2007. Sialic acid utilization by bacterial pathogens. Microbiology 153:2817–2822. [DOI] [PubMed] [Google Scholar]
- Sievers F, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 7:539.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skurnik M, Bengoechea JA. 2003. The biosynthesis and biological role of lipopolysaccharide O-antigens of pathogenic Yersiniae. Carbohydr Res. 338:2521–2529. [DOI] [PubMed] [Google Scholar]
- Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr. 1998. Microbial complexes in subgingival plaque. J Clin Periodontol. 25:134–144. [DOI] [PubMed] [Google Scholar]
- Spaulding EH, Rettger LF. 1937a. The Fusobacterium genus: I. Biochemical and serological classification. J Bacteriol. 34:535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding EH, Rettger LF. 1937b. The Fusobacterium genus: II. Some observations on growth requirements and variation. J Bacteriol. 34:549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tame JR, et al. 1994. The structural basis of sequence-independent peptide binding by OppA protein. Science 264:1578–1581. [DOI] [PubMed] [Google Scholar]
- Tamura K, et al. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 28:2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waack S, et al. 2006. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 7:142.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SG, Dessen A. 2014. Structure of a bacterial alpha2-macroglobulin reveals mimicry of eukaryotic innate immunity. Nat Commun. 5:4917.. [DOI] [PubMed] [Google Scholar]
- Woodson JD, Escalante-Semerena JC. 2004. CbiZ, an amidohydrolase enzyme required for salvaging the coenzyme B12 precursor cobinamide in archaea. Proc Natl Acad Sci U S A. 101:3591–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Kathariou S. 1995. Differentiation of epidemic-associated strains of listeria-monocytogenes by restriction-fragment-length-polymorphism in a gene region essential for growth at low-temperatures (4-degrees-C). Appl Environ Microbiol. 61:4310–4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39:W347–W352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.