

Abstract
Genomic architecture is the framework within which genes and regulatory elements evolve and where specific constructs may constrain or potentiate particular adaptations. One such construct is evident in phages that use a headful packaging strategy that results in progeny phage heads packaged with DNA until full rather than encapsidating a simple unit-length genome. Here, we investigate the evolution of the headful packaging phage Sf6 in response to barriers that impede efficient phage adsorption to the host cell. Ten replicate populations evolved faster Sf6 life cycles by parallel mutations found in a phage lysis gene and/or by large, 1.2- to 4.0-kb deletions that remove a mobile genetic IS911 element present in the ancestral phage genome. The fastest life cycles were found in phages that acquired both mutations. No mutations were found in genes encoding phage structural proteins, which were a priori expected from the experimental design that imposed a challenge for phage adsorption by using a Shigella flexneri host lacking receptors preferred by Sf6. We used DNA sequencing, molecular approaches, and physiological experiments on 82 clonal isolates taken from all 10 populations to reveal the genetic basis of the faster Sf6 life cycle. The majority of our isolates acquired deletions in the phage genome. Our results suggest that deletions are adaptive and can influence the duration of the phage life cycle while acting in conjunction with other lysis time-determining point mutations.
Keywords: experimental evolution, deletions, bacteriophage, holin, Shigella flexneri
Introduction
The evolutionary relationships between genomic architecture and life history traits are important for understanding the molecular basis of adaptation. For bacteriophages—with their rapid generation times, straight-forward propagation methods, and highly organized genomes—many life history traits have been readily characterized, making phages useful study systems for understanding genome structure and life history trait evolution. As a result, phages have been used extensively, not only as model systems in molecular biology (Hendrix 1983; Cairns et al. 2007) and structural biology (Chiu et al. 1997; Johnson and Chiu 2007; Bhardwaj et al. 2014; Fokine and Rossmann 2014), but they were also among the first organisms studied using experimental population biology (Chao et al. 1977; Levin et al. 1977). Although some studies on life history traits and tradeoffs within bacteriophages have been done, many facets remain elusive (Goldhill and Turner 2014), and novel phage infection and replication mechanisms continue to be discovered.
Phages and other viruses can often utilize multiple host receptors, and this ability is important for viral evolution and host tropism. This phenomenon has been characterized in a variety of study systems, explaining how the host ranges of viruses as diverse as human viruses and bacteriophages evolve (Morona and Henning 1984; Drexler et al. 1991; Tufaro 1997). Host range is often determined by the same viral structures that determine rate of host cell acquisition—a key life history trait in viruses.
Some virion structural proteins are responsible for host recognition and transfer of the phage genome. Shigella flexneri phage Sf6 is a close relative of the well-known Salmonella phage P22. Its structural proteins include the tailspike, tail needle, portal, two “plug” proteins, and three internal head proteins (gp11, gp12, and gp13). Sf6 initially binds lipopolysaccharide (LPS) through interactions with the tailspike protein (Muller et al. 2008), and then it binds to an outer membrane protein (“Omp”) (Parent et al. 2014; Porcek and Parent 2015). OmpA is the preferred secondary receptor for Sf6, but it can also infect using OmpC when OmpA is removed (Parent et al. 2014). In the absence of both OmpA and OmpC, Sf6 can still infect, albeit at much lower efficiency, possibly by using another, unidentified receptor (Parent et al. 2014; Porcek and Parent 2015). The Sf6 protein(s) responsible for interactions with these secondary receptors has yet to be determined. In P22, initial host attachment is similar to Sf6; tailspike proteins bind and hydrolyze the LPS (Iwashita and Kanegasaki 1976). While work by the Seckler laboratory has suggested that LPS is sufficient for P22 genome ejection in vitro (Andres et al. 2010, 2012), this process is rather slow, and recent work suggests that an Omp is likely also required for release of the internal proteins (Jin et al. 2015). The P22 tail needle and internal proteins are then ejected into the host cell before DNA transfer (Israel 1977; Jin et al. 2015). This is likely also the case in Sf6.
After successful genome ejection, Sf6 is potentially subject to host defenses, including restriction and CRISPR systems. However, S. flexneri lacks restriction-modification systems, and although some Shigella strains possess a CRISPR sequence, many S. flexneri strains do not possess an intact cas gene. It was suggested that this reflects a degrading CRISPR system resulting from mobile genetic disruptions (Yang et al. 2015).
The morphogenetic functions of Sf6 are homologous to those in P22, and the overall structure of the two virions is highly similar (Casjens et al. 2004; Parent et al. 2012). The Sf6 genome is mosaic, and genetic synteny is conserved with other lambdoid phages. Of significance, the lysogenic control functions, the replication proteins, and the general phage recombination systems are conserved between Sf6, P22, and phage λ. The overall pathways of development, including genome circularization, transcription regulation, and DNA replication of all three phages are therefore likely to be highly similar. Two regions of the Sf6 genome warrant specific mention. The antigen conversion genes, which are expressed from the prophage of P22 and Sf6, are nonhomologous and have been described as morons (Cumby et al. 2012). Second, the nin (N-independent) genes of λ are very divergent, both in number and sequence, in the lambdoid phage group. These genes are not essential in the laboratory, they do not appear to be independently mobile, and it was suggested they have arisen from multiple illegitimate recombination events (Casjens et al. 2004). Their functions in phage growth are incompletely understood. However, the Sf6 nin region uniquely contains an intact mobile genetic element: IS911. This IS element is common in Shigella chromosomes (Soldati and Piffaretti 1991) and likely transposed into the ancestral prophage genome of the S. flexneri serotype 3a strain 3–19 (Gemski et al. 1975) where it disrupted one of the nin genes (gene 54) before Sf6 was first isolated. Although small parasitic elements (homing endonucleases, introns, inteins, etc.) are found in many phage genomes (though not Sf6), active transposase elements seem to be rare. Of well-characterized phages, only P1 harbors an active IS element (Iida et al. 1978; Lobocka et al. 2004).
Experiments used to study phage evolution have typically included a variety of model systems. Examples include phages φ6 (Burch and Chao 2000), φX174 and its relatives (Doore and Fane 2016), T7 (Bull and Molineux 2008), and λ (Meyer et al. 2012). Experimental evolution studies using these systems have uncovered important trends in phage evolution. Sf6 has two distinct features important for studying the evolution of phage genetic architecture. The first is “headful packaging” (Eppler et al. 1991; Casjens et al. 2004), where the phage head is filled with DNA until it is “full”; more DNA than a unit genome length is packaged, thus generating progeny that each contain differing genomic architecture as a result of terminal genomic redundancy. The second is that the Sf6 genome harbors a mobile parasitic genetic element that may exert a replication cost.
After 20 rounds of serially passaging 10 independent populations of phage Sf6 on ompA−C−S. flexneri, we found highly parallel evolution of phages. By selecting for faster Sf6 growth on this strain, we anticipated that, in addition to mutants with faster intracellular development, we would observe mutants with an improved rate of Sf6 adsorption and genome ejection. This second group of mutants might be comparable to mutants of phage λ that efficiently adsorb to OmpF when the usual receptor, LamB, is missing (Meyer et al. 2012). However, we saw no changes in structural proteins, and as a result, the mutant phages are not expected to display changes in the rate of cell adsorption. Instead, all of the mutant phages displayed faster life cycles through three distinct types of mutations. The first includes mutations in noncoding regions that affect the lytic-lysogeny control switch (Hendrix 1983; Ptashne 1992; Oppenheim et al. 2005; Svenningsen and Semsey 2014). The second includes various amino acid substitutions in the “holin” protein, which is important in the timing of phage-induced cell lysis (Wang et al. 2000). The third includes rearrangements of genomic architecture: all 10 populations evolved large deletions, and 74% of all clonal isolates tested have this trait.
Materials and Methods
Strains/Media
The strains used were wild-type S. flexneri and its ompA−C− null derivative (Morona et al. 1994; Parent et al. 2014). The ancestral Sf6 phage used was a spontaneous clear mutant that contains a frameshift mutation in gene 39, coding for the prophage repressor (Casjens et al. 2004). Purified phage were stored in a 10 mM Tris (pH 7.6) and 10 mM MgCl2 buffer. LB-Lennox (10 g tryptone, 5 g yeast extract, 5 g NaCl per liter) containing 50 μg/ml kanamycin and 10 μg/ml tetracycline was used for passaging the phage with ompA−C− null S. flexneri, and LB-Lennox without antibiotics was used in the remaining experiments. LB agar (Invitrogen) was used in all plating experiments.
Evolution Scheme
Ten populations of bacteriophage Sf6 were established by infecting separate isogenic ompA−C− null S. flexneri cultures. Ten flasks containing LB-Lennox were seeded as a 1:50 dilution from a single overnight culture. Cultures were incubated for 90 min at 37°C with shaking, and then an individual plaque obtained from an ancestral Sf6 population was added to each flask. Infected cells were incubated and shaken at 200 rpm in a New Brunswick incubator shaker (Excella E24) until the culture cleared. Chloroform was then added to complete lysis, and the phage lysate was centrifuged at 8,000×g for 10 min to remove cell debris. Clarified lysates were stored overnight at 4°C in between passages in the presence of saturating chloroform to prevent growth, and with 10 mM MgCl2 to maintain the stability of the phage. Holding the cell line constant, 19 additional passages were performed by transferring a portion of the previous lysate to pre-incubated cells at a final phage concentration of 0.1% of the culture volume (e.g., corresponding to a 1:1,000 dilution from the previous lysate). Plaques were then isolated from the final lysate (after passage 20) for each of the 10 populations. Final phage stocks were further purified as described (Parent et al. 2012).
As we were expecting to find mutants that adsorbed and infected cells faster, we initially performed Sanger sequencing of several genes encoding the structural proteins from isolates that had the fastest life cycles. No mutations were found. We then tried to increase the likelihood of capturing such mutants by including a “wash” selection step. Phage-infected cells from the 20th passage of Population 1 were quickly centrifuged 5-min post-infection to wash away unadsorbed phage. Cells were then resuspended in fresh media and the culture was allowed to clear. Ten isolated plaques were screened after this wash selection step. No mutations were found in structural genes from these isolates (designated with a “w” in table 1).
Table 1.
Distribution of nin Region Deletions and Gene 61 Mutations
| Isolate | Deletion | Gene 61 | Isolate | Deletion | Gene 61 | Isolate | Deletion | Gene 61 |
|---|---|---|---|---|---|---|---|---|
|
Population 1
|
Population 3
|
Population 7
|
||||||
| 1 | + | − | 1 | − | + | 1 | + | − |
| 2 | + | − | 2 | + | − | 2 | − | + |
| 3 | + | − | 3 | − | + | 3 | + | + |
| 4 | + | − | 4 | − | + | 4 | + | − |
| 5 | + | − | 5 | − | + | 5 | + | − |
| 6 | + | − | 6 | − | + | 6 | + | − |
| 7 | + | − | 7 | + | − | 7 |
+
|
−
|
| 8 | + | − | 8 | + | + |
Population 8
|
||
| 9 | + | − | 9 | − | + | 1 | + | − |
| 10 | + | − | 10 |
−
|
−
|
2 | + | + |
| 1w | + | − |
Population 4
|
3 | + | + | ||
| 2w | + | − | 1 | + | − | 4 | + | + |
| 3w | + | − | 2 | + | − | 5 | + | + |
| 4w | + | − | 3 | + | − | 6 | + | + |
| 5w | + | − | 4 | + | + | 7 | + | + |
| 6w | + | − | 5 | + | + | 8 |
+
|
+
|
| 7w | + | − | 6 | + | − |
Population 9
|
||
| 8w | + | − | 7 | + | − | 1 | − | + |
| 9w | + | − | 8 | + | − | 2 | − | + |
| 10w |
+
|
−
|
9 | + | − | 3 | + | + |
|
Population 2
|
10 |
+
|
+
|
4 | + | + | ||
| 1 | − | + |
Population 5
|
5 | + | − | ||
| 2 | − | + | 1 | − | − | 6 | + | − |
| 3 | + | − | 2 | + | − | 7 |
+
|
+
|
| 4 | − | + | 3a | − | − |
Population 10
|
||
| 5 | + | + | 4 |
+
|
−
|
1 | − | + |
| 6 | − | + |
Population 6
|
2 | − | + | ||
| 7 | − | + | 1 | + | − | 3 | + | − |
| 8 | − | + | 2 | − | + | 4 | + | + |
| 3 | + | + | ||||||
| 4 | + | + | ||||||
Note.—The suffix “w” refers to isolates obtained after a wash step, see “Materials and Methods” section.
aancestral genotype.
Sequencing and Analysis
For Illumina sequencing, genomes were extracted by treating individual phage lysates with RNase A (Sigma–Aldrich) and DNase I (Roche), each at a 1 μg/ml final concentration. Samples were incubated at 37°C for 1 h, and then the DNase was inactivated by incubating at 75°C for 10 min. The samples were then adjusted to contain 0.1 μg/ml proteinase K (Roche) and 0.5% SDS, and were incubated at 55°C for 1 h. Samples were then extracted once with phenol:chloroform (Amresco) and a second time with chloroform. DNA was isolated by ethanol precipitation.
The complete genomes of select isolates (tables 1 and 2) were sequenced in two runs using the Michigan State University Research Technology Support Facility. Libraries were prepared using the Illumina TruSeq Nano Library Preparation Kit and loaded on either a standard or Nano MiSeq flow cell. Sequencing was done using a 2×150 paired end format. FastQ data were analyzed using breseq versions 0.23 and 0.26.0 (Deatherage and Barrick 2014). For the remaining isolates of interest, gene 61 amplicons were Sanger sequenced and the results aligned and analyzed using BLAST (Altschul et al. 1990) and/or the program MEGA (Tamura et al. 2007). To detect nin deletions, PCR and gel electrophoresis were used for screening amplicon sizes. The primers used were: 5′-CGCAGTCCAAGAAGAAAGG-3′ (forward) and 5′-AGACGACCTGAATAAACCCATG-3′ (reverse).
Table 2.
Isolates Selected for Biological Characterization
| Isolate a |
Deletion
|
Gene 61
|
Noncoding
|
|||||
|---|---|---|---|---|---|---|---|---|
| Size (bp) | Location b | Gene Product | Gp61 | Location | NCS | Location | Seq. | |
| 1-3 | 1683 | 32871–34553 | 54–56 | None | – | None | – | I |
| 1-5 | 3428 | 31676–35103 | 50–58 | None | – | None | – | I |
| 2-3 | 1245 | 32875–34119 | 54–56 | None | – | A->G | 27157 | I |
| 2-4 | None | – | – | G88D | 36725, G->A | None | – | I |
| 2-8 | None | – | – | K6T | 36479, A->C | None | – | I |
| 3-1 | None | – | – | I46T | 36599, T->C | ND | ND | S |
| 3-3 | None | – | – | N70S | 36671, A->G | ND | ND | S |
| 3-4 | None | – | – | L67I | 36661, C->A | ND | ND | S |
| 3-5 | None | – | – | S69I | 36668, G->T | ND | ND | S |
| 3-7 | 4006 | 30436–34441 | 46–56 | None | – | None | – | I |
| 3-9 | None | – | – | G66R | 36658, G->C | ND | ND | S |
| 3-10 | None | – | – | None | – | A->G | 27177 | I |
| 4-5 | 2412 | 31979–34390 | 51–56 | I46T | 36599, T->C | None | – | I |
| 5-1 | None | – | – | None | – | T->G | 27147 | I |
| 6-3 | 2516 | 31661–34176 | 50–54 | A28V | 36545, C->T | ND | ND | S |
| 6-4 | 1239 | 32876–34124 | 54–56 | A23S | 36529, G->T | ND | ND | S |
| 7-2 | None | – | – | A55T | 36626, G->A | None | – | I |
| 7-3 | 3995 | 30641–34635 | 46–54 | I82V | 36706, A->G | ND | ND | S |
| 8-1 | 3075 | 31477–34551 | 49–56 | None | – | None | – | I |
| 8-6 | 2320 | 32177–34496 | 51–56 | Y73D | 36679, T->G | None | – | I |
| 8-8 | 3773 | 31520–35292 | 50–58 | Y73D | 36679, T->G | None | – | I |
| 9-1 | None | – | – | Y73D | 36679, T->G | ND | ND | S |
| 9-5 | 3114 | 31223–34336 | 48–56 | None | – | None | – | I |
aOur naming scheme indicates population number, followed by isolate number. For example, 1–10 corresponds to Population 1, plaque 10.
bLocation refers to nucleotide positions within AF547987 and defines the first and last nucleotides of the deletion, I, Illumina whole genome sequencing; S, Sanger sequencing of selected regions; ND, not determined.
Lysis Experiments
Lysis experiments were performed on a Molecular Devices FilterMax F5 plate reader using 96-well plates. An overnight culture of ompA−C− null S. flexneri cells was diluted 1:10 in broth and added to each well. Cells were infected at an multiplicity of infection (MOI) of ∼0.2 and the plates were then incubated at 37°C for 3.5 h. Absorbance at 595 nm was measured every 5 min with vigorous shaking before each read.
Burst Size Calculations
One-step growth experiments of Sf6 on ompA−C− null S. flexneri cells were performed as previously described (Parent et al. 2014). Infected cells were incubated at 37°C for 90 min, and the final burst size was determined by plating on WT S. flexneri.
Statistical Analysis
The general linear model was constructed in R 3.2.3 (site The R Foundation for Statistical Computing http://www.R-project.org).
qPCR of Sf6 Late Gene
For qPCR, a Roche Lightcycler 480 was used. RNA was extracted using a Promega Reliaprep RNA Cell Miniprep System, and cDNA was made using Applied Biosystem’s High Capacity Reverse Transcription Kit. Primers were constructed with Primer3Plus within Sf6 genes 1 (5′-AAGACTTCGCGTTGATACCC-3′, 5′-GTGCTCATCGGTGATGTTTC-3′), 5 (5′-TGCAACAGCAGACCAAACAG-3′, 5′-AAAGCGTAACCGTCACATCG-3′), 44 (5′-GACGCTGATGACGGCTATCA-3′, 5′-GCGTGTCGTCGATGTAAAGC-3′), and 61 (5′-GTTTGCAATGGCGTACCTTC-3′, 5′-CACATCGTTGCGTCGATTAC-3′) and were used with an in-house SYBR Green master mix as described (Bryant et al. 2008).
Restriction Enzyme Digests to Show Terminal Redundancy
Single restriction enzyme digests used BamHI HF and PstI HF (New England Biolabs) at 37°C for 15 min. Gel electrophoresis used 0.7% agarose in TAE and was run for 90 min at a constant 80 V. The ladder used was 1 kb Plus (Invitrogen).
Results
Serial Evolution and Resulting Evolved Populations
We conducted a phage evolution experiment for 10 replicate populations in which the phage, but not the host, were serially passaged on ompA−C− null Shigella, a host to which Sf6 only slowly adsorbs (Parent et al. 2014) (fig. 1). We started the populations of Sf6 from clones of a common ancestor and evolved each for 20 serial rounds of culture growth, corresponding to >100 generations of phage progeny (Parent et al. 2014). By the 20th passage, we observed faster clearing of the host cultures than in earlier passages, indicating that the evolved phage populations more quickly lysed the available hosts. To investigate the genetic basis of the faster phage life cycle, we made high-titer preparations from single plaque isolates obtained from the lysate of the 20th passage for each of the 10 populations. We attempted to choose diverse isolates, but in general, there was little discernable difference in plaque morphology. In total, 82 plaque isolates from the final 10 lysates were tested further.
Fig. 1.—
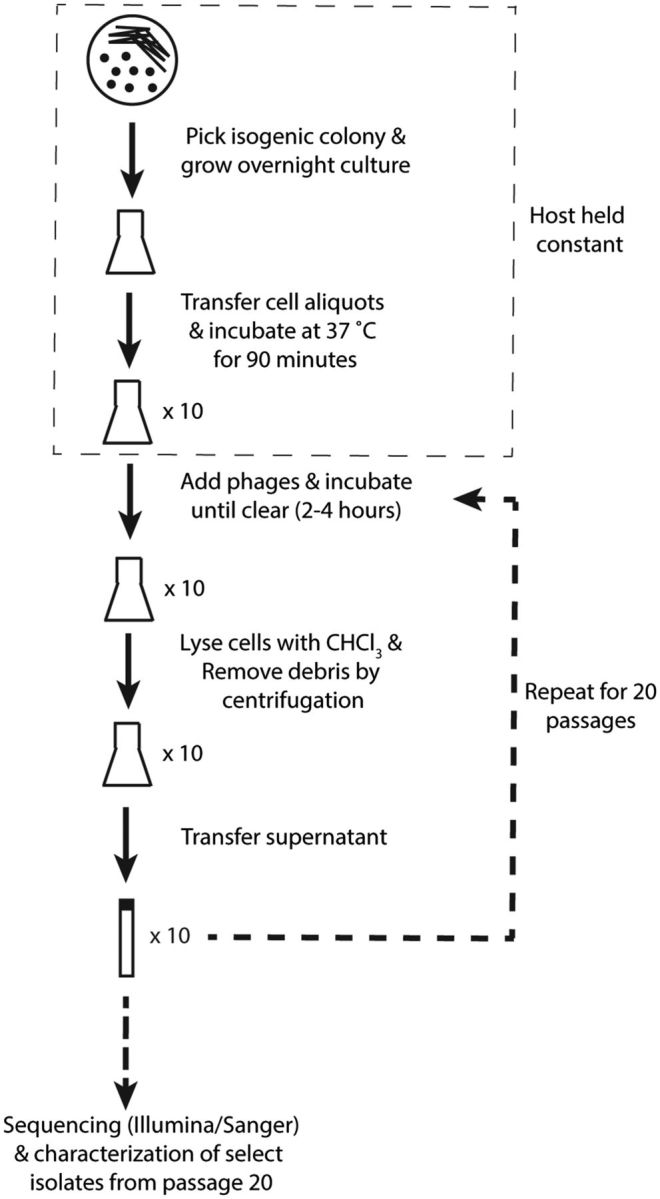
Sf6 serial evolution. A cartoon schematic of the evolution scheme is shown.
All but one isolate had a faster life cycle phenotype than the ancestor. We performed whole-genome, Illumina sequencing on 25 barcoded isolates selected from several populations. As a control, the ancestral phage genome was sequenced, and no mutations were found relative to the reference genome (GenBank accession AF547987). PCR amplification and Sanger sequencing in selected regions were used to characterize the remaining 57 isolates.
Parallel Evolution at the Gene Level
The most common mutation type, found in 61 out of the 82 isolates from all 10 populations, included a deletion affecting a specific region of the Sf6 genome. Each deletion removed part or all of genes 46–58 (table 1), and the observed deletion lengths ranged from ∼1.2 to 4 kb. This region is homologous to a nonessential region in phage lambda, called nin, for “N-independent”, and we maintain this nomenclature here. The second most common type, found in 37 of the 82 isolates and 8 out of 10 populations, comprised 13 different nonsynonymous mutations in gene 61, which encodes for holin (table 2). The least common type, found in 3 of the 25 fully sequenced isolates that arose in three different populations (table 2), were mutations in noncoding regions that likely affect the activity of the major early promoter PR. Mutations in this region of related phages, including P22, λ, and 434 (the latter having 100% nucleotide identity to Sf6 in this region), have been intensively studied (Ptashne 1992; Oppenheim et al. 2005; Svenningsen and Semsey 2014) both in terms of promoter activity determinants and lysis-lysogeny control and were not further investigated here. We observed no other mutation—synonymous or nonsynonymous—outside these three types, strongly suggesting that the observed parallel mutation types were adaptive (Wichman et al. 1999). Notably, only 1 of the 82 isolates, from Population 5, had the ancestral genotype (designated as “a” in table 1).
For the majority of Sf6 isolates (61 of the 82) either from fully sequenced genomes or characterized by PCR screens of selected areas, we observed only one mutation (counting a deletion as a single mutation) per genome. Twenty isolates, from 8 of the 10 populations, contained two mutations per genome (tables 1 and 2), and no isolates contained more than two mutations. Therefore, determining the genotype/phenotype relationship for the majority of evolved Sf6 isolates was straightforward. From the initial 82 isolates, we focused on 23 that were genotypically unique for biological characterization (table 2).
Sf6 does not adsorb efficiently to the ompA−C− null Shigella host, and despite specific attempts to isolate mutants that adsorbed faster (see “Materials and Methods” section), we did not find any mutations in structural genes. Perhaps such mutations do not confer as large an effect on the overall phage life cycle as those analyzed here, or perhaps multiple mutations are required and further evolution would have been necessary for them to become prevalent in the population.
Holin Variants Result in Phage with Faster Life Cycles via Earlier Lysis
Of the phages containing nonsynonymous mutations in gene 61 (codes for holin, or “gp61”), some also had nin region deletions. We examined 9 isolates, from 4 of the 10 populations, that contained only a gene 61 mutation to determine the biological effect of the various amino acid substitutions (see supplementary table ST1, Supplementary Material online). The time to lysis for each variant was compared with the ancestor using the ompA−C− null S. flexneri host (fig. 2). A low MOI, to allow multiple cycles of cell growth before culture lysis, was used in order to enhance assay sensitivity. The initial increase in absorbance at 595 nm was due to growth of uninfected cells, but it is followed by a decrease that corresponds to culture lysis. All nine gp61 variants resulted in faster culture lysis than the ancestral phage (supplementary table ST1, Supplementary Material online).
Fig. 2.—

gp61 variants display faster times to lysis. Representative growth curves for the ancestral Sf6 and two holin variants grown on the S. flexneri ompA−C− host are shown. Arrows indicate the time at which lysis begins. Three technical and three biological replicates were performed with each phage.
Faster cell lysis decreases the number of progeny released (i.e., “burst size”) in phage lambda (Shao and Wang 2008). Therefore, we assayed the burst size of each Sf6 gp61 variant in a single cycle of growth on ompA−C− null S. flexneri. The burst size was calculated by titering and normalizing to the number of input infected cells, as described previously for ancestral Sf6 (Parent et al. 2014). All nine gp61 holin mutants yielded a decreased burst compared with the ancestor (supplementary table ST1, Supplementary Material online). Our results suggest that Sf6, like other phages, faces an antagonistically pleiotropic trade-off between burst size and lysis time (Abedon et al. 2003; Heineman and Bull 2007; Shao and Wang 2008).
Type I holins, such as λ S105, have an experimentally determined topology consisting of three transmembrane alpha helices (Grundling et al. 2000; Young 2014). Sf6 and λ S105 holins differ by only a single amino acid (residue 103), and therefore, the topology of the two proteins is likely indistinguishable. We mapped all 13 gp61 amino acid substitutions onto the predicted topology of Sf6 holin and found that the majority were present in any one of the three transmembrane helices or a periplasmic loop that connects the second and third transmembrane helix (fig. 3). Most Sf6 gp61 variants found contain different amino acid residues from those described in λ (Wang et al. 2000; Shao and Wang 2008), but they all likely function similarly in that the end result is earlier lysis of the infected cell. Lysis timing is a function of the aggregation of two proteins termed “holin” and “antiholin”. These proteins have the same genetic locus but result from alternative start codons. Holes form in the cytoplasmic membrane when holin/antiholin aggregates trigger a small drop in the membrane potential (White et al. 2011). Our mutants (as well as those described in λ) affect both holin and anitholin and could affect either the aggregation step or sensitivity to changes in the membrane potential.
Fig. 3.—

Holin topology. The PSI-PRED prediction server was used to predict the topology of holin using the ancestral Sf6 sequence as the input (McGuffin et al. 2000). The 13 different amino acid substitutions are shown. The N-terminus is in the periplasmic space, and the C-terminus is in the cytoplasm.
Deletions Also Result in Phage with Faster Life Cycles via Earlier Lysis
Alignment of deletions in our isolates to the Sf6 reference genome (Casjens et al. 2004) show that all deletions occur within the nin region and overlap an area containing the IS911 insertion sequence (fig. 4). Bacterial insertion sequences are autonomous transposable elements with the potential to indiscriminately disrupt genes, often causing fitness costs (Chandler and Fayet 1993). IS911 is a member of the IS3 family and excises itself from a genome during nonreplicative transposition (Chandler and Fayet 1993). It can also be deleted by homologous recombination over direct repeats in flanking DNA. Examination of sequences flanking each deletion revealed that 4 of the 10 populations contained deletions flanked by direct repeats, suggesting that they may be products of homologous recombination. Direct repeats have allowed the isolation of deletion mutants in other phage genomes (Studier et al. 1979; Kroger and Hobom 1982; Kong and Masker 1994). However, 9 of the 13 deletions sequenced were likely mediated by IS911 auto-excision. Repair of the resulting dsDNA break usually requires loss of some flanking DNA, which explains the variety of deletions observed. It is logical to conclude that removal of IS911 is beneficial to rapid Sf6 growth in an environment with high host-cell density (such as the adaptations here) and that selection strongly favors phages that lose this molecular parasite.
Fig. 4.—

Overlay of deletions. Representative isolates containing deletions are shown mapped onto the ancestral genome. Every deletion removes the entire IS911 element.
We developed a PCR-based deletion polymorphism assay using primers just outside the Sf6 nin region in order to rapidly screen for deletions using agarose gels. The largest amplicon corresponds to the presence of the entire nin region (the ancestor), and the shortest amplicon corresponds to the largest deletion (fig. 5A). The assay was then used to qualitatively probe the presence of phage containing deletions at the population level. After 20 serial passages, all 10 populations contain at least one deletion type, and several populations contain multiple types (fig. 5B).
Fig. 5.—

Deletion polymorphism and population diversity. Agarose gels (1%) stained with ethidium bromide show changes in PCR product length resulting from nin region deletions. (A) Individual isolates clearly demonstrate different product lengths. The longest amplicon corresponds to the ancestral genotype, which contains the entire nin region. The shortest amplicon corresponds to the isolate with the longest deletion. (B) Distribution of deletion phage within mixed populations for each of the 10 lineages after the 20th passage. Timeline of deletion emergence and selective sweeps in representative populations: (C) Population 3 and (D) Population 8.
We then asked whether multiple deletion types were maintained by clonal interference or if particular deletions outcompeted others throughout the evolutionary time of our experiment. We therefore tracked the emergence of deletions as a function of evolutionary time in representative populations. For example, in Population 3, one type of deletion was detected as early as passage 10, and by passage 20, there had been a clear selective sweep (fig. 5C). In Population 8, deletions were obvious by passage 5, and by passage 10, the ancestral genome was undetectable (fig. 5D). However, unlike Population 3, Population 8 did not show a selective sweep. Instead, a mixture of deletion types co-existed, an indicator of high diversity, even after >100 generations of phage growth. The presence of multiple deletion types may be due to clonal interference (Gerrish and Lenski 1998), which can increase the time before one lineage sweeps through a population (Maddamsetti et al. 2015). Alternatively, the Population 8 lineages may have accumulated other mutations, resulting in frequency dependence. We have no evidence that helps distinguish between these possibilities.
IS911 deletions result in a faster Sf6 life cycle. Five isolates, containing different deletions but no other mutation (table 2), were chosen for lysis time and burst size assays. All mutants displayed faster lysis and smaller bursts when compared with the ancestral Sf6 (supplementary table ST2, Supplementary Material online).
As deletions were common when Sf6 was passaged on ompA−C− null S. flexneri, we asked if deletions would also be selected for when Sf6 was passaged on a wild-type host. We used the same serial passaging protocol for two Sf6 populations evolving on ompA+, ompC+S. flexneri for 10 passages. Using the PCR polymorphism assay described earlier, we observed the emergence of a deletion in one of the two populations (data not shown). Therefore, deletions of IS911 from the Sf6 genome are not confined to the ompA−C− null strain and may be generally adaptive in this particular environment.
Faster Lysis Due to Holin Variation and IS911 Deletions Are Additive
We compared lysis times and burst sizes of all 23 genotypically unique isolates—including double mutants. Data were normalized so that the ancestral phage had a relative lysis time and burst of 1. Lysis time was positively correlated with burst size (fig. 6). When examining the effect of two mutations per genome, isolates containing both a holin amino acid substitution and an IS911 deletion were fastest of all, indicating that both changes contribute to faster lysis and appear additive. Overall, the high degree of parallelism at the genotype and phenotype levels strongly suggests that both the holin substitutions and deletion mutations are selectively advantageous, independently conferring increased fitness relative to the common ancestral phage. Previous reports have shown that phage evolution can lead to faster lysis time when holin is affected (Abedon et al. 2001, 2003; Bull et al. 2004; Shao and Wang 2008). However, it is not obvious how large genomic deletions in Sf6 directly cause faster time to lysis.
Fig. 6.—

Relative lysis time and burst size of unique isolates. The data were normalized so that the ancestral phage represents a relative lysis time of 1. Lysis time was positively correlated with burst size (general linear model, F1,22= 66.2, P < 0.001). Asterisks indicate isolates containing both a gene 61 variation and a deletion, but where the same gene 61 variation was not isolated as a single mutation.
Nin Region Deletions Do Not Result in Earlier Transcription of Late Genes in Sf6
To understand the molecular basis of the Sf6 deletions, we compared the genetic architecture of ancestral Sf6, the evolved Sf6 mutants, and phage λ, which is a well-established molecular biology model system. Genomic comparisons suggest that there are 30–50 genes in the entire nin region gene pool with individual phages containing a subset of ∼10 genes (Juhala et al. 2000), resulting in considerable diversity. Sf6 has 10 nin genes, and two appear nonfunctional—one resulting from a premature nonsense codon and the other resulting from disruption with the IS911 element (Casjens et al. 2004).
In λ, the nin region houses several regulatory elements, and their presence or absence affects transcription regulation and timing (Gottesman and Weisberg 2004). Nin deletions lead to full-length early transcripts without the need for synthesizing the N transcription anti-terminator protein (hence the designation of nin, for N-independent). This, in turn, leads to earlier production of the second anti-terminator Q protein, which results in earlier transcription of late genes compared with λ having a full-length genome (Costantino et al. 1990). We developed a qPCR assay to determine transcription timing of the late genes using the ancestral Sf6 and two deletion isolates that have the fastest and slowest life cycles. We focused on three late genes: gene 1 (small terminase subunit), gene 5 (coat protein), and gene 61 (holin). We also included a fourth gene: gene 44 (DNA replication initiation protein), which is transcribed earlier than nin genes and serves as a control.
Aliquots of phage-infected ompA−C− null S. flexneri were taken every 10-min post-infection from 0 to 30 min at 37°C. Single step growth curves of the deletion isolates (data not shown) and the ancestor (Parent et al. 2014) indicated that, at these times, cells have not lysed. These times are also within the range of λ late gene expression (Liu et al. 2013). RNA was extracted, and cDNA was generated for each sample. There was no obvious difference in the onset of transcription for the ancestor relative to the nin region deletion isolates for the genes tested (fig. 7). Although perhaps not statistically significant, after 10-min infection, there were slightly fewer late gene transcripts from the deletion isolates than from the ancestral phage. Close inspection of the nin region sequences in all of our deletion mutants revealed that a predicted transcriptional terminator was retained. This is in contrast to λ, where selection for N-independence has to remove the homologous terminator. We are thus left with the question: if Sf6 IS911 deletions do not induce earlier transcription of late genes, why are these deletions adaptive?
Fig. 7.—

Sf6 deletions do not change late transcriptional regulation. Results were normalized to the relative concentration of transcripts of the ancestor for each gene. Three biological replicates for the ancestor and two isolates were used; each biological replicate had four technical replicates.
Nin Region Deletions Result in Sf6 Progeny with Increased Terminal Redundancy
DNA packaging strategy (discrete vs. headful) is a critical difference between phages λ and Sf6. Phage λ packages discrete copies of its genome, and isolates with deletions simply result in phage with shorter genomes packaged into capsids (Parkinson and Huskey 1971; Feiss et al. 1977). There is an apparent balance over the control of genome length in λ to maximize phage fitness. Phages with shorter genomes are adaptive as they can be both replicated and transcribed faster (Costantino et al. 1990). Yet, if the genome becomes too short, there are fitness costs due to inefficient packaging (Feiss et al. 1977; Feiss and Siegele 1979). Unlike λ, Sf6 packages by a headful mechanism (Casjens et al. 2004; Leavitt et al. 2013); even in the ancestral particles, more than a discrete genome is packaged, and there is therefore a terminal redundancy. As a result of headful packaging, Sf6 deletion mutants should package the same total length of DNA, and the terminal redundancies will therefore increase. Consequently, the genetic architecture of deletion mutant phage should differ from that of the ancestor.
To confirm that deletion mutants still package by a headful mechanism, DNA was extracted from both the ancestral Sf6 and deletion isolates and electrophoresed on a low percentage agarose gel. Small differences (∼0.4 kb) in genome lengths can be detected (Jin et al. 2015), and we should readily detect 1.2–4 kb changes if the deletion mutants packaged shorter genomes. As expected, all had the same genome length as the Sf6 ancestor (data not shown), confirming that headful packaging was still occurring. The extracted DNA was then analyzed by restriction enzyme digests to detect changes in genomic architecture relative to ancestral Sf6. Isolates with deletions produced altered restriction digestion patterns when compared with the wild-type ancestor (fig. 8). Taken together, these experiments clearly demonstrate that Sf6 deletion mutants are still headful packagers and that they have altered genomic architecture and increased terminal redundancies.
Fig. 8.—

Effect of terminal redundancy on Sf6 progeny. Agarose gel (1%) stained with ethidium bromide shows restriction digestion pattern changes between ancestral Sf6 and two representative deletions.
Discussion
Our data suggest that Sf6 can evolve a faster life history trait adaptation through three distinct types of mutations: lytic/lysogeny control, holin gene mutations, and deletion-mediated terminal redundancy increases. The first two have been extensively characterized in other systems (Lewis et al. 1998; Wang et al. 2000), and we fully expect that the comparable Sf6 mutants studied here exert their effects by similar mechanisms. The third mechanism, which was the most common in our experiments, corresponds to large genomic deletions, and to our knowledge, this has not been previously reported in experimental evolution studies of headful packaging phage. Sf6 deletion mutants display both faster time to lysis and smaller bursts, which is suggestive of a compressed life cycle. Surprisingly, no mutations were found in any Sf6 structural genes. If beneficial mutations affecting cell entry arose anytime during the course of our study, they likely did not increase fitness enough to be detectable in our sample size of 82 isolates. Rather, it is likely that our selection scheme favored faster life cycle adaptations.
Several hypotheses can be offered to explain how deletions in Sf6 are beneficial to the phage life cycle. (1) IS911 itself is highly deleterious to phage development: IS911 is transcribed in the opposite orientation to Sf6 late genes, which can lead to phage gene transcription interference, or transposase activity may be detrimental. In either case, removal of IS911 would increase Sf6 fitness. (2) Deletions within the nin region could be beneficial by simply removing “extraneous” information that would otherwise need to be replicated and transcribed, slowing down the phage life cycle. (3) A longer terminal redundancy could increase the efficiency of genome circularization after infection, as observed in the related phage, P22 (Botstein and Matz 1970). Genome circularization is an essential early step in P22 development and is therefore likely to play the same role in Sf6, which has an erf homolog (“erf” = essential recombination function) (Casjens et al. 2004). (4) Removing large portions of the genome effectively increases the relative density of essential genes in replicating DNA that express potentially fitness-limiting functions such as structural proteins or the holin. These and other ideas are not mutually exclusive, and their effects could even be additive.
Evolutionary Implications of Deletions in Sf6
We created a model to estimate the extent to which deletions affect genetic architecture with increased terminal redundancy in Sf6 progeny (see supplementary information and supplementary fig. S1, Supplementary Material online). The large deletions result in duplications of entire genes within the packaged DNA of progeny phage (supplementary fig. S1, Supplementary Material online). Although all phage progeny are “clonal” descendants of a common parent, the model shows that terminal redundancy causes progeny to hold uniquely packaged DNA. This observation has some implications for subsequent phage and host evolution. As the total length of the discrete genome is reduced, deletion mutants can more readily acquire additional genetic information by recombination and remain able to propagate as infective particles. Therefore, deletion mutants could acquire new DNA and transfer this information to the next host.
Our studies show a highly parallel and novel mode of deletion-mediated adaptation and shed light on a previously unreported area of phage genetics. Genomic architecture is the framework within which genes and regulatory elements evolve. Understanding the genetics of headful packaging phage is important for understanding the evolution and ecology of many phages, their hosts, and complex biomes that allow ready genetic exchange between phages and hosts (Lepage et al. 2013; Penades et al. 2015).
Supplementary Material
Acknowledgments
Thanks to friends who discussed data and inspired us: Drs Richard E. Lenski at Michigan State University for experimental evolution discussions, as well as Sherwood R. Casjens at the University of Utah, and Bentley A. Fane at the University of Arizona for phage biology discussions. Thanks to Dr Monique Floer and graduate student Mohita Tagore (Michigan State University) for allowing us to use their qPCR system and providing advice on experimental design and interpretation. This material is based upon work supported by National Institutes of Health, R01GM110185 to K.N.P., and the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1424871, awarded to A.R.B.
Literature Cited
- Abedon ST, Herschler TD, Stopar D. 2001. Bacteriophage latent-period evolution as a response to resource availability. Appl Environ Microbiol. 67:4233–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedon ST, Hyman P, Thomas C. 2003. Experimental examination of bacteriophage latent-period evolution as a response to bacterial availability. Appl Environ Microbiol. 69:7499–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Andres D, et al. 2010. Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J Biol Chem. 285:36768–36775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres D, et al. 2012. Tail morphology controls DNA release in two Salmonella phages with one lipopolysaccharide receptor recognition system. Mol Microbiol. 83:1244–1253. [DOI] [PubMed] [Google Scholar]
- Bhardwaj A, Olia AS, Cingolani G. 2014. Architecture of viral genome-delivery molecular machines. Curr Opin Struct Biol. 25:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botstein D, Matz MJ. 1970. A recombination function essential to the growth of bacteriophage P22. J Mol Biol. 54:417–440. [DOI] [PubMed] [Google Scholar]
- Bryant GO, et al. 2008. Activator control of nucleosome occupancy in activation and repression of transcription. PLoS Biol. 6:2928–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Molineux IJ. 2008. Predicting evolution from genomics: experimental evolution of bacteriophage T7. Heredity 100:453–463. [DOI] [PubMed] [Google Scholar]
- Bull JJ, Pfennig DW, Wang IN. 2004. Genetic details, optimization and phage life histories. Trends Ecol Evol. 19:76–82. [DOI] [PubMed] [Google Scholar]
- Burch CL, Chao L. 2000. Evolvability of an RNA virus is determined by its mutational neighbourhood. Nature 406:625–628. [DOI] [PubMed] [Google Scholar]
- Cairns J, Stent GS, Watson JD. 2007. Phage and the origins of molecular biology. Centennial ed. Cold Spring Harbor (NY: ): Cold Spring Harbor Laboratory Press. [Google Scholar]
- Casjens S, et al. 2004. The chromosome of Shigella flexneri bacteriophage Sf6: complete nucleotide sequence, genetic mosaicism, and DNA packaging. J Mol Biol. 339:379–394. [DOI] [PubMed] [Google Scholar]
- Chandler M, Fayet O. 1993. Translational frameshifting in the control of transposition in bacteria. Mol Microbiol. 7:497–503. [DOI] [PubMed] [Google Scholar]
- Chao L, Levin BR, Stewart FM. 1977. A complex community in a simple habitat: an experimental study with bacteria and phage. Ecology 58:369–378. [Google Scholar]
- Chiu W, Burnett RM, Garcea RL. 1997. Structural biology of viruses. New York: Oxford University Press. p. 484. [Google Scholar]
- Costantino N, Zuber M, Court D. 1990. Analysis of mutations in the ninR region of bacteriophage lambda that bypass a requirement for lambda N antitermination. J Bacteriol. 172:4610–4615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumby N, Davidson AR, Maxwell KL. 2012. The moron comes of age. Bacteriophage 2:225–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol Biol. 1151:165–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doore SM, Fane BA. 2016. The microviridae: diversity, assembly, and experimental evolution. Virology 491:45–55. [DOI] [PubMed] [Google Scholar]
- Drexler K, Dannull J, Hindennach I, Mutschler B, Henning U. 1991. Single mutations in a gene for a tail fiber component of an Escherichia coli phage can cause an extension from a protein to a carbohydrate as a receptor. J Mol Biol. 219:655–663. [DOI] [PubMed] [Google Scholar]
- Eppler K, Wykoff E, Goates J, Parr R, Casjens S. 1991. Nucleotide sequence of the bacteriophage P22 genes required for DNA packaging. Virology 183:519–538. [DOI] [PubMed] [Google Scholar]
- Feiss M, Fisher RA, Crayton MA, Egner C. 1977. Packaging of the bacteriophage lambda chromosome: effect of chromosome length. Virology 77:281–293. [DOI] [PubMed] [Google Scholar]
- Feiss M, Siegele DA. 1979. Packaging of the bacteriophage lambda chromosome: dependence of cos cleavage on chromosome length. Virology 92:190–200. [DOI] [PubMed] [Google Scholar]
- Fokine A, Rossmann MG. 2014. Molecular architecture of tailed double-stranded DNA phages. Bacteriophage 4:e28281.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemski P, Jr., Koeltzow DE, Formal SB. 1975. Phage conversion of Shigella flexneri group antigens. Infect Immun. 11:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish PJ, Lenski RE. 1998. The fate of competing beneficial mutations in an asexual population. Genetica 102-103:127–144. [PubMed] [Google Scholar]
- Goldhill DH, Turner PE. 2014. The evolution of life history trade-offs in viruses. Curr Opin Virol. 8:79–84. [DOI] [PubMed] [Google Scholar]
- Gottesman ME, Weisberg RA. 2004. Little lambda, who made thee? Microbiol Mol Biol Rev. 68:796–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Bläsi U, Young R. 2000. Biochemical and genetic evidence for three transmembrane domains in the class I holin, λ S. J Biol Chem. 275:769–776. [DOI] [PubMed] [Google Scholar]
- Heineman RH, Bull JJ. 2007. Testing optimality with experimental evolution: lysis time in a bacteriophage. Evolution 61:1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix RW. 1983. Lambda II. Cold Spring Harbor Laboratory: Cold Spring Harbor NY [Google Scholar]
- Iida S, Meyer J, Arber W. 1978. The insertion element IS1 is a natural constituent of coliphage P1 DNA. Plasmid 1:357–365. [DOI] [PubMed] [Google Scholar]
- Israel V, et al. 1977. E proteins of bacteriophage P22. J. Virol. 23:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwashita S, Kanegasaki S. 1976. Enzymic and molecular properties of base-plate parts of bacteriophage P22. Eur J Biochem. 65:87–94. [DOI] [PubMed] [Google Scholar]
- Jin Y, et al. 2015. Bacteriophage P22 ejects all of its internal proteins before its genome. Virology 485:128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Chiu W. 2007. DNA packaging and delivery machines in tailed bacteriophages. Curr Opin Struct Biol. 17:237–243. [DOI] [PubMed] [Google Scholar]
- Juhala RJ, et al. 2000. Genomic sequences of bacteriophages HK97 and HK022: pervasive genetic mosaicism in the lambdoid bacteriophages. J Mol Biol. 299:27–51. [DOI] [PubMed] [Google Scholar]
- Kong D, Masker W. 1994. Deletion between direct repeats in T7 DNA stimulated by double-strand breaks. J Bacteriol. 176:5904–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroger M, Hobom G. 1982. Structural analysis of insertion sequence IS5. Nature 297:159–162. [DOI] [PubMed] [Google Scholar]
- Leavitt JC, Gilcrease EB, Wilson K, Casjens SR. 2013. Function and horizontal transfer of the small terminase subunit of the tailed bacteriophage Sf6 DNA packaging nanomotor. Virology 440:117–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepage P, et al. 2013. A metagenomic insight into our gut’s microbiome. Gut 62:146–158. [DOI] [PubMed] [Google Scholar]
- Levin BR, Stewart FM, Chao L. 1977. Resource-limited growth, competition, and predation: a model and experimental studies with bacteria and bacteriophage. Am Nat. 111:3–24. [Google Scholar]
- Lewis RJ, Brannigan JA, Offen WA, Smith I, Wilkinson AJ. 1998. An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J Mol Biol. 283:907–912. [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang H, Gu Z, Roberts JW. 2013. High-resolution view of bacteriophage lambda gene expression by ribosome profiling. Proc Natl Acad Sci U S A. 110:11928–11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobocka MB, et al. 2004. Genome of bacteriophage P1. J Bacteriol. 186:7032–7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddamsetti R, Lenski RE, Barrick JE. 2015. Adaptation, Clonal Interference, and Frequency-Dependent Interactions in a Long-Term Evolution Experiment with Escherichia coli. Genetics 200:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L. J., Bryson K., Jones D. T. 2000. The PSIPRED protein structure prediction server. Bioinformatics. 4:404–5. [DOI] [PubMed] [Google Scholar]
- Meyer JR, et al. 2012. Repeatability and contingency in the evolution of a key innovation in phage lambda. Science 335:428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morona R, Henning U. 1984. Host range mutants of bacteriophage Ox2 can use two different outer membrane proteins of Escherichia coli K-12 as receptors. J Bacteriol. 159:579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morona R, Mavris M, Fallarino A, Manning PA. 1994. Characterization of the rfc region of Shigella flexneri. J Bacteriol. 176:733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller JJ, et al. 2008. An intersubunit active site between supercoiled parallel beta helices in the trimeric tailspike endorhamnosidase of Shigella flexneri Phage Sf6. Structure 16:766–775. [DOI] [PubMed] [Google Scholar]
- Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S. 2005. Switches in bacteriophage lambda development. Annu Rev Genet. 39:409–429. [DOI] [PubMed] [Google Scholar]
- Parent KN, et al. 2014. OmpA and OmpC are critical host factors for bacteriophage Sf6 entry in Shigella. Mol Microbiol. 92:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent KN, Gilcrease EB, Casjens SR, Baker TS. 2012. Structural evolution of the P22-like phages: comparison of Sf6 and P22 lapsid and virion architectures. Virology 427:177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JS, Huskey RJ. 1971. Deletion mutants of bacteriophage lambda. I. Isolation and initial characterization. J Mol Biol. 56:369–384. [DOI] [PubMed] [Google Scholar]
- Penades JR, Chen J, Quiles-Puchalt N, Carpena N, Novick RP. 2015. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 23:171–178. [DOI] [PubMed] [Google Scholar]
- Porcek NB, Parent KN. 2015. Key residues of S. flexneri OmpA mediate infection by bacteriophage Sf6. J Mol Biol. 427:1964–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptashne M. 1992. A genetic switch. 2nd ed. Cold Spring Harbor: Blackwell Scientific Publications. [Google Scholar]
- Shao Y, Wang IN. 2008. Bacteriophage adsorption rate and optimal lysis time. Genetics 180:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldati L, Piffaretti JC. 1991. Molecular typing of Shigella strains using pulsed field gel electrophoresis and genome hybridization with insertion sequences. Res Microbiol. 142:489–498. [DOI] [PubMed] [Google Scholar]
- Studier FW, Rosenberg AH, Simon MN, Dunn JJ. 1979. Genetic and physical mapping in the early region of bacteriophage T7 DNA. J Mol Biol. 135:917–937. [DOI] [PubMed] [Google Scholar]
- Svenningsen SL, Semsey S. 2014. Commitment to lysogeny is preceded by a prolonged period of sensitivity to the late lytic regulator Q in bacteriophage lambda. J Bacteriol. 196:3582–3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol Evol. 24:1596–1599. [DOI] [PubMed] [Google Scholar]
- Tufaro F. 1997. Virus entry: two receptors are better than one. Trends Microbiol. 5:257–258. [DOI] [PubMed] [Google Scholar]
- Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol. 54:799–825. [DOI] [PubMed] [Google Scholar]
- White R, et al. 2011. Holin triggering in real time. Proc Natl Acad Sci U S A. 108:798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wichman HA, Badgett MR, Scott LA, Boulianne CM, Bull JJ. 1999. Different trajectories of parallel evolution during viral adaptation. Science 285:422–424. [DOI] [PubMed] [Google Scholar]
- Yang C, et al. 2015. Polymorphism of CRISPR shows separated natural groupings of Shigella subtypes and evidence of horizontal transfer of CRISPR. RNA Biol. 12:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. 2014. Phage lysis: three steps, three choices, one outcome. J Microbiol. 52:243–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.