

Abstract
One of the largest multigene families in Metazoa are the tyrosine kinases (TKs). These are important multifunctional proteins that have evolved as dynamic switches that perform tyrosine phosphorylation and other noncatalytic activities regulated by various allosteric mechanisms. TKs interact with each other and with other molecules, ultimately activating and inhibiting different signaling pathways. TKs are implicated in cancer and almost 30 FDA-approved TK inhibitors are available. However, specific binding is a challenge when targeting an active site that has been conserved in multiple protein paralogs for millions of years. A cassette domain (CD) containing SH3–SH2–Tyrosine Kinase domains reoccurs in vertebrate nonreceptor TKs. Although part of the CD function is shared between TKs, it also presents TK specific features. Here, the evolutionary dynamics of sequence, structure, and phosphorylation across the CD in 17 TK paralogs have been investigated in a large-scale study. We establish that TKs often have ortholog-specific structural disorder and phosphorylation patterns, while secondary structure elements, as expected, are highly conserved. Further, domain-specific differences are at play. Notably, we found the catalytic domain to fluctuate more in certain secondary structure elements than the regulatory domains. By elucidating how different properties evolve after gene duplications and which properties are specifically conserved within orthologs, the mechanistic understanding of protein evolution is enriched and regions supposedly critical for functional divergence across paralogs are highlighted.
Keywords: tyrosine kinase, gene duplication, intrinsic disorder, protein evolution, allostery, evolutionary dynamics
Introduction
Protein kinases are critical to intra- and extracellular signaling and act as important regulators of a wide variety of functions in a proteome by providing specific phosphorylation. In human, >8,000 proteins are known to be phosphorylated (Gnad et al. 2011). While most phosphorylations are catalyzed by the more ancient Ser/Thr kinase (STK) (Jin and Pawson 2012), the tyrosine kinase (TK) family has undergone a vast expansion in early Metazoa (Manning, Plowman et al. 2002), followed by different lineage-specific expansions. In particular, after Amphioxus diverged from the Chordate lineage, many TKs have at least duplicated in number (Putnam et al. 2008) leading to a repertoire of close homologs in vertebrates. It is estimated that ∼2% of human genes code for >500 different kinases (Manning, Whyte et al. 2002). TKs regulate many biochemical activities and participate in numerous cell signaling pathways involved in cell growth, division, migration and survival (Mano 1999; Serfas and Tyner 2003; Colicelli 2010; Bononi et al. 2011; Okada 2012). Given that kinases are key members of signaling cascades, phosphorylation must proceed with accuracy. In humans, kinase phosphorylation going awry is associated with cancer [for instance, ABL1 (Greuber et al. 2013), ITK (Hussain et al. 2011)] and immunodeficiency (for instance, BTK in B-cells; Hussain et al. 2011). To date, the US Food and Drug Administration (FDA) has approved 28 small-molecule kinase inhibitors, many of them targeting TKs (Wu et al. 2015). Almost all were developed for cancer treatment, binding with different degrees of specificity. These inhibitors are predominantly bound in, or close to, the active site, either in a kinase’s active or inactive conformation (Wu et al. 2015). Since the kinase domain’s active site must catalyze phosphorylation, the binding pocket is conserved, making kinase-specific binding of drugs hard to achieve. To develop kinase specific drugs, different approaches to inhibit a constitutively active kinase are being explored that target other areas of the kinase instead of the active site region. Some approaches aim to inhibit the kinase by targeting a region that is important for the allosteric effect (Gavrin and Saiah 2013; Cowan-Jacob et al. 2014), but where most of the allosterically important regions are located and how these regions evolve among different TKs is not known.
Tyrosine Kinases and Allostery
Tyrosine kinases are often allosterically regulated proteins that propagate conformational or vibrational changes from an effector site to an active site (Kornev and Taylor 2015). Kinases have evolved to be transiently activated to function as dynamic switches (Taylor et al. 2012). Thus, they have at least one inactive state and one active state, but numerous conformations may be present as a conformational ensemble. The allosteric modulation of the tyrosine kinase domain (TyrK) involves different regions of the protein. Some allosteric effectors are post-translational modifications, while others stem from intra- or intermolecular interactions often mediated through surface exposed interfaces. The interfaces involved in allosteric effects frequently include linker regions. For instance, the binding “cap” in ABL blocks catalysis (Corbi-Verge et al. 2013), regulatory domains such as SH3 domains (PXXP motifs binders) cause inhibition, and SH2 domains can activate or inhibit depending on the interface of binding (Nagar et al. 2003). Interactions with other domains, such as F-actin domains in the ABL family, can also allosterically regulate kinase activity (Woodring et al. 2005). Phosphorylation of the kinase itself can induce disorder-order transitions, ultimately activating or inhibiting the kinase. Phosphorylation may block catalysis, for example, a phosphorylated Tyr residue in the C-terminal tail of SRC proteins can prevent catalysis by binding to the SH2 domain (Okada 2012), or a phosphorylation in the activation loop (A-loop) is required for catalysis in some TKs (Yamaguchi and Hendrickson 1996). The A-loop is a highly dynamic segment in the middle of TyrK. Further, allosteric modulation might occur via assembly/disassembly of hydrophobic residues, called spines, at the core of TyrK linking the conformational dynamics of both lobes (Taylor et al. 2012). Lastly, structurally disorder residues, that is, residues with low propensity to form stabilizing contacts in 3D space, play a major role in the overall stability and ability to form intra- and intermolecular contacts of a protein (Galea et al. 2008; Boehr et al. 2009; Uversky 2011). Thus, the amount and location of disorder fragments may be involved in the allosteric regulation via disorder-order transitions modulated by phosphorylation and through biomolecular contacts, in regions far from the active site. Structural disorder has been found experimentally for various TK paralogs in PDB (Berman et al. 2000). For the cassette domain (CD) region, structural disorder has been experimentally identified, for example, in the A-loop, in the long linker connecting SH2–TyrK domains, and in loops within the SH2 domains, but the disordered regions vary depending on conformation and protein.
The Cassette Domain
In addition to the TyrK domain that allows for phosphorylation activity, TKs have been found within a variety of domain architectures (Jin and Pawson 2012). Besides TyrK, the domain architecture often contains domains involved in regulating the kinase activity and in recognizing phosphorylation substrates. TKs are further divided into receptor or nonreceptor TKs. Among the nonreceptor TKs are five kinase families [ABL, SRC, TEC, CSK, and FRK (a.k.a. SRC-like)] (fig. 1A) that have undergone further expansion in vertebrates (D’Aniello et al. 2008). Most members of these five kinase families share a larger domain architecture, namely the SH3 (fig. 1B), SH2 (fig. 1C), and TyrK (fig. 1D and E) domains. The three domains together are frequently referred to as the CD. Despite shared domains, functional and regulatory divergence has been reported, together with different cellular locations (Sato et al. 2009; Kim et al. 2014), substrate specificities (al-Obeidi et al. 1998; Deng et al. 2014), and signaling pathways (Liu and Nash 2012; Gocek et al. 2014). Some of this functional divergence can be explained by the additional domains, sequence divergence, and structural effects, but the extent of divergence in the allosteric kinase networks is still an open question.
Fig. 1.—

Domain architectures and folds of human CD-containing tyrosine kinase (TK) families. (A) Consensus domain architectures for five nonreceptor TK families (ABL, TEC, SRC, FRK, and CSK) with a common cassette domain (CD). The different CD regions are colored as follows (from N- to C-term): linker 1 (L1): gray, SH3 domain: green, linker 2 (L2): dark blue, SH2 domain: orange, linker 3 (L3): cyan, TyrK domain (N- and C-lobe): purple, and linker 4 (L4): red. Additional regions outside the CD are colored in black. M-like symbols denote fatty acid modifications involved in membrane anchoring and allostery. Green circles denote PXXP motifs (SH3-binders) and orange stars denote phospho-Tyrosines (p-Tyr, SH2-binders) involved in regulation of the activation loop (A-loop) in the C-lobe and/or the C-term tail. (B and C) 3D structures of individual domains: (B) SH3 domain (5-6 β-strands) colored as in (A) (ABL1, PDB id:1opl; Nagar et al. 2003) and (C) SH2 domain (α-helix, 3 β-strands, α-helix) colored as in (A) (ABL1, PDB id:1opl; Nagar et al. 2003). (D and E) 3D structures of the TyrK domain, inactive and active states. On top, cartoon representations illustrate the intramolecular interactions between domains and linkers. Functional regions involved in regulation and catalysis are color-coded and labeled. (D) Inactive state (SRC, PDB code:2src; Xu et al. 1999) and (E) Active state (SRC, PDB code:1y57; Cowan-Jacob et al. 2005).
Here, we take an evolutionary approach to studying the potentially dynamic regions in the CD of 17 nonreceptor TKs, predominantly focusing on vertebrates. This study does not explicitly study dynamics from actual 3D models of protein structure, but utilizes high-quality sequence data to extract information. Through an approximation gained from predicting sites where intrinsic disorder is present based on the primary sequences alone, we can infer what regions may be involved in providing dynamic effects for regulation and allostery. Further, we approximate secondary structure and the presence of phosphorylation sites by predictions. We hypothesize that regions of structural disorder and phosphorylation sites are less conserved among paralogs than within orthologs. Further, we hypothesize that secondary structure is conserved across paralogs as structure is more conserved than sequence and maintaining the fold is important for the catalytic function. Thus, clade-specific signatures of structural disorder and phosphorylation within orthologs but not amongst paralogs may indicate functional, in particular, regulatory and allosteric divergence, while the main fold of the CDs remains conserved. To test these hypotheses, we investigate the evolutionary dynamics of structural disorder, phosphorylation, and secondary structure among the 17 different CD-containing TK clades. While this analysis shines new light on how structural disorder and phosphorylation evolve in an extended kinase family with numerous orthologs, it also highlights clade-specific regions that are of potential allosteric importance and opens up new avenues for informed drug development of kinase-specific inhibitors.
Results
Phylogenetic Reconstruction
Two different phylogenies were built for the CD region. The first phylogeny was built with sequences that had a minimum of 70% sequence identity and at least 90% query coverage to 1 of the 17 human CD regions (supplementary table S1, Supplementary Material online). This phylogeny and its corresponding multiple sequence alignment contain 543 bilaterian sequences and will be referred to as 70-90. The second phylogeny was built with sequences that had a minimum of 50% sequence identity and at least 90% query coverage to 1 of the 17 human CD regions (supplementary table S2, Supplementary Material online). This phylogeny and its corresponding multiple sequence alignment contain 655 sequences ranging from mammals to the choanoflagellate Monosiga brevicollis (fig. 2A and supplementary figs. S1 and S2, Supplementary Material online) and will be referred to as 50-90. Both trees define the 17 different CD-containing paralogs, with minor disagreement within the SRC-A subfamily regarding FYN and FGR being sister clades. Some disagreement can be expected as the sequence composition changes for the different sets (supplementary table S3, Supplementary Material online). Mostly, there are expanded clades in the 50-90 tree: adding M. brevicollis and invertebrates to the CSK, ABL, and FRK families, and adding invertebrates to TEC and SRC families. Additional vertebrates, with the majority being ray-finned fish, are added to MATK, FRK, and to the TEC family, where an additional sister clade to BLK (called BMX) is formed containing mammals, reptiles, and lobe-finned fish, but no ray-finned fish. The FGR clade also remains devoid of ray-finned fish (fig. 2B). At 30% sequence identity, no additional vertebrate proteins were found in any of the 17 clades under analysis compared with 50-90. While tree 50-90 gives a more ancestral view of the TKs, the MSA for this more divergent set is much longer (length of MSA: 1,061 positions, ungapped sites: 72) compared with the less divergent 70-90 set (length of MSA: 644 positions, ungapped sites: 204) for the same CD region. Tree 70-90 consists predominantly of vertebrate sequences but does go back far enough to include some invertebrate sequences in the ABL family (supplementary fig. S1, Supplementary Material online).
Fig. 2.—

Protein phylogeny and distribution of vertebrates per clade. (A) Circular representation of the consensus tree of a Cassette Domain (CD) protein phylogeny under the cut-offs of 70% sequence identity and 90% CD coverage of human proteins (70-90 set) obtained with MrBayes (Ronquist et al. 2012). Branch lengths represent sequence divergence. Posterior probabilities per node are shown as circles following the color code on the left. Nodes with posterior probability < 0.5 are unresolved. Protein families are colored as follows: CSK: yellow, TEC: blue, ABL: red and SRC–A: light green, and SRC-B dark green. Different paralogs within the protein families are labeled. For detailed phylogenies, see supplementary figures S1 and S2, Supplementary Material online; (B) Number of sequences per vertebrate taxa for the paralogs in (A) (0: white, 1: light orange, 2: dark orange, 3: brown). The black circles denote sequences that are missing in the 70-90 set, but present in the 50-90 set (at 50% sequence identity and 90% CD coverage cut-offs). Color-schemes account for protein families (on top) as described in A, and species groups (on the right): ray-finned fish: dark blue, lobe-finned fish: cyan, amphibian: green, reptiles and birds: pink, nonplacental mammals: brown, placental mammals (nonprimates): orange and primates: black.
The 70-90 tree is mostly well-resolved with only weak branch support within clades of highly similar or identical sequences, such as in ABL and SRC-A families. Moreover, discrepancies were identified when comparing our phylogenetic trees to the common species tree. Some species, such as Xenopus tropicalis and Latimeria chalumnae, do not follow the common species tree in all clades. In some cases, this may be due to lineage-specific indels or due to lack of resolution in that region of the phylogenetic tree.
Further, for each of the 17 different paralogs in vertebrates, the sub-tree from tree 70-90 and its corresponding block of the multiple sequence alignment were extracted. The resulting number of sequences per paralog is ABL1 (35), ABL2 (36), CSK (36), FGR (24), FYN (48), HCK (30), LYN (34), SRC (38), YES (38), BLK (27), BTK (31), FRK (23), ITK (28), LCK (28), MATK, (21), TEC (29), TXK (26). Invertebrate proteins were discarded from the remaining paralogs versus orthologs analysis. The block from the complete 70-90 multiple sequence alignment that corresponds to a paralogous clade is not re-aligned, allowing for site specific comparison between the different clades, and consequently, all sub-trees are from the same phylogeny enabling site-specific comparison across clades.
Sequence Conservation across Paralogs and within Orthologs
Pairwise sequence identities for the human proteins in the different clades show that CSK paralogs are the most divergent (53% identical), and ABL paralogs are the most evolutionary constrained (91% identical), followed by SRC-A paralogs. Sequence redundancy (100% identity) within orthologs is high for ABL1, ABL2, FYN, SRC, and YES kinases (37%, 36%, 27%, 11%, and 11%, respectively), showing a phylogenetic subtree with very short branch lengths and poorly supported nodes. The most divergent human CD pairs in the data set (MATK-TXK and MATK-TEC) are 33% identical.
Comparisons of sequence conservation patterns within orthologs and between the 17 paralogs of the same family are generally in agreement (supplementary fig. S3, Supplementary Material online). Site-specific amino acid evolutionary rates (SEQ) were estimated from the tree and alignment sets from the 17 paralogous groups (supplementary fig. S4, Supplementary Material online). Despite an overall high conservation, rapidly evolving sites are found involving residue substitutions as well as indels. Sites with rapid SEQ are located at the end of C-lobe in the catalytic domain and in regulatory regions such as the SH2 domain and linker L3 (fig. 3B). In the highly conserved clades ABL1 and ABL2, three sequences (two from ABL1 and one from ABL2) cause elevated evolutionary rates in the TyrK domains.
Fig. 3.—

Conservation profiles of structural predictions for tyrosine kinase paralogs. (A) Profiles of conservation fractions per site for the 17 paralogous groups showing shared and divergent distributions of structural propensities and phosphorylations. Fractions are obtained based on binary predictions mapped per alignment site for structural disorder (red), secondary structure (blue), and phosphorylation sites (black). In all cases gaps are included in the calculation. (B) Profiles of the fraction of gapped positions for the 17 paralogous groups combined with sequence conservation per site for each group. Normalized SEQ rates are codified into three categories: slow evolving sites (blue), nearly neutral (white), and fast evolving sites (green) for rates <0, between 0 and 1 or > 1, respectively. On the right side of the panels, a color guide per family as in figure 2 is included.
Conservation of Structural Disorder
Structural disorder propensities were predicted and mapped onto the MSA in the phylogenetic context (fig. 4A and B and supplementary fig. S6A, Supplementary Material online). While the graphical representation illustrates predicted continuous disorder propensity and provides an overview of how disorder propensity is changing between sequences and clades, a quantitative measure is needed. Establishing the number of residues with disorder propensities <0.4 as ordered allows us to quantify the fraction of order versus disordered residues in the different proteins (supplementary fig. S5, Supplementary Material online). By comparing the distribution of the fraction of disordered residues per protein and per CD in all versus all clades, statistically significant differences are obtained for the full-length ABL paralogs and for the CD region in MATK (fig. 5). In both cases, these outliers are more disordered than the others. Experimental evidence support high amounts of disorder in ABL proteins that present an extremely long disordered region between TyrK and F-actin domains (de Oliveira et al. 2015) and in MATK proteins that use a noncatalytic mechanism of inhibition by binding SRC proteins in different active conformations (Chong et al. 2006), suggesting their enhanced conformational flexibility.
Fig. 4.—

Sequence-based predictions for the cassette domain region of tyrosine kinases. (A) Cartoon representation of the 70-90 CD-based phylogenetic tree (colored according to fig. 2). Horizontal width represents sequence divergence. Paralogs are labeled with the number of taxa per group in brackets. (B–D) Heat maps for predicted structural traits and phosphorylation plotted in the order of the phylogenetic tree. The heat maps show sequence-based predictions mapped to their corresponding residue sites in the multiple sequence alignment (543 taxa in rows and 644 alignment positions in columns, gaps in the alignment are colored in black). Vertical thick lines lining the heat maps are color-coded by TK family as in figure 2 and positioned to reflect the correspondence between tree and heatmap. (B) Continuous structural disorder propensities by IUPred (Dosztányi, Csizmok, et al. 2005a) colored according to the gradient below the heat map. The color gradient from blue to white to red mirrors the disorder propensity gradient from low (blue) to high (red), with white being the boundary between order and disorder. (C) Secondary structure predictions by PSIPRED (Jones 1999) displaying loop (white), α-helix (gradient of purple) and β-strand (gradient of orange). Gradients are based on the confidence values of the predictions (lighter colors for lower supported predictions). (D) Sites predicted to be phosphorylated by NetPhos (Blom et al. 1999) using a 0.75 cut-off (predicted p-Ser/p-Thr in green and p-Tyr in purple). Above the heat maps, a consensus domain architecture is shown to delimitate Pfam domain regions colored as in figure 1 and on the top-left a cartoon representation of the CD. For larger heatmap representations, see supplementary figs. S6 and S7, Supplementary Material online.
Fig. 5.—

Distribution of structural disorder across Tyrosine Kinase paralogs. Box plots of the distribution of structural disorder per paralogous group in (A) full-length proteins, and (C) in cassette domains (CDs). Paralogs are colored according to the family color scheme from figure 2 and numbered following the order of the cartoon tree shown in the box. Structural disorder is based on IUPred (Dosztányi, Csizmok, et al. 2005a) using a 0.4 cut-off to define binary states of order or disorder. (B and D) Statistical significance networks based on pairwise Mann–Whitney test with Bonferroni correction. Pairs with P value >0.05 are not significantly different in means and are shown as linked nodes: (B) significance inference of disorder distribution in full-length proteins and (D) in CDs.
However, a similar amount of disordered residues does not necessarily mean that the locations of disorder in different orthologs and paralogs are conserved. In fact, in the entire MSA 70-90, no disordered site is conserved across all paralogs: only 1 and 9 site(s) are found to be disorder in at least 75% and 50% of sequences, respectively. On the clade level, these numbers are higher and thus, structural disorder is not conserved across all paralogs.
Based on the disorder predictions, we calculated the fraction of conserved disorder per alignment position, including gapped sites, to generate a profile of disorder conservation and location across the alignment per paralogous group (fig. 3). These profiles indicate that the conserved regions of disorder overlap between clades suggesting that some regions are prone to be disordered. However, not all clades have conserved disorder in all disorder prone regions. Conserved disorder is particularly high in MATK proteins, where disorder accumulates at the beginning and end of the regulatory domains plus at the end of the catalytic domain. The end of the catalytic domain is also in the vicinity of a known allosteric pocket in other TKs such as ABL1 (Hantschel et al. 2003) (fig. 3A). FRK has conserved structural disorder at the end of SH3 domains and in the N-lobe of the TyrK domain and presents a divergent profile of conserved disorder when compared with the other clades (fig. 3A). In the TEC family, all paralogs except TXK present similar disorder conservation in SH2 and SH3 while the TyrK domain is devoid of conserved disorder. In the BTK clade conserved disorder is also predicted in L3, between the regulatory and catalytic domains (fig. 3A). Pairs of paralogs from the SRC family also share similar disorder conservation profiles along the CDs, apart from BLK that has less conserved disorder. Other clades such as FYN and FGR have less disorder in L1, beginning of SH3, and L3 (although FGR has gained disorder at the end of the CD). YES clade has less disorder at the beginning of both SH3 and SH2 domains (fig. 3A). Conservation of disorder propensities within ABL paralogs varies in SH2 and L3 (with conserved disorder in ABL1 but not in ABL2) (fig. 3A).
Conservation of Secondary Structure Elements
The predicted secondary structures for all proteins were mapped onto the MSA (fig. 4C and supplementary fig. S6B, Supplementary Material online). Most secondary structure appears conserved but some regions are changing secondary structure element, for example, at the junction of L3 and the TyrK domain, a region that rapidly switches from helix to strand is located. Profiling the fraction of any secondary structure per site indicates that secondary structure is mostly conserved, but some regions do change in whether they are structured or not across the different kinases in domains and linkers (fig. 3A), especially at the end of SH3, the beginning/end of N-lobe and the central region of the C-lobe including the activation loop in TyrK.
Conservation of Functional Domains
While the starting point for this study was the CD architecture, not all sequences included in our data sets have all three domains according to the Pfam predictions, despite the >90% coverage. Domain conservation and domain loss imply protein function conservation and divergence, respectively. In total, a set of 6 different Pfam domains has been predicted in the CD regions of the 70-90 proteins. The catalytic domain (TyrK) is found in all proteins, mostly as a complete domain, but sometimes as a partial domain and, in rare, cases it is broken due to indels (i.e., two Pfam blocks matching different regions of the HMM profiles) (supplementary fig. S5, Supplementary Material online). The SH2 domain, centrally located in the CD region, was predicted for all sequences. Prediction of the SH3 domain reveals a divergent set: four variants of the SH3 domain (including a bacterial version (SH3_3) in H9GIP7_ANOCA) were predicted by Pfam (supplementary fig. S5, Supplementary Material online). Thus, SH3 shows the highest domain dynamics in the CD region. Also, if we look at the 50-90 tree and the BMX clade that was not present in the 70-90 tree, these sequences are not predicted to have the SH3 domain although the divergent sequence region seems to remain.
Conservation of Phosphorylation Patterns
Phosphorylation sites (phospho-sites) were predicted for all proteins and mapped onto the MSA (fig. 4D, supplementary figs. S6C and S7, Supplementary Material online). Profiling the fraction of phospho-sites predicted with a score ≥0.75, reveals that some sites are highly conserved, but many are clade-specific (fig. 3A). 37 predicted phospho-sites are found in all sequences in at least one clade, but no predicted phospho-site is found across all clades. The predicted phospho-sites are found in the TyrK domain followed by the SH2 domain and linkers. The SH3 domain has few predicted phospho-sites, particularly in CSK, and none in ABL1 and ABL2 (supplementary fig. S6C, Supplementary Material online). 59 predicted phospho-sites are conserved across >90% of sequences in one or more clades. 95 predicted phospho-sites are conserved across >70% of sequences in one or more clades (fig. 6).
Fig. 6.—

Shared and clade-specific conserved phosphorylations patterns across tyrosine kinase paralogs. (A) From the 70-90 multiple sequence alignment (keeping original enumeration), 95 sites with conserved predicted phosphorylation sites within paralogous groups in at least 70% of their taxa are shown. Locations are denoted as linker regions (L1, L2, L3, L4) or domain regions (SH3, SH2, and Tyrosine Kinase (TyrK) domains). Phosphorylation site conservation is based on a prediction score ≥0.75. The Activation loop (A-loop) and the C-tail of the catalytic domain are labeled. On the left, a family color-scheme based on figure 2 is included. Sites located in a domain are colored in gray. In red, sites with conserved phosphorylation predictions not experimentally verified in humans (Predicted), in yellow, sites with conserved phosphorylation predictions experimentally verified in humans (Predicted & PhosphoSitePlus), and in blue, sites of experimental human phospho-sites but predictions are not predicted/conserved across vertebrates (PhosphoSitePlus). Experimental phosphorylations were extracted from PhosphoSitePlus (Hornbeck et al. 2015), as of 4 May 2016. For more detailed information, see supplementary table S4, Supplementary Material online.
At the 0.75 cut-off, 35.3%, 20.6%, and 16.5% of all Ser, Thr, and Tyr residues are predicted to be phosphorylated, p-Ser, p-Thr, and p-Tyr, respectively (supplementary fig. S8, Supplementary Material online). Known phospho-sites identified with low-throughput experiments and/or at least five high-throughput experiments were extracted from the PhosphoSitePlus database (Hornbeck et al. 2015). In total, 147 experimentally validated sites were found for the CD regions of the 17 human TK sequences (23 p-Ser, 17 p-Thr, and 107 p-Tyr), distributed at 60 alignment sites in 70-90. That 147 sites map to 60 alignment sites means some sites are phosphorylated in more than one clade. Above, we noted that a higher percentage of Ser is predicted as p-Ser and that the lowest fraction of phosphorylated predictions is found for p-Tyr. While these percentages are for the entire protein set and not directly comparable to the human sequences only, it should be noted that the total frequency of Ser is higher than for Tyr (supplementary fig. S7, Supplementary Material online). Thus, the number of experimentally validated sites for Ser, Thr, and Tyr, follow the opposite pattern from the predicted phosphorylation sites. For the 147 experimentally validated phospho-sites in the CD region, the phosphorylation prediction scores range from 0.37 to 0.99, and only 45 sites are above the 0.75 cut-off. Comparing the phosphorylation predictions for experimentally validated phospho-sites versus those at any cut-off, present similar bimodal distributions, where data accumulates around intermediate or high scores (supplementary fig. S9, Supplementary Material online), complicating the selection of a valid cut-off. Based on the 0.75 cut-off, it should be noted that many of the proteins studies here are enriched in tyrosine phospho-sites, while some paralogs seem p-Tyr independent. For instance, predicted phospho-sites in MATK clade are restricted to serine and threonine residues with a couple of exceptions (supplementary fig. S7A, Supplementary Material online).
Last, in agreement with the lack of universally conserved predicted phospho-sites, no experimentally validated phospho-sites are found across all human proteins from the 17 clades included here.
Evolutionary Rates across Paralogs
Evolutionary rates for amino acid sequence substitutions (SEQ), and for transitions between disorder and order (DOT), secondary structure and loop (SLT), and presence/absence of a predicted phosphorylation site (PT) were calculated for the entire 70-90 MSA (fig. 7A). Differential locations of rapidly/slowly transitioning sites are found for the properties under analysis. When splitting the data set into clades (supplementary fig. S4, Supplementary Material online), more specific structural patterns were revealed. The correlation coefficients for clade-specific SEQ (fig. 8A) reveal higher correlation for closely related paralogs (i.e., those belonging to the same TK family) with exceptions. For instance, YES lost correlation with many SRC paralogs but still keeps some significant correlation with TEC paralogs (BTK and TEC paralogs). The correlation coefficients for clade-specific SLT (fig. 8B) reveal high overlap across all clades, in general, and especially between members of the same family. The correlation coefficients for clade-specific DOT (fig. 8C) reveal a similar trend as for SEQ. The correlation coefficients for clade-specific PT (fig. 8D) reveal a mosaic of correlations, significantly higher within TEC family and SRC family (with the exception of the SRC paralog that is more divergent even within its subfamily). In FRK, PT correlates significantly with all paralogs except for ABL family and YES. Correlation profiles of CSK paralogs are low in almost all pairwise comparisons. ABL2 is not significantly correlated with other paralogs outside the family and is poorly correlated with ABL1. ABL1 paralogs, on the contrary, still keep moderate correlation with TXK and SRC paralogous groups with the exception of the SRC paralog.
Fig. 7.—

Evolutionary rates of sequence and structural traits in closely related Tyrosine Kinases. (A) Normalized evolutionary rates per site mapped onto the 70-90 alignment sites for amino acid sequence (SEQ) in green versus binary traits of disorder-order transitions (DOT) in orange, secondary structure elements—loop transitions (SLT) in blue, and phosphorylation transitions (PT) in pink. All evolutionary rates were normalized with a mean of zero and standard deviation of 1 (negative rates for slow evolving sites and positive rates for fast evolving sites). Gray-shaded areas delimitate Pfam domain regions and are labeled as SH3, SH2, and Tyrosine kinase (TyrK) domains. L1, L2, L3 and L4 stand for linkers between domains from N-term to C-term; (B–E) Evolutionary rates mapped onto a representative 3D structure of human ABL1 (homology model of the CD region based on 1opl; Nagar et al. 2003) for easier visualization and interpretation: (B) SEQ, (C) DOT, (D) SLT, and (E) PT.
Fig. 8.—

Correlation matrices of evolutionary rates per site of sequence or different structural traits across vertebrate paralogs. Pairwise paralog correlation coefficients of evolutionary site-specific rate profiles for the CD region, 404 nongapped alignment sites, comparable across all paralogs, are included out of the original 644 alignment sites, obtained with the R package corrgram for (A) sequence transitions (SEQ), (B) secondary structure-loop transitions (SLT), (C) disorder-order transitions (DOT), and (D) phosphorylation transitions (PT). Pairs with Pearson correlations significantly different from zero are labeled with asterisks (“***” for P value <5e-4, “**” for P value < 5e-3, and “*” for P value 5e-2). The color gradient goes from blue (rho = −1) to white (rho = 0), to red (rho = 1). On the left and the bottom of the matrices, a color guide per family as in figure 2 is included.
Additional correlation analysis was performed for the individual domains [SH3 (supplementary fig. S10, Supplementary Material online), SH2 (supplementary fig. S11, Supplementary Material online) and TyrK (supplementary fig. S12, Supplementary Material online)]. To quantify the different correlations of the domain-specific SEQ, DOT, SLT and PT to each other and to the entire CD region, Fisher transformation of the pairwise clade correlation coefficients to Z-scores was applied to the matrices from figure 8 and supplementary figures S10–S12, Supplementary Material online. The Z-score distributions for the different transition rates revealed that SLT correlates the most across clades for both CD regions and individual domains (fig. 9) when secondary structure predictions are based on default PSIPRED (supplementary fig. S12, Supplementary Material online). For the entire CD region, PT rates correlate the least across clades while DOT and SEQ present similar intermediate correlations (fig. 9). Within domains, all rates have different correlations for SH3 and TyrK, but in SH2, clade-specific SEQ correlations are similar to both DOT and PT (fig. 9B and C). Clade-specific SLT for the SH3 and SH2 domains have similar correlations while the correlations are significantly lower for TyrK. Clade-specific SEQ for the SH3 and SH2 domains have similar correlations while the correlations are significantly higher for TyrK. Clade-specific DOT for the SH2 and TyrK domains have similar correlations while DOT is significantly less correlated for the SH3 domain. Clade-specific PT for the SH2 and TyrK domains have similar correlations while PT is significantly better correlated for the SH3 domain.
Fig. 9.—
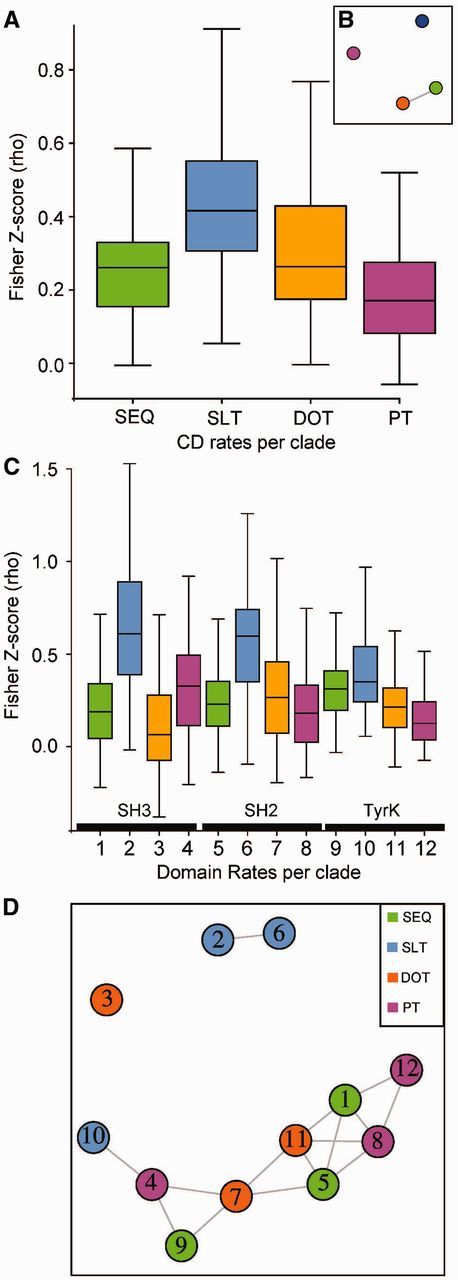
Distributions of correlation scores across clades for evolutionary rates of sequence and structural traits. Fisher transformation of correlation coefficient values (rho) into Z-scores using R software and psych library (Revelle 2015). Only nongapped sites across all groups are included in the correlation analysis. All pairwise paralog Z-scores are plotted together (sample size = 272) for the different evolutionary rates of sequence (SEQ: green), secondary structure-loop transitions (SLT: blue), disorder-order transitions (DOT: orange), and phosphorylation transitions (PT: magenta). (A) Z-scores based on the CD region correlations of paralog rates from figure 8, and (C) Z-scores based on individual domain correlations of paralog rates from supplementary figures S10–S12, Supplementary Material online. Statistical significance networks based on pairwise Mann–Whitney test with Bonferroni correction. Pairs without statistically significant differences in means (P value >0.05) are shown as linked nodes: (B) for CD-based evolutionary rates and (D) for domain-based rates for SH3, SH2 and tyrosine kinase (TyrK) domains.
Discussion
Protein Phylogenies
TKs predate Metazoa because they are found in choanoflagellates and Metazoa (Manning et al. 2008; Pincus et al. 2008). At the beginning of vertebrates, TKs underwent a major expansion. In the human genome, kinases are one of the largest protein families. When constructing the data sets and building phylogenetic trees, our aim was to identify proteins with high CD coverage (90%) ensuring that, in most proteins, the CD region had maintained its complete domain architecture and with relatively low sequence divergence allowing a high-quality multiple sequence alignment. Thus, this study does not attempt to identify a complete set of TKs or even all CD containing proteins. Including more divergent sequences reduces the quality of the alignment and the resulting phylogenetic tree. For instance, if we compare the multiple sequence alignment of the 543 sequences in the 70-90 set to the 655 sequences in the 50-90 set, the alignment has doubled in length and the number of ungapped sites is reduced to about a third. Most of the additional vertebrate sequences in 50-90 are from ray-finned fish. In fact, the fish kinomes for these TKs are diverging from the rest of vertebrates: different families have expanded, some are missing, and others show an elevated divergence rate compared with the rest of the sequences in the clade, for example, in the TEC family (fig. 2). Altogether, this can significantly alter the signaling networks in ray-finned fish. While human and mouse kinomes have been broadly studied, little is known about fish kinomes which are also relevant to a broad range of experiments in health and disease. For instance, Zebrafish (Danio rerio) is a popular model organism of human cancers, TK signaling, and pharmaceutical development (Challa and Chatti 2013; Gibert et al. 2013). Preliminary drafts of the Zebrafish kinome (Challa and Chatti 2013; Rakshambikai et al. 2014) show that a large number of recent kinase gene duplicates are retained, but lack a human counterpart (Wlodarchak et al. 2015). Further efforts towards understanding divergent signaling networks in fish are needed to make them efficient model organisms for kinase signaling and cancer.
Protein Structure, Disorder, Phosphorylation and Functional Divergence
Structural and functional divergence is expected to be low within orthologs. Structure enables function, so when structure is not conserved it signals functional divergence. Although we think of kinases as first and foremost performing phosphorylation, this is far from their only function and far from the only aspect in which they may functionally diverge. Kinases are intricately regulated through allosterism and phosphorylation that invoke conformational changes (Huse and Kuriyan 2002). As kinases evolved they have gained specificity in which protein sequence motifs they can phosphorylate, on which proteins, and under what conditions. The CD region in different TKs is conformationally dynamic along the different stages of substrate recognition, catalysis, and release, but not necessarily in a manner shared by all paralogs. Disorder provides conformational flexibility (Uversky 2011). We find that regions of disorder are commonly only conserved on a clade-specific level, where two paralogs that shared a last common ancestor sometime between the beginning of Metazoa and the beginning of vertebrates have diversified in location of disordered regions, even if the overall amount remains rather constant. This suggests that high evolutionary dynamics of disorder may be important for rapidly rewiring regulatory properties affecting activation and inhibition, but perhaps not the primary function “to phosphorylate” in the TK family. We also observe that regions prone to disorder are fluctuating across paralogs suggesting that while secondary structure is kept mostly constant, disorder may be used to fine-tune, for example, stability, flexibility, and domain–domain or protein–protein interactions. Kinases also have noncatalytic functions mediated through different types of interactions (Kung and Jura 2016). MATK (Chong et al. 2006), LYN (Katsuta et al. 1998), FYN (Chapman et al. 2012), and LCK (Rossy et al. 2013) can allosterically regulate signaling cascades via protein–protein interactions in their active state (Kung and Jura 2016). Structural disorder can potentially facilitate these scaffolding functions (activation/inhibition/complex formation). Altogether, kinases are highly multifunctional.
The structural dynamics of TyrK domains are well-studied, and some disordered regions have been experimentally verified. Our findings show that, although TyrK domains present some flexible regions, SH3 and SH2 domains accumulate more disorder that is less conserved across paralogs. SH2 and SH3 mediate specific protein–protein interactions that largely define specificity in cellular signaling transduction. SH2 promote, for example, protein recruitment and complex assembly (Liu and Nash 2012). Further, SH3 is a highly versatile and promiscuous binder (Agrawal and Kishan 2002; Maffei et al. 2015), known to act as a scaffold for the N-terminal disorder region in SRC (Maffei et al. 2015). In this scenario of divergent regulatory mechanisms, it is reasonable that structural disorder plays a role. In paralogs, disorder conservation and rapid DOT are differentially located at many functionally relevant areas (supplementary fig. S14, Supplementary Material online), especially at interfaces of inhibition in SH2 (the helix interacting with C-lobe), L3, and in SH3 domains. Still, some regions of conserved disorder and slow DOT within orthologs but not across all paralogs, are found (for instance, at the SH3 interface of inhibition with N-lobe in ITK) suggesting their functional relevance and potential as allosteric pockets for drug design.
TKs’ different roles in signaling cascades are often highly interconnected via protein–protein interactions and/or phosphorylations, regulating each other (fig. 10). Remarkably, CSK, MATK, and FRK are inhibitors of signaling cascades that function as tumor suppressors while the rest of TKs are activators of signaling cascades that function as oncogenes as shown to promote cell growth, division, migration and survival (Mano 1999; Serfas and Tyner 2003; Colicelli 2010; Bononi et al. 2011; Okada 2012). FRK proteins interact with CSK (Varjosalo et al. 2013). CSK orchestrates the inhibition of SRC paralogs (Okada 2012) which, in contrast, exert an activation effect on TEC paralogs (Qiu and Kung 2000) and on ABL paralogs (Colicelli 2010). Sequence divergence at regions such as the A-loop, determines the different catalytic efficiencies in TKs (Joseph et al. 2013), while sequence heterogeneity of linkers L3 play another important role regulating catalysis via allosteric coupling (Register et al. 2014). The differential disorder predicted between paralogs in the same family such as MATK-CSK and BLK-LCK agrees with reported noncatalytic activities for MATK (Chong et al. 2006) and LCK (Rossy et al. 2013) and their accumulated fractions of disorder after the gene duplications. MATK, with the highest amount of disorder per CD region (fig. 5) inhibits SRC via a unique, experimentally verified, scaffolding mechanism by binding multiple active conformations (Chong et al. 2006). Thus, mutation-driven conformational selection, where certain amino acid substitutions enable functional adaptation by shifting the conformational ensemble, could have contributed to the increased structural flexibility conferring a selective advantage to the performance of such a novel regulatory mechanism. In the TyrK domain from MATK, a conserved disorder region is located at the helical C-lobe, usually a structurally constrained region in TKs. Overall, MATK has numerous sites with conserved structural disorder but is also the most divergent paralogous group at the sequence level.
Fig. 10.—

Functional evidence of cross-talk in the tyrosine kinase family. Enriched protein–protein interaction (PPI) network obtained from String v. 10 (Szklarczyk et al. 2015) including evidence from experiments and databases for 17 human TKs (nodes) connected by 67 edges, shaping their modes of action color-coded as default (see legend). Nodes are color-coded by TK family (as in fig. 2). The PPI network was used to highlight proteins (nodes with stars) known to be involved in the human immune system: (A) BCR signaling pathway (GO:0050853, 6 proteins) and (B) TCR signaling pathway (GO:0055082, 6 proteins).
Bellay et al. classified functional disordered sites within yeast orthologs as constrained disorder and flexible disorder based on disorder and sequence conservation (Bellay et al. 2011). Constrained disordered sites had at least 50% conserved disorder and sequence and were found to have, for example, protein chaperone and RNA binding functions. Flexible disordered sites also had at least 50% conserved disorder but with sequence conservation <50% and were found to be involed in signaling and multifunctionality (Bellay et al. 2011). We applied a similar classification scheme for each of the different paralogous clades, defining constrained disorder to sites with at least 70% of sequences at the site predicted to be disordered and SEQ is negative. Flexible disorder was defined for sites that have at least 70% of sequences predicted to be disordered but positive SEQ. Disorder prone regions, containing both constrained and flexible disorder, accummulate predominantly in two regions in the SH3 domain (around alignment sites 40 and 100), in the first half of the SH2 domain, in L3, and towards the end of the CD (supplementary fig. S15, Supplementary Material online). MATK has the largest amount (23%) of functional disorder; 68 sites are classified as constrained disorder and 34 sites are classified as flexible disorder. Second is ABL1 and third is LCK with 13% and 11% functionally disordered sites, respectively. Thereafter, arranged in decreasing order of total amount of functional disorder are BTK (11%), SRC (9%), TEC (9%), HCK (8%), FGR (8%), ABL2 (6%), ITK (5%), FYN (5%), YES (4%), LYN (3%), CSK (3%), FRK (2%), TXK (1%), and BLK (1%) (supplementary fig. S15, Supplementary Material online). These results show major changes in functional disorder between closely related TKs, for example, MATK versus CSK, ABL1 versus ABL2, and LCK versus LYN, indicating that even subtle changes in disorder propensities may be important for divergence and specialization after gene duplication.
Experimental Evidence of Divergent Tyrosine Phosphorylation Patterns
Both conserved tyrosine and tyrosine predicted to be phosphorylated follow clade-specific patterns. Of 43 alignment sites with predicted p-Tyr, 13 were experimentally verified to be phosphorylated in humans and consistently predicted p-Tyr in at least 70% of the orthologs (supplementary table S5, Supplementary Material online). Although none of them are 100% conserved across all clades, alignment sites 200, 201, 369, 500, and 600 are predicted and experimentally verified across most clades as p-Tyr (fig. 6), suggesting shared mechanisms of protein regulation. For instance, alignment site 500 holds a p-Tyr located in the A-loop of the TyrK domain which is a conserved activation mechanism in all but CSK paralogs (Okada 2012) (amino acid substitutions to Arg in MATK and Ser in CSK).
Many regulatory mechanisms of TK function are unique to a paralogous group or family, where a conserved amino acid in the corresponding site in other clades cannot be phosphorylated. In some cases, the amino acid in a corresponding site compensates the electrostatic effect of the missing phosphorylation with Asp and Glu residues. In others, the phosphorylated residue switches from Tyr to Ser/Thr. While Ser/Thr may still be phosphorylated, phosphorylation will likely be performed by another kinase, and consequently, a Tyr to Ser/Thr substitution can alter the protein–protein interaction and signaling network. Divergent activation mechanisms are shown for the SH3 domain and L3 linker, where the p-Tyr causes the disruption of interdomain interfaces, preventing a self-inhibited conformation. Auto-phosphorylation of a conserved Tyr in SH3 of TEC paralogs (alignment site 45) prevent TEC family self-inhibition (Qiu and Kung 2000). SRC activates ABL paralogs at another SH3 site (alignment site 27), or in L3 (alignment site 254). These ABL sites are experimentally verified (Colicelli 2010) but p-Tyr predictions are not conserved in >70% of orthologs. FRK proteins are inhibitors of signaling cascades and present another activation mechanism by p-Tyr at L3 (alignment site 264) and L4 (alignment site 639), supported by experimental data in PhosphoSitePlus (fig. 6). Another mechanism of inhibition in TKs at L4 is carried out by CSK proteins at the C-tail of SRC paralogs (alignment site 632; Okada 2012). Notably, L4 is depleted of Tyr residues outside SRC and FRK families except for a few proteins in the CSK clade.
It is commonly known that disordered regions can undergo disorder to order transitions upon phosphorylation (Bah and Forman-Kay 2016). With both clade-specific disordered regions and phosphorylated sites, an even greater route to functional diversification or specialization can be envisioned.
Correlation of Evolutionary Rates across Paralogs: SEQ, DOT, SLT, and PT
We find that over the entire CD region, SLT is fluctuating less than DOT, while DOT and SEQ are similar, and PT is fluctuating more than the other rates (fig. 9A). This reinforces that phosphorylation (PT) is more clade-specific, and SLT is more conserved (fig. 8).
On the domain level, SLT fluctuates less than SEQ, DOT, or PT among clades. Secondary structure is expected to be conserved as the structural fold of domains tend to be conserved (fig. 9B). Also, to test for bias inferred from the PSIPRED secondary structure predictor that relies on a PSI-BLAST profile and thus assumes that the predicted property is conserved, unlike the other predictors used here, we also ran PSIPRED with a single sequence. With the PSIPRED single sequence, the PSI-BLAST step is avoided and the evolutionary enforcement of structural conservation is relieved. Indeed, the PSIPRED predictions from single sequences are more variable (supplementary fig. S13, Supplementary Material online). Still, it can be argued that PSIPRED default’s assumption that secondary structure is conserved is the better option as it is generally accepted that structure is conserved and since that is the benchmarked version found to be ∼80% accurate (Bryson et al. 2005). In contrast, structural disorder has not been shown to be conserved across paralogs and thus, we cannot assume that structural disorder is a conserved trait, although many predictors make that assumption.
We used one such predictor, DISOPRED2, to predict structural disorder for this data set, but at the given cut-off, there was only a small amount of disordered sites. Thus, we used IUPred (Dosztányi, Csizmók, et al. 2005a, 2005b), which uses an energy function to estimate disorder propensity and does not assume that disorder is conserved. Reported accuracy varies from 62% (Di Domenico et al. 2013) to 85% (Fukuchi et al. 2012) based on the intended cut-off (0.5). At this cut-off, 124 proteins had no disordered sites. The IUPred disorder prediction accuracy was previously shown to improve if the cut-off was reduced to 0.4 (Fuxreiter et al. 2007; Xue et al. 2009). At cut-off 0.4, all proteins in this study had some disordered sites as expected based on available PDB structures (Ogawa et al. 2002; Nagar et al. 2003; Wang et al. 2015). For these reasons, a cut-off of 0.4 was used to infer disordered sites. Regarding phosphorylation predictions, all retrieved experimental phospho-sites were identified when no score cut-off was applied (supplementary fig. S9, Supplementary Material online). With a cut-off of 0.5, many experimental phospho-sites are identified, but also sites that are not experimental phospho-sites are identified. Many of these are likely to be false positives, but some are also likely yet-to-be identified as phospho-sites. This also suggests that the sequence motifs used by classifiers like NetPhos may need to be updated with phosphoproteomics data to increase their sensitivity.
Based on the predictions from these methods that are not perfect but widely used, we find that domain-specific trends of evolutionary dynamics for secondary structure, intrinsic disorder, and phosphorylation are at play. These transition rates of structural and regulatory properties add to the traditionally site-specific amino acid substitution rates, SEQ, and can be developed further to infer functionally divergent proteins and sites. However, in this limited data set, not all properties evolve the same way in the different domains: DOT and PT are similar for SH2 and TyrK domains, while SLT and SEQ are similar for SH3 and SH2 domains. Also, the four rates in the SH3 and TyrK domains, respectively, are all significantly different from each other. In the SH2 domain, SEQ is not different from DOT or PT (fig. 9). Further studies are needed to verify if these indicate general trends or not.
The most structurally dynamic domain is SH3, enriched with clade-specific disordered regions. This domain also has high evolutionary dynamics. The SH3 domain may be dispensable in some cases or able to diverge beyond Pfam’s sequence-based HMM recognition. Importantly, the BMX proteins that were only present in the 50-90 set and not in the 70-90 set, do not have a recognizable SH3 domain according to Pfam. For the rest, four different versions of SH3 are found in our data set; one is even supposed to be bacterial according to the Pfam prediction. A bacterial SH3 is unreasonable in this context and we propose that this is rather a divergent SH3 domain that has started to resemble a bacterial SH3 domain. Further, this implies that one SH3 domain might easily converge on another SH3 domain variant at high sequence divergence. Thus, differentiating SH3 domain variants may lead to misinterpretations of domain ancestry.
Evolution of Disorder in TKs Involved in the Immune Response in Vertebrates
Some TKs such as CSK, SRC, YES, and LYN are ubiquitously expressed in most tissues (Kim et al. 2014), while others (SRC-B paralogs and TEC paralogs) are more restrained to hematopoietic cell lineages. In blood cells, TKs cross-talk between them (fig. 10) and activate the immune response via T-cells (TCR signaling pathways; Smith-Garvin et al. 2009) and B-cells (BCR signaling pathway; Dal Porto et al. 2004). Particularly, SRC-B paralogs activate TEC paralogs in immune cells (fig. 10).
An interesting observation from the results is that high disorder propensities can accumulate in similar regions in pairs from different TK families that are not close paralogs (fig. 3). Although TKs are distinct types of molecular switches (Bradshaw 2010), DOT for TEC and SRC paralogs that are known to interact in a signaling pathway, correlate better with each other than with their closer sister groups, for example, pairs BTK-LYN or TEC-LYN (figs. 8 and 10). This may suggest a mechanism of recognition and binding through conformational flexibility. Allosteric networks have been described individually for SRC paralogs (Foda et al. 2015) and TEC paralogs (Joseph et al. 2010). Future efforts are required to elucidate if conserved conformational flexibility is coevolving in TK binding interfaces, or if the accumulated conformational flexibility is a result of convergent evolution, perhaps promoting rewiring of signaling networks through conformational mimicry.
Concluding Remarks
The importance of protein sequence and structure conservation features is well recognized. It is only in recent years that the role of protein structural dynamics on evolutionary time-scales has received some consideration (Marsh and Teichmann 2014). Thus, our understanding of the evolutionary dynamics of conformational flexibility is still in its early stage.
Studying where structural dynamic profiles overlap and differ along evolution is important for elucidating mechanisms of protein evolution, especially after gene duplication. It can improve detection of amino acid sites (and substitutions) that dictate (and disrupt) function. Since structurally disordered regions often have high sequence divergence rates, recognizing conserved sequence motifs that indicate functional importance is challenging (Babu et al. 2012). Further, structurally disordered regions that are not conserved across homologs may be dismissed as not functionally important (van der Lee et al. 2014), but the evolutionary context in which it is not conserved is imperative for making that call. The evolutionary dynamics of structural disorder is informative. If structural disorder is not conserved across paralogs, it may not be of universal importance to all homologs, but it could be to certain paralogs. If structural disorder is rapidly changing within orthologs, it may not be important for function. However, when structural disorder is highly conserved within orthologs, it may contribute to their functional specificity. It should be noted that conserved disorder in disorder prone regions with high evolutionary dynamics between paralogous clades, may have conserved disorder for different reasons in different paralogs. The site-specific rates for different clades suggest that the evolutionary dynamics of disorder/order is high, but this is under the conditions investigated here. If this is a general trend remains to be tested on a larger data set, but these results warrant a word of caution in considering disorder as a conserved feature when aiming to predict it. Simultaneous prediction of secondary structure and structural disorder informed by the evolutionary context but with different evolutionary constraints may be beneficial. This concept is further supported by a recent large-scale study where sites with conserved disorder and secondary structure were the most constrained at the sequence level (Ahrens et al. 2016).
Finally, in addition to the gallery of predicted disorder propensities, secondary structure, and phosphorylation mapped in an evolutionary and MSA context, 3D models with mapped disorder conservation and transition rates that highlight kinase specific regions are provided (Supplementary Material online). We hope that these broad and integrative findings are of use to the kinase expert and that these inspire new avenues of experimental hypothesis testing on TK divergence while also guiding the identification of potential hotspots that can be targeted by rational drug development.
Methods
Identification of Orthologs
The canonical sequences of 17 human nonreceptor TKs that contain the CD region were retrieved from the human reference proteome (UniProtKB/Swiss-Prot, release 07/2014). The sequences were trimmed to reflect the CD region based on the Pfam envelope boundaries (from N-term to C-term, a SH3_1 (PF00018, HMM length: 48), a SH2 (PF00017, HMM length: 77), and a TyrK domain (PF07714, Pkinase_Tyr, HMM length: 259), adding 10 extra residues in both N-term and C-term, when present (supplementary table S1, Supplementary Material online). BLASTp (Altschul et al. 1990) (version 2.2.26) was performed against a database of 213 (of which 59 are metazoan and 36 are vertebrate) canonical complete reference proteomes (UniProtKB/Swiss-Prot, release 07/2014) with a 30% minimum sequence identity for each of the 17 CDs. The best 1,000 hits per run were filtered to produce two data sets with a minimum of 90% coverage for at least one of the human CDs, but that differ in sequence divergence. For sets 70-90 and 50-90, a pairwise sequence identity of at least 70% and 50% to at least one human CD was required, respectively. When the same protein was found in multiple runs, the largest range was included. The canonical reference proteomes only have one protein representative per gene, but although Uniprot entries’ redundancy has been removed, sequence redundancy is present (100% identical pairs from different species). Sequence identifiers for all sequences are given in supplementary table S2, Supplementary Material online.
Phylogenetic Reconstruction
Protein sequences from the different sets (70-90 and 50-90) were aligned with MAFFT (Katoh and Toh 2008) v7.123. For each multiple sequence alignment (MSA) a phylogenetic tree was reconstructed with MrBayes (Ronquist et al. 2012) v.3.2.2 using an amino acid mixture model, a four category gamma distribution, and invariant sites. For both runs, JTT (Jones et al. 1992) was the primary substitution matrix chosen by MrBayes and the models applied were confirmed by Prottest v. 3.4 (Darriba et al. 2011). For each phylogeny, two MCMC runs with four chains each were performed for a given number of generations (70 and 50 million generations for the 70-90 and 50-90 sets, respectively), sampling every 100 generations. Upon completion, the average standard deviation of split frequencies was 0.0067 with a maximum of 0.0815 for 70-90, and 0.0089 with a maximum of 0.1481 for 50-90. After discarding the first 25% of trees (default burn-in phase), consensus trees were summarized using the 50% majority rule, followed by midpoint rooting in FigTree.
Sequence-Based Predictions
For data set 70-90, predictions were obtained for full-length proteins and mapped on its corresponding site in the 70-90 MSA. Structural disorder was predicted under two different scenarios (considering protein’s evolutionary context or not): using DISOPRED2 (Ward et al. 2004) with default parameters and database, or using IUPred (Dosztányi, Csizmok, et al. 2005) version 1.0 with the “long” option. Two matrices were generated, one based on the continuous disorder propensity and one binary. A default 0.5 cut-off for DISOPRED and IUPred, as well as a 0.4 cut-off for IUPred, were applied to infer binary states of order (0) versus disorder (1) (Fuxreiter et al. 2007). The resulting disorder matrices were used for analyzing the evolutionary dynamics of structural disorder to order transitions (DOT). Secondary structure was predicted using PSIPRED (Jones 1999) version 3.4 using single protein sequences or sequence profiles built with a filtered database (Uniref90, as of April 2015). Two types of matrices were generated, one reflecting the confidence of the predicted state and one binary. The binary conversion was simplified by classifying helix/strand as one state (1) and loop as the other (0). The resulting matrices were used for analyzing the evolutionary dynamics of secondary structure elements to loop transitions (SLT). Phosphorylation of Ser, Thr, and Tyr residues were predicted using NetPhos (Blom et al. 1999) v 3.1 and a cut-off for the binary conversion was set to 0.75 [values ≥0.75 are predicted to be phosphorylated (1), and all others are not (0)]. The resulting matrices were used for analyzing the evolutionary dynamics of phosphorylation transitions (PT). To validate the obtained phosphorylation patterns, experimentally verified phosphorylation sites (true positives) were retrieved from PhosphoSitePlus (Hornbeck et al. 2015) (as of 4 May 2016) using “by protein accession” search option for each human “canonical” entry included in the protein data sets up to a total of 147 protein residues within the 17 CDs (involving 60 alignment sites in the 70-90 set), where only sites with strong phosphorylation evidence were considered (at least 5 high-throughput experiments and/or 1 low-throughput experiment). Protein domains for all sequences were predicted based on Pfam 27 (Finn et al. 2014).
Conservation Per Site
For the conservation analysis, the fractions of structural disorder, secondary structure elements (either α-helices or β-strands) and predicted phosphorylation sites per alignment position were calculated (gaps included) based on their binary matrices of predicted traits. Sequence conservation was graphically represented using WebLogos (Crooks et al. 2004) for each of the vertebrate clades where amino acids are colored according to their chemical properties: polar amino acids (G,S,T,Y,C,Q,N) are green, basic (K,R,H) blue, acidic (D,E) red and hydrophobic (A,V,L,I,P,W,F,M) amino acids are black (supplementary fig. S3, Supplementary Material online).
Amino Acid Substitution Rates
Amino acid substitution rates per site (SEQ) for the 70-90 set (MSA and its corresponding tree for 70-90; for the entire data set or per paralogous group) were estimated with the empirical Bayesian method in Rate4Site (Mayrose et al. 2004). The model of evolution used was based on the JTT amino acids substitution model (Jones et al. 1992) with a 16 category gamma distribution. The results were normalized as Z-scores, with a mean equal to 0 and a standard deviation of 1: negative sites with evolutionary rates slower than average and positive sites with evolutionary rates faster than average. Further classification into three discrete states defined sites with (i) slow rates (values < 0), (ii) nearly neutral rates (between 0 and 1), and (iii) fast rates (values > 1) (fig. 3B).
Evolutionary Transitions of Structural and Phosphorylation Traits: DOT, SLT, and PT
Transition rates per site were estimated based on the phylogenetic tree (for the entire data set or per paralogous group) and binary matrices of predictions for structural disorder, secondary structure, and phosphorylation sites for data set 70-90. Evolutionary analyses of structural properties were conducted by the GLOOME version (Cohen et al. 2010) of Rate4Site. GLOOME was adapted to analyze trends of different structural properties using binary information with equal substitution rates in Rate4Site (an implemented Q matrix weight transitions the following way: 1->1 or 0->0 are equal to −1, and transitions 0->1 or 1->0 are equal to 1) with a gamma distribution including 6 discrete categories. Similarly to SEQ, the empirical evolutionary rates per alignment site are normalized as Z-scores (mean equal to 0 and standard deviation of 1).
Graphical Representations of Predictions and 3D Mapping of Different Transition Rates
Raw prediction values of structural disorder and secondary structure or binary states for phosphorylation sites were mapped onto the MSAs and visualized as heat maps using iTOL (Letunic and Bork 2007). Site-specific rates and conservation were also visualized in 3D, mapped onto a homology model. The homology model was built with Modeller (Webb and Sali 2014) based on PDB id 1opl (Nagar et al. 2003), chain A, adding 5 residues at the N-term and removing 9 residues at the C-term to show the CD. The figures were generated using the open source version of PYMOL (DeLano 2002).
Statistical Analysis
The correlation analysis was carried out using the corrgram function (R software; Wright 2015; The R Core Team 2012) to identify overlapping/distinct patterns of the different normalized transition rates across paralogs. Pearson correlation coefficients were plotted into a lower triangular matrix (for simplicity), in addition to their significance levels (inferred with t-tests). Further, correlation coefficients were converted into Z-scores by Fisher transformation (R library psych; Revelle 2015). When normality could not be assumed, significance inference was carried out using Mann–Whitney test with Bonferroni correction (Mann and Whitney 1947; Dunn 1961).
Protein Networks
Functional evidence of protein–protein interactions (PPI) across human TKs was retrieved from STRING database, version 10 (Szklarczyk et al. 2015), filtering only experiments and databases. The network, including 17 nodes connected by 67 edges (average node degree = 7.88, clustering coefficient = 0.84), has significantly more interactions than expected, that is, the network is enriched in PPI (P value = 0) more than expected for a random set of proteins of similar size, drawn from the whole human genome. Such an enrichment is an indicative that these TKs are, at least partially, biologically connected.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the Instructional & Research Computing Center (IRCC) at Florida International University for providing HPC computing resources that have contributed to the research results reported within this article, web: http://ircc.fiu.edu.
Literature Cited
- Agrawal V, Kishan KVR. 2002. Promiscuous binding nature of SH3 domains to their target proteins. Protein Pept Lett. 9:185–193. [DOI] [PubMed] [Google Scholar]
- Ahrens J, Dos Santos HG, Siltberg-Liberles J. 2016. The nuanced interplay of intrinsic disorder and other structural properties driving protein evolution. Mol Biol Evol. 33:2248–2256. [DOI] [PubMed] [Google Scholar]
- al-Obeidi FA, Wu JJ, Lam KS. 1998. Protein tyrosine kinases: structure, substrate specificity, and drug discovery. Biopolymers 47:197–223. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Babu MM, Kriwacki RW, Pappu RV. 2012. Structural biology. Versatility from protein disorder. Science 337:1460–1461. [DOI] [PubMed] [Google Scholar]
- Bah A, Forman-Kay JD. 2016. Modulation of intrinsically disordered protein function by post-translational modifications. J Biol Chem. 291: 6696–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellay J, et al. 2011. Bringing order to protein disorder through comparative genomics and genetic interactions. Genome Biol. 12:R14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, et al. 2000. The Protein Data Bank. Nucleic Acids Res. 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. 1999. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 294:1351–1362. [DOI] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. 2009. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 5:789–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bononi A, et al. 2011. Protein kinases and phosphatases in the control of cell fate. Enzyme Res. 2011:329098.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw JM. 2010. The Src, Syk, and Tec family kinases: distinct types of molecular switches. Cell Signal. 22:1175–1184. [DOI] [PubMed] [Google Scholar]
- Bryson K, et al. 2005. Protein structure prediction servers at University College London. Nucleic Acids Res. 33:W36–W38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challa AK, Chatti K. 2013. Conservation and early expression of zebrafish tyrosine kinases support the utility of zebrafish as a model for tyrosine kinase biology. Zebrafish 10:264–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Yoder AN, Houtman JCD. 2012. Non-catalytic functions of Pyk2 and Fyn regulate late stage adhesion in human T cells. PLoS One 7:e53011.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong Y-P, et al. 2006. C-terminal Src kinase-homologous kinase (CHK), a unique inhibitor inactivating multiple active conformations of Src family tyrosine kinases. J Biol Chem. 281:32988–32999. [DOI] [PubMed] [Google Scholar]
- Cohen O, Ashkenazy H, Belinky F, Huchon D, Pupko T. 2010. GLOOME: gain loss mapping engine. Bioinformatics 26:2914–2915. [DOI] [PubMed] [Google Scholar]
- Colicelli J. 2010. ABL tyrosine kinases: evolution of function, regulation, and specificity. Sci Signal. 3:re6.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbi-Verge C, et al. 2013. Two-state dynamics of the SH3-SH2 tandem of Abl kinase and the allosteric role of the N-cap. Proc Natl Acad Sci U S A. 110:E3372–E3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan-Jacob SW, et al. 2005. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure 13:861–871. [DOI] [PubMed] [Google Scholar]
- Cowan-Jacob SW, Jahnke W, Knapp S. 2014. Novel approaches for targeting kinases: allosteric inhibition, allosteric activation and pseudokinases. Future Med Chem. 6:541–561. [DOI] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, Brenner SE. 2004. WebLogo: a sequence logo generator. Genome Res. 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Aniello S, et al. 2008. Gene expansion and retention leads to a diverse tyrosine kinase superfamily in amphioxus. Mol Biol Evol. 25:1841–1854. [DOI] [PubMed] [Google Scholar]
- Dal Porto JM, et al. 2004. B cell antigen receptor signaling 101. Mol Immunol. 41:599–613. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira GAP, Rangel LP, Costa DC, Silva JL. 2015. Misfolding, aggregation, and disordered segments in c-Abl and p53 in human cancer. Front Oncol. 5:97.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. 2002. The PyMOL Molecular Graphics System, Version 1.7 Schrödinger, LLC.
- Deng Y, et al. 2014. Global analysis of human nonreceptor tyrosine kinase specificity using high-density peptide microarrays. J Proteome Res. 13:4339–4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico T, Walsh I, Tosatto SCE. 2013. Analysis and consensus of currently available intrinsic protein disorder annotation sources in the MobiDB database. BMC Bioinformatics 14(Suppl 7):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztányi Z, Csizmok V, Tompa P, Simon I. 2005a. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21:3433–3434. [DOI] [PubMed] [Google Scholar]
- Dosztányi Z, Csizmók V, Tompa P, Simon I. 2005b. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 347:827–839. [DOI] [PubMed] [Google Scholar]
- Dunn OJ. 1961. Multiple comparisons among means. J Am Stat Assoc. 56:52–64. [Google Scholar]
- Finn RD, et al. 2014. Pfam: the protein families database. Nucleic Acids Res. 42:D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA. 2015. A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat Commun. 6:5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuchi S, et al. 2012. IDEAL: intrinsically disordered proteins with extensive annotations and literature. Nucleic Acids Res. 40:D507–D511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuxreiter M, Tompa P, Simon I. 2007. Local structural disorder imparts plasticity on linear motifs. Bioinformatics 23:950–956. [DOI] [PubMed] [Google Scholar]
- Galea CA, Wang Y, Sivakolundu SG, Kriwacki RW. 2008. Regulation of cell division by intrinsically unstructured proteins; intrinsic flexibility, modularity and signaling conduits. Biochemistry 47:7598–7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrin LK, Saiah E. 2013. Approaches to discover non-ATP site kinase inhibitors. Med Chem Commun. 4:41–51. [Google Scholar]
- Gibert Y, Trengove MC, Ward AC. 2013. Zebrafish as a genetic model in pre-clinical drug testing and screening. Curr Med Chem. 20:2458–2466. [DOI] [PubMed] [Google Scholar]
- Gnad F, Gunawardena J, Mann M. 2011. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 39:D253–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gocek E, Moulas AN, Studzinski GP. 2014. Non-receptor protein tyrosine kinases signaling pathways in normal and cancer cells. Crit Rev Clin Lab Sci. 51:125–137. [DOI] [PubMed] [Google Scholar]
- Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. 2013. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer 13:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O, et al. 2003. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112:845–857. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, et al. 2015. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43:D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. 2002. The conformational plasticity of protein kinases. Cell 109:275–282. [DOI] [PubMed] [Google Scholar]
- Hussain A, et al. 2011. TEC family kinases in health and disease-loss-of-function of BTK and ITK and the gain-of-function fusions ITK-SYK and BTK-SYK. FEBS J. 278:2001–2010. [DOI] [PubMed] [Google Scholar]
- Jin J, Pawson T. 2012. Modular evolution of phosphorylation-based signalling systems. Philos Trans R Soc Lond B Biol Sci. 367:2540–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D, Taylor W, Thornton J. 1992. The rapid generation of mutation data matrices from protein sequences. Comput Appl Biosci. 8:275–282. [DOI] [PubMed] [Google Scholar]
- Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 292:195–202. [DOI] [PubMed] [Google Scholar]
- Joseph RE, et al. 2013. Activation loop dynamics determine the different catalytic efficiencies of B cell- and T cell-specific tec kinases. Sci Signal. 6:ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph RE, Xie Q, Andreotti AH. 2010. Identification of an allosteric signaling network within Tec family kinases. J Mol Biol. 403:231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Toh H. 2008. Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform. 9:286–298. [DOI] [PubMed] [Google Scholar]
- Katsuta H, Tsuji S, Niho Y, Kurosaki T, Kitamura D. 1998. Lyn-mediated down-regulation of B cell antigen receptor signaling: inhibition of protein kinase C activation by Lyn in a kinase-independent fashion. J Immunol. 160:1547–1551. [PubMed] [Google Scholar]
- Kim M-S, et al. 2014. A draft map of the human proteome. Nature 509:575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS. 2015. Dynamics-driven allostery in protein kinases. Trends Biochem Sci. 40:628–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung JE, Jura N. 2016. Structural basis for the non-catalytic functions of protein kinases. Structure 24:7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128. [DOI] [PubMed] [Google Scholar]
- Liu BA, Nash PD. 2012. Evolution of SH2 domains and phosphotyrosine signalling networks. Philos Trans R Soc Lond B Biol Sci. 367:2556–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei M, et al. 2015. The SH3 domain acts as a scaffold for the N-terminal intrinsically disordered regions of c-Src. Structure 23:893–902. [DOI] [PubMed] [Google Scholar]
- Mann HB, Whitney DR. 1947. On a test of whether one of two random variables is stochastically larger than the other. Ann Math Stat. 18:50–60. [Google Scholar]
- Manning G, Plowman GD, Hunter T, Sudarsanam S. 2002. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 27:514–520. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. [DOI] [PubMed] [Google Scholar]
- Manning G, Young SL, Miller WT, Zhai Y. 2008. The protist, Monosiga brevicollis, has a tyrosine kinase signaling network more elaborate and diverse than found in any known metazoan. Proc Natl Acad Sci U S A. 105:9674–9679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mano H. 1999. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 10:267–280. [DOI] [PubMed] [Google Scholar]
- Marsh JA, Teichmann SA. 2014. Parallel dynamics and evolution: protein conformational fluctuations and assembly reflect evolutionary changes in sequence and structure. Bioessays 36:209–218. [DOI] [PubMed] [Google Scholar]
- Mayrose I, Graur D, Ben-Tal N, Pupko T. 2004. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol. 21:1781–1791. [DOI] [PubMed] [Google Scholar]
- Nagar B, et al. 2003. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell 112:859–871. [DOI] [PubMed] [Google Scholar]
- Ogawa A, et al. 2002. Structure of the carboxyl-terminal Src kinase, Csk. J Biol Chem. 277:14351–14354. [DOI] [PubMed] [Google Scholar]
- Okada M. 2012. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 8:1385–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Letunic I, Bork P, Lim WA. 2008. Evolution of the phospho-tyrosine signaling machinery in premetazoan lineages. Proc Natl Acad Sci U S A. 105:9680–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, et al. 2008. The amphioxus genome and the evolution of the chordate karyotype. Nature 453:1064–1071. [DOI] [PubMed] [Google Scholar]
- Qiu Y, Kung HJ. 2000. Signaling network of the Btk family kinases. Oncogene 19:5651–5661. [DOI] [PubMed] [Google Scholar]
- Rakshambikai R, Srinivasan N, Gadkari RA. 2014. Repertoire of protein kinases encoded in the genome of zebrafish shows remarkably large population of PIM kinases. J Bioinform Comput Biol. 12:1350014.. [DOI] [PubMed] [Google Scholar]
- Register AC, Leonard SE, Maly DJ. 2014. SH2-catalytic domain linker heterogeneity influences allosteric coupling across the SFK family. Biochemistry 53:6910–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelle W. 2015. psych: procedures for psychological, psychometric, and personality research. http://cran.r-project.org/package=psych. Version release: 2015-08-30.
- Ronquist F, et al. 2012. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61:539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossy J, Owen DM, Williamson DJ, Yang Z, Gaus K. 2013. Conformational states of the kinase Lck regulate clustering in early T cell signaling. Nat Immunol. 14:82–89. [DOI] [PubMed] [Google Scholar]
- Sato I, et al. 2009. Differential trafficking of Src, Lyn, Yes and Fyn is specified by the state of palmitoylation in the SH4 domain. J Cell Sci. 122:965–975. [DOI] [PubMed] [Google Scholar]
- Serfas MS, Tyner AL. 2003. Brk, Srm, Frk, and Src42A form a distinct family of intracellular Src-like tyrosine kinases. Oncol Res. 13:409–419. [DOI] [PubMed] [Google Scholar]
- Smith-Garvin JE, Koretzky GA, Jordan MS. 2009. T cell activation. Annu Rev Immunol. 27:591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, et al. 2015. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Keshwani MM, Steichen JM, Kornev AP. 2012. Evolution of the eukaryotic protein kinases as dynamic molecular switches. Philos Trans R Soc Lond B Biol Sci. 367:2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The R Core Team. 2012. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Uversky VN. 2011. Intrinsically disordered proteins from A to Z. Int J Biochem Cell Biol. 43:1090–1103. [DOI] [PubMed] [Google Scholar]
- van der Lee R, et al. 2014. Classification of intrinsically disordered regions and proteins. Chem Rev. 114:6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varjosalo M, et al. 2013. Interlaboratory reproducibility of large-scale human protein-complex analysis by standardized AP-MS. Nat Methods 10:307–314. [DOI] [PubMed] [Google Scholar]
- Wang Q, et al. 2015. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife 4:e06074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. 2004. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 337:635–645. [DOI] [PubMed] [Google Scholar]
- Webb B, Sali A. 2014. Comparative protein structure modeling using MODELLER. Curr Protoc Bioinforma. 47:5.6.1–5.6.32. [DOI] [PubMed] [Google Scholar]
- Wlodarchak N, Tariq R, Striker R. 2015. Comparative analysis of the human and zebrafish kinomes: focus on the development of kinase inhibitors. Trends Cell Mol Biol. 10:49–75. [PMC free article] [PubMed] [Google Scholar]
- Woodring PJ, Hunter T, Wang JYJ. 2005. Mitotic phosphorylation rescues Abl from F-actin-mediated inhibition. J Biol Chem. 280:10318–10325. [DOI] [PubMed] [Google Scholar]
- Wright K. 2015. https://cran.r-project.org/web/packages/corrgram/index.html. Version release: 2015-02-13.
- Wu P, Nielsen TE, Clausen MH. 2015. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 36:422–439. [DOI] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. 1999. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell 3:629–638. [DOI] [PubMed] [Google Scholar]
- Xue B, Oldfield CJ, Dunker AK, Uversky VN. 2009. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 583:1469–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hendrickson WA. 1996. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature 384:484–489. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.