

Abstract
A comprehensive and reliable eukaryotic tree of life is important for many aspects of biological studies from comparative developmental and physiological analyses to translational medicine and agriculture. Both gene-rich and taxon-rich approaches are effective strategies to improve phylogenetic accuracy and are greatly facilitated by marker genes that are universally distributed, well conserved, and orthologous among divergent eukaryotes. In this article, we report the identification of 943 low-copy eukaryotic genes and we show that many of these genes are promising tools in resolving eukaryotic phylogenies, despite the challenges of determining deep eukaryotic relationships. As a case study, we demonstrate that smaller subsets of ∼20 and 52 genes could resolve controversial relationships among widely divergent taxa and provide strong support for deep relationships such as the monophyly and branching order of several eukaryotic supergroups. In addition, the use of these genes resulted in fungal phylogenies that are congruent with previous phylogenomic studies that used much larger datasets, and successfully resolved several difficult relationships (e.g., forming a highly supported clade with Microsporidia, Mitosporidium and Rozella sister to other fungi). We propose that these genes are excellent for both gene-rich and taxon-rich analyses and can be applied at multiple taxonomic levels and facilitate a more complete understanding of the eukaryotic tree of life.
Keywords: fungal phylogeny, eukaryotic phylogeny, single-copy genes, phylogenomics
Introduction
A eukaryotic tree of life provides the evolutionary framework for many facets of life sciences, including investigation of evolutionary origin and history of developmental and physiological characteristics, inferences of structural and functional relatedness, understanding of ecological interactions, translational medicine, and crop improvements. Methodological advances in sequencing and phylogenetic reconstruction have led to a substantial progress toward the goal of reconstructing the tree of life (e.g., Ciccarelli et al. 2006). Recent molecular phylogenies supported the classification of eukaryotes into five supergroups (Adl et al. 2012; Katz 2012): (1) Amoebozoa (e.g., the free-living model organism Dictyostelium discoideum and the anaerobic parasite Entamoeba histolytica which infects millions of humans), (2) Archaeplastida (e.g., green plants, red algae, and glaucophytes), (3) Excavata (mostly single-cell free-living heterotrophs and parasites, such as Giardia, which causes giardiasis, and Trichomonas, which causes trichomoniasis), (4) Opisthokonta (e.g., animals and fungi), and (5) SAR, which consists of stramenopiles (e.g., diatoms and oomycetes, characterized by tripartite tubular hairs on one of the flagella), alveolates (e.g., apicomplexans like Plasmodium, ciliates like Tetrahymena, dinoflagellates, and other protists that have cortical alveoli), and Rhizaria (a diverse set of protists without clearly shared cellular or molecular characteristics). These supergroups probably resulted from the deepest divergences in the extant eukaryotic tree of life. However, current understanding of eukaryotic phylogeny is still rather incomplete; the eukaryotic diversity is only sparsely represented in tree of life studies. Uncertainties still exist regarding relationships within and among major eukaryotic lineages (Parfrey et al. 2006), although many of which are being addressed by recent studies (e.g., Parfrey et al. 2010; Katz and Grant 2014).
Previous studies suggest that increasing both gene and taxon sampling densities are important to improve the accuracy of phylogenetic inference (Rokas et al. 2003, Rokas and Carroll 2005; Hedtke et al. 2006; Jeffroy et al. 2006; Townsend and Lopez-Giraldez 2010): the sampling of more genes provides much greater resolving power than traditional single gene-scale phylogenetics, thus overcoming stochastic errors; the sampling of more taxa reduces systematic errors, such as Long-Branch Attraction (LBA), which might be greatly amplified with increased gene sampling and could lead to highly supported yet incorrect topologies in phylogenomic analyses (Jeffroy et al. 2006). No matter which strategy is adopted, marker genes must be carefully chosen to avoid the violation of the orthology assumption; such violation might be due to gene duplication and/or horizontal gene transfer (HGT) and result in incongruence between gene phylogeny and species phylogeny. To examine the orthology of selected marker genes, it is a common practice to compare supported branches in gene phylogenies with established organismal relationships (Philippe et al. 2004, 2005, 2009; Rodriguez-Ezpeleta et al. 2007a).
The marker genes used in recent eukaryotic phylogenetic studies mainly include previously identified universal markers (e.g., rDNA genes) and single-copy genes selected from targeted taxonomic groups, both with limitations. The number of known universal marker genes is small and a few of them (e.g., eEF1α—Keeling and Inagaki 2004 and α-tubulin—Simpson et al. 2008) have recently been shown to have complex evolutionary histories, rendering them non-orthologous. Recent phylogenomic studies included over one hundred genes (e.g., Philippe et al. 2005; Aguileta et al. 2008), but the gene selection usually focused on the organisms being studied. Hence, studies of different taxon groups have had very different sets of marker genes; for example, the 146 genes used in a study of animal phylogeny (Philippe et al. 2005) and the 246 genes used in a study of fungal phylogeny (Aguileta et al. 2008) shared only 35 common genes. This is not surprising because different genes are often suitable for different phylogenetic questions (Townsend 2007; Townsend and Lopez-Giraldez 2010). More importantly, phylogenomic studies usually adopted different approaches to identify orthologous genes, and often included transcriptome datasets, which have uneven coverage of the gene space. In any case, the lack of common threads among analyses of different organisms hinders the integration of multiple studies into a comprehensive eukaryotic tree of life.
Recent studies have suggested the importance of developing additional phylogenetic markers for eukaryotic phylogeny (Yoon et al. 2008; Tekle et al. 2010). New phylogenetic markers, as independent dataset, provide valuable opportunity to evaluate existing phylogenetic hypotheses. In addition, it is of great interest to identify a common set of genes that are suitable for analyzing organismal relationships in different parts of the eukaryotic phylogeny; such marker genes would more easily allow the assembly of a robust and complete eukaryotic tree of life. Here we report the identification of 943 low-copy genes that are widely distributed and well conserved across major eukaryotic groups. We demonstrate that subsets of these genes can yield a robust hypothesis of eukaryotic phylogeny and provide tests for possible biases by removing the most rapidly evolving sites, as well as constant sites and singletons (Cox et al. 2008; He et al. 2014; Burki et al. 2016). Furthermore, we have identified two smaller subsets: one subset consisting of 52 genes that can construct relatively robust phylogenetic relationships with taxon variation, the other consisting of 20 genes that can provide the power necessary to resolve fungal relationships at various evolutionary depths. The marker genes we present here are promising tools for both gene-rich and taxon-rich analyses, and have the potential to greatly improve our understanding of the eukaryotic tree of life.
Materials and Methods
Identification of Marker Genes
To identify marker genes for eukaryotic phylogeny, we screened OrthoMCL-DB (Chen et al. 2006) (version 4), which delineated putative groups of orthologous genes (orthogroups) from 88 eukaryotic and 50 prokaryotic genomes. The taxon sampling in OrthoMCL-DB was biased toward well-established clades, with 51 out of the 88 species belonging to animals, fungi, and green plants. While this bias largely reflects the phylogenetic distribution of completed eukaryotic genome projects, over-sampling of specific taxonomic groups is unnecessary for our goal to identify widely distributed low-copy eukaryotic genes and also will greatly increase the computational burden of subsequent analyses. In addition, the remaining 37 species are from 18 genera, indicating redundancy at species level. To obtain a more balanced and manageable representation of the eukaryotic diversity, we selected, respectively, 7, 4, and 4 representative species from animals, fungi, and green plants, as well as one species from each of the remaining 18 genera. In total, 33 species were selected as representatives of all five eukaryotic supergroups (supplementary table S1, Supplementary Material online).
The complete list of 116,536 orthogroups was downloaded from OrthoMCL-DB. 1,291 orthogroups that contain genes from at least 75% of the 33 species were retained. For these orthogroups, phylogenetic analysis was performed as follows: protein sequences from the 33 representative species were extracted and aligned using MUSCLE v3.8 with default settings (Edgar 2004); conserved alignment blocks were selected using Gblocks v0.91b (Castresana 2000) with “Allowed Gap Positions” set to “all” and “Minimum Number of Sequences for a Flank Position” set to half of the number of sequences (the same parameters were used throughout this study); then Maximum Likelihood (ML) analysis was performed using RAxML v7.2.8 (Stamatakis 2006) with the “PROTGAMMALG” option and 100 bootstrap replicates. The LG model (Le and Gascuel 2008) was used because it is the best fit model for the vast majority of single-genes as determined by ProtTest v3 (Darriba et al. 2011). The resulting phylogenetic trees were carefully examined (both manually and computationally using custom Perl scripts, which are available at https://github.com/EukaryotesGBE/EukayotesGBE; other scripts are similarly available) for gene duplication shared by multiple organisms. If a gene tree suggested duplication(s) before the divergence of eukaryotic supergroups, additional phylogenetic analyses including bacterial homologs were performed. Paralogous clades derived from duplication(s) in early eukaryotes were analyzed separately. An orthogroup was retained if the resulting phylogeny showed no evidence for duplication or only evidence for terminal duplication(s) (Class I genes), or a few duplications that are shared by closely related organisms (Class II genes). As a result, 348 orthogroups were deemed unsuitable because they showed shared duplications among a relative large number of taxa analyzed here. Finally, we obtained 943 potential marker genes, both version4 and version5 (the latest one) orthoMCL ID for them are shown in supplementary table S2, Supplementary Material online.
To characterize the potential resolving power of the remaining 943 marker genes, we calculated their per-site phylogenetic informativeness by following the procedure outlined previously (Townsend 2007). In brief, we performed Bayesian analysis of the 33 representative species using dataset Euk-S33G138 (see below) and converted the resulting phylogeny into a chronogram using r8s v1.71 (Sanderson 2003) with the “PL” method and “TN” algorithm. Based on the chronogram, site-specific evolutionary rates for each gene were estimated using rate4site v.3.2 (Mayrose et al. 2004) with the ML method. Per-site phylogenetic informativeness was then calculated according to the previously described equation (Townsend 2007).
Supermatrix Datasets for Eukaryotic Phylogeny
To evaluate the performance of the 943 marker genes in resolving eukaryotic phylogeny with taxon variation, we also included in this study 9 additional species besides the 33 species retrieved from OrthoMCL database. The nine species include two haptophytes (Emiliania huxleyi from JGI, Pavlovales sp. From NCBI), two Rhizaria (Bigelowiella natans from JGI, Reticulomyxa filose from NCBI), and one from each of the Apusozoa (Thecamonas trahens from BROAD), cryptophytes (Guillardia theta from JGI), Excavata (Naegleria gruberi from JGI), glaucophytes (Cyanophora paradoxa from NCBI), and red algae (Porphyridium purpureum from NCBI) (supplementary table S1, Supplementary Material online). We downloaded the genome assemblies of seven species, and generated in-house de novo transcriptome assembly for C. paradoxa and Pavlovales sp. using RNA-seq reads retrieved from the Short Read Archive database in NCBI. HaMStR (versoin 13) (Ebersberger et al. 2009) was used to identify the orthologs of the 943 genes from these genomic and transcriptomic assemblies.
We generated a series of datasets for phylogenetic analyses using the supermatrix approach (table 1). Throughout this study, supermatrix datasets are named by the targeted taxon group (e.g., “Euk” for eukaryotes, and “Fun” for fungi), the number of species (e.g., “S33” for 33 species), and number of genes (e.g., “G68” for 68 genes). Depending on the species included in supermatrix datasets, the names of them start with either “Euk-S33” (the 33 species from OrthoMCL database), “Euk-S35” (the 33 species plus 2 Rhizaria), “Euk-S42” (the 33 species plus all 9 additional eukaryotes), or “Euk-S40” (the 42 species in “Euk-S42” minus 2 fast-evolving Excavates, Giardia lamblia and Trichomonas vaginalis) (see detailed gene and species composition of each dataset in table 1 and supplementary table S1, Supplementary Material online).
Table 1.
Summary of Supermatrix Datasets
| Dataset | Number of Species * | Number of Genes * | Number of Positions | Percentage of Gaps |
|---|---|---|---|---|
| Euk-S33G478 | 478 | 129,463 | 16.79 | |
| Euk-S33G478-sub1 to -sub7 | 33 | 24–144 | 17,518–18,067 | 11.32–22.86 |
| Euk-S33G465 | 465 | 142,013 | 17.40 | |
| Euk-S33G465-sub1 to -sub7 | 19-140 | 19,534-19,869 | 13.18–21.17 | |
| Euk-S42G478 | 478 | 121,817 | 17.95 | |
| Euk-S42G478-sub1 to -sub7 | 42 | 24-144 | 16,758-17,453 | 13.65–24.38 |
| Euk-S42G465 | 465 | 133,064 | 17.98 | |
| Euk-S42G465-sub1 to -sub7 | 19-140 | 18,551-19,367 | 14.04–22.38 | |
| Euk-S39G20 | 39 | 20a | 9,819 | 11.07 |
| Euk-S39G25 | 25b | 13,886 | 10.12 | |
| Fun-S114G24 | 112 | 24c | 16,106 | 13.07 |
| Euk-S33G52 | 33 | 30,938 | 13.89 | |
| Euk-S35G52 | 35 | 52 | 30,700 | 14.73 |
| Euk-S40G52 | 40 | 30,645 | 15.99 | |
| Euk-S42G52 | 42 | 30,202 | 16.96 |
a MCM2-9, MLH1/4, MSH2/6, SMC1-6, DMC1, RAD51.
bWith the addition of RPA1, RPB1, RPC1, eIF1A, eIF5B.
c MCM2-7, MLH1-4, MSH1-6, SMC1-6, DMC1, RAD51.
*See supplementary tables S1, S2, S3, S5, and S7, Supplementary Material online for the complete list of species, genes and Support Information online for important lineages in each dataset.
First, we selected the most conserved genes (average protein sequence identity ≥40%) from the two classes and constructed four smaller datasets using different thresholds for alignment length, including: Euk-S33G68 (68 Class I genes, alignable region ≥400 aa), Euk-S33G138 (138 Class I genes, alignable region ≥300 aa), Euk-S33G78 (78 Class II genes, alignable region ≥400 aa), and Euk-S33G139 (139 Class II genes, alignable region ≥300 aa). It should be noted that Euk-S33G68 and Euk-S33G78 are subsets of Euk-S33G138 and Euk-S33G139, respectively. Furthermore, these gene sets were also tested with the species sets S35/40/42. Second, we assembled four large datasets, Euk-S33G478, Euk-S33G465, Euk-S42G478, and Euk-S42G465, consisting of all 478 Class I genes and all 465 Class II genes, respectively, for both the 33 and 42 eukaryotic species sets. Lastly, we created 14 subsets (Euk-S33G478-sub1 to -sub7 and Euk-S33G465-sub1 to -sub7) from the two large datasets; genes in each class were ranked in descending order of their alignment length and average identity, and divided into seven similar sized subsets. The 14 subsets of genes were implemented with four taxon subsets (S33, S35, S40, and S42), thus resulting in 56 supermatrix subsets.
Besides these gene-rich datasets, we performed additional analyses on a subset of the 943 orthogroup genes for taxon-rich analysis (see supplementary table S3, Supplementary Material online for their orthoMCL ID). Among the orthogroups are members of recA/RAD51, SMC, MCM, MLH, and MSH gene families (a total of 26 genes, supplemental table S3, Supplementary Material online) that had been characterized in detail phylogenetically and found to be orthologous among fungi, animals and plants and are thus likely excellent phylogenetic markers (e.g., Lin et al. 2006, 2007; Surcel et al. 2008; Liu et al. 2009). To further assess the copy number of these 26 genes in eukaryotes, we downloaded genomic sequences of an expanded taxon set of 230 species, including 118 fungi (with the early divergent fungal relatives Rozella, Mitosporidium, and two other Microsporidia), 83 animals and related protists, and 29 other eukaryotes (see supplementary table S4, Supplementary Material online for detailed sources of data). We also included the transcriptomic data of Glomus intraradices, which is the only representative of the early-branching fungal lineage Glomeromycota with a genome-scale dataset (supplementary table S4, Supplementary Material online). The sequences of the 26 genes in well annotated genomes (e.g., human and Arabidopsis) were collected from the literatures and used as queries to retrieved their homologs from the 231 species by exhaustive homolog searches using an in-house developed program called “Phoenix” (see Supplementary Methods online).
Although the 26 genes are ancient paralogs that existed before the divergence of animals and plants, they have been maintained as single-copy for most of the histories of major eukaryotic lineages. As reported previously, the descendants of each of the ancient paralogs form a subfamily (Lin et al. 2006, 2007; Surcel et al. 2008). For each gene family, preliminary ML analysis was performed using RAxML v7.2.8 (Stamatakis 2006) with the “PROTGAMMALG” option to assign genes into subfamilies. The reliability of topologies was evaluated by 100 bootstrap replicates. Subfamilies were further analyzed using the same ML approach to test for orthology (all ML trees of gene families and individual genes are available at https://github.com/EukaryotesGBE/EukayotesGBE or upon request). The LG model was found to be the best fit model for all selected marker genes using ProtTest v3 (Darriba et al. 2011).
Careful examination the 26 genes in 231 species (see detailed data in supplementary table S4, Supplementary Material online) revealed that most of the 26 genes are widely distributed in eukaryotes, but MLH2/3 and MSH1/3/4/5 are often absent outside animal, plant and fungi lineages; on the other hand, MCM8/9 are absent from most fungi species. Thus we adopted somewhat different subsets of the 26 genes for analyses of eukaryotes and fungi, respectively (see supplementary tables S3 and S4, Supplementary Material online for detailed information).
We first assembled one dataset to study the eukaryotic phylogeny (case study Ia), Euk-S39G20, which contains members of the RAD51/SMC/MCM/MSH/MLH gene families that are shared by the supergroups of eukaryotes (supplemental tables S3 and S4, Supplementary Material online). To further investigate certain difficult relationships (e.g., the position of red algae), we also generated an additional dataset (Euk-S39G25; case study Ib) by including five commonly used phylogenetic markers (i.e., RPA1, RPB1, RPC1, eIF1A, and eIF5B, with functions in transcription and translation) to the dataset Euk-S39G20 for eukaryotic phylogeny (see table 1, supplementary tables S1 and S3, Supplementary Material online for their supermatrix, species, and gene information, respectively). The S39 species set differs from S33 described earlier in this section in two ways: (1) the S33 species set included as many protists as possible from the orthoMCL database, but still had greater representation of vertebrate animals, green plants and Ascomycotes (a major lineage of fungi); (2) the S39 species analysis aimed to represent the supergroups more evenly, with fewer animals, plants, and Ascomycota, but more representatives of other lineages of fungi, Amoebozoa, Rhodophyta and Stramenopiles, than the S33 set (supplemental table S1, Supplementary Material online).
To study fungal phylogeny (case study II) and to test whether genes of the 26 genes are also suitable for a taxon-rich analysis, we constructed a dataset Fun-S114G24 (see table 1 for matrix information, supplementary tables S3 and S5, Supplementary Material online for genes and species).
To test the monophyly of Excavata using a much smaller subset than Class I (478 genes) or Class II (465 genes), we also generated the Euk-G52 gene set by combining the genes used in case studies Ia and II with the best-performing subset of Class I genes (Euk-S40G478-sub1, introduced in results below). There are 30 genes using in either case studies Ia or II (not including MLH2 due its absence from the orthoMCL database; supplemental table S3, Supplementary Material online), and 24 genes in Euk-S40G478-sub1, with 2 genes found in both sets, thus the combined set had 52 genes (see supplementary table S3, supplementary Material online for detailed information of the 52 genes). Euk-G52 was further implemented with four species sets to evaluate its performance with taxon variation, thus resulting in four supermatrix datasets, Euk-S33G52, Euk-S35G52, Euk-S40G52, and Euk-S42G52.
For paralogs derived from terminal duplications, we compared each of the recent paralogs with an HMM profile built from the alignment of the gene; the copy with the highest score was considered the most conserved one and was included in the dataset. For paralogs derived from duplications shared by two or more species in our study, all paralogous copies were discarded. All datasets, alignments, trees, custom scripts, and other materials (including data that are not shown) used in this study are at Supplementary Data, or upon request.
Finally, we would like to summarize all the datasets constructed in this study. In the 943 total-gene analysis, we constructed 4 large supermatrices (Euk-S33G478, Euk-S33G465, Euk-S42G478, and Euk-S42G465). In analyses of subsets of the 943-genes, 4 intermediate-sized gene sets (Euk-G68/138/78/139) were implemented with 4 species sets, resulting in 16 supermatrices (Euk-S33G68 and so on). Then we divided 943 genes into 14 small gene sets and implemented with 4 taxa set, thus resulting in 56 small supermatrices with similar amino acids numbers. In the analysis using genes from 26 orthogroups, we constructed 2 supermatrices (Euk-S39G20 and Euk-S39G25) to investigate eukaryotic phylogeny, and one supermatrix to study fungal phylogeny (Fun-S114G24). Finally, we constructed a relatively small subset Euk-G52 implemented with S33/35/40/42, respectively.
Site Stripping Analysis
To investitage the impact of fast-evolving sites, constant sites, and singletons on the statistical support for the trees, we selected 24 representative supermatrices for site stripping analysis, including 4 largest ones: Euk-S33/42-G478/465, 16 median ones: Euk-G68/78/138/139 implemented with four taxa (S33/35/40/42) variations, and 4 small ones: Euk-S33/35/40/42-G52. For each supermatrix, sites were categorized according to their rates of evolution using the TIGER software (Cummins and McInerney 2011), with total bin number set to 20. Two classes of supermatrices were constructed: -slow1 matrices which excluded only BIN20 sites, and -slow2 matrices which excluded both BIN19 and BIN20 sites. Supermatrices without constant sites and singletons were also constructed, termed -rm_C/S. Detailed information and support values on major lineages can be found in supplementary table S8.
Phylogenetic Analysis of Supermatrix Datasets
For all supermatrix datasets, multiple sequence alignments of the protein sequences of individual marker genes were prepared using MUSCLE v3.8 with default settings (Edgar 2004). Columns containing nongap character from only one sequence were removed and conserved alignment blocks were selected using Gblocks v0.91 (Castresana 2000). Filtered single-gene alignments were concatenated using a custom Perl script. ML analyses were performed using RAxML v7.2.8 with the “PROTGAMMALG” option (Stamatakis 2006). Support values for topologies were estimated from 100 bootstrap replicates. PhyloBayes v3.3b (Lartillot et al. 2009) was used for the Bayesian analyses under the CAT + GAMMA model (Quang et al. 2008). Each analysis consisted of two independent chains of at least 15,000 cycles in total. The first 5,000 cycles of each chain were discarded as burn-in and consensus tree was computed from the remaining 10,000 cycles with one tree sampled from every 10 cycles. All Bayesian analyses were checked for convergence using the largest discrepancy between the two chains <0.1 as the criterion. In the analysis of the fungal phylogeny, alternative placements of Microsporidia were evaluated by the approximately unbiased (AU) test, the Kishono–Hasegawa (KH) test, the Shimodaira–Hasegawa (SH) test, and the weighted version of the latter two tests (wKH test and wSH test). For each of the alternative placement of Microsporidia, the most likely tree was built and the site-likelihood values were calculated using RAxML v7.2.8 under PROTGAMMALG model (Stamatakis 2006). All trees were compared collectively using Consel v0.1k (Shimodaira and Hasegawa 2001).
Results
Identification of 943 Candidate Marker Genes for Eukaryotic Phylogeny
We analyzed orthogroups from OrthoMCL-DB (Chen et al. 2006) and identified 943 low-copy genes that have wide phylogenetic distribution and high potential for being orthologous in eukaryotes (see Materials and Methods). These genes are present in more than 75% of 33 species representing major eukaryotic lineages, and most of them (898/943) are present in all 5 eukaryotic supergroups (supplementary table S2, Supplementary Material online). According to their evolutionary patterns, we divided the 943 genes into two classes: Class I included 478 genes which were single-copy or only showed terminal duplication(s); Class II included 465 genes which had experienced a limited number of duplication(s) shared by related species (e.g. duplication shared by vertebrates, possibly due to the 1/2R whole-genome duplication—WGD).
We then compared individual phylogenetic trees of the 943 genes with a reference eukaryotic phylogeny that has emerged from recent studies (Burki et al. 2008; Hampl et al. 2009; Parfrey et al. 2010; Katz and Grant 2014); these phylogenomic studies using gene- or taxon-rich approaches all revealed the same groupings regarding the 33 representative species (supplementary fig. S1, Supplementary Material online). Because individual genes usually contain limited phylogenetic information, single-gene phylogenies often have poorly or incorrectly resolved relationships. One common solution is to focus only on the well supported branches (Philippe et al. 2011). Following this strategy, we found that only 8.5% of the supported bipartitions (bootstrap [BS] ≥ 70%) in single-gene trees were incongruent with the reference phylogeny. Many of the incongruences are regarding relationships that are known to be difficult (e.g., the monophyly of Ecdysozoa and Excavata). Most supported single-gene trees were able to recover well-accepted clades such as animals, fungi, green plants, Stramenopiles, Apicomplexa, and Euglenozoa (supplementary fig. S1, Supplementary Material online). Moreover, Class I and II genes showed highly similar levels of support for almost all bipartitions in the reference phylogeny. We also analyzed phylogenetic informativeness profiles of the 943 genes and found that most genes carry phylogenetic signals for both ancient and relatively recent relationships in eukaryotes (supplementary table S2, Supplementary Material online). These results suggest that the genes we identified are likely suitable for the analysis of deep eukaryotic phylogeny.
Gene-Rich Analyses of the 33 Representative Eukaryotes Using the 943 Candidate Markers
To evaluate the resolving power of these candidate phylogenetic marker genes, we first analyzed four supermatrix datasets consisting of the most conserved and longest genes in Class I (Euk-S33G68 and Euk-S33G138) and Class II (Euk-S33G78 and Euk-S33G139) (table 1; see Materials and Methods for details of dataset construction). The ML analyses had obvious misplacement of fast-evolving species (e.g., the grouping of the Microsporidia with Excavata; data not shown), likely due to the LBA artefact. Therefore, for eukaryotic datasets we only report results from Bayesian analysis with CAT model, which is more robust to LBA (Baurain et al. 2007, 2010; Rodriguez-Ezpeleta et al. 2007b).
Bayesian analyses of the four datasets revealed the same topology (fig. 1A) only that Euk-S33G139 suggested a slightly different placement of Toxoplasma gondii. Strikingly, the topology was in complete agreement with the reference eukaryotic phylogeny shown in supplementary figure S1, Supplementary Material online, with the only exception of the grouping of Trichoplax adhaerens and Nematostella vectensis. All relationships received very strong support, including deep ones such as the monophyly of Amoebozoa, Excavata, Opisthokonta, Aveolates, Stramenophiles, and the grouping of green plants with red algae, as well as the branching order among these major clades. Particularly, the supergroup Excavata here encompasses both free-living and parasitic protists with diverse characteristics, such as the presence of chloroplasts in some and the lack of mitochondria in others, although previously the paraphyly of Excavata has been a frequent finding in both gene-scale and genome-scale analyses (Parfrey et al. 2006; Simpson et al. 2006; Yoon et al. 2008).
Fig. 1.—

Bayesian analyses of eukaryotic phylogeny with 33 representative species. (A) An unrooted Bayesian tree estimated from Euk-S33G68/138/78/139. (B) The tree estimated from Euk-S33G478. The topologies were estimated by Phylobayes using CAT model. The five eukaryotic supergroups are colored as following; red, Amoebozoa; black, Opisthokonta; green, Archaeplastida; blue, SAR; and brown, Excavata. Posterior Probability (PP) support values are shown for each nodes. Black dots indicate 100% PP support. In (A), black dots indicate nodes receiving 100% support from all four datasets. Dashes indicate the lack of support for the relationship from the relevant dataset(s).
We also analyzed the total sets of all 478 Class I (Euk-S33G478) and 465 Class II genes (Euk-S33G465), respectively, from the 33 representative eukaryotic species. The two resulting Bayesian phylogenies (fig. 1B and supplementary fig. S2A, Supplementary Material online) are again highly congruent with the reference eukaryotic phylogeny shown in supplementary figure S1, Supplementary Material online. Almost all relationships were recovered with maximum support, with the monophyly of Excavata being the only exception. We further created multiple smaller datasets (Euk-S33G478-sub1 to -sub7 and Euk-S33G465-sub1 to -sub7) by dividing genes in each class into seven similar sized subsets based on their alignable region length and identity. Similarly, most of the clades received very strong support in the sub dataset analyses, whereas the monophyly of Excavata was still not recovered (table 2). Altogether, our results strongly support the utility of the genes identified here as phylogenetic markers in studying the eukaryotic tree of life, with smaller subsets of the markers for Excavata.
Table 2.
Support for Major Eukaryotes Clades in the Analyses of Subsets of Euk-S33G478 and Euk-S33G465
|
Euk-S33G478
|
Euk-S33G465
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sub1 | sub2 | sub3 | sub4 | sub5 | sub6 | sub7 | sub1 | sub2 | sub3 | sub4 | sub5 | sub6 | sub7 | |
| Animals | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| fungi | 100 | 100 | nm* | 56 | 100 | 100 | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 99 |
| Opisthokonta | 100 | 100 | nm | 99 | 100 | 100 | 100 | 100 | 100 | 96 | 100 | 100 | 100 | 100 |
| Amoebozoa | 100 | 100 | nm | nm | 100 | nm | 50 | 50 | 100 | 100 | 99 | 100 | nm | 50 |
| Archaeplastida | nm | 58 | nm | 100 | nm | nm | nm | 85 | 99 | nm | 84 | nm | nm | nm |
| Excavata | nm | Nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm |
| Alveolates | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Stramenopiles | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Alveolates + Stramenopiles | 100 | 100 | 63 | 100 | nm | 100 | nm | 100 | 100 | 99 | 99 | 100 | 95 | 100 |
Note.—nm - not monophyletic.
Selection of a Moderate Number of Marker Genes for Taxon-Rich Analyses
The 943 orthogroups include a few commonly used universal markers (e.g., elongation factors and ATPase subunits) and recently reported markers for taxon-rich analyses of the eukaryotic phylogeny (Tekle et al. 2010). We reasoned that the 943 orthogroups also encompass other promising marker genes for taxon-rich analyses. As a case study, we elected to focus on a subset of the 943 orthogroups and performed extensive analyses at different depths of eukaryotic phylogeny. We found that among the 943 orthogroups are members of five gene families, including recA/RAD51, MSH, MLH, SMC, and MCM, totalling 26 ancient paralogous genes (supplemental table 3, Supplementary Material online). Previous phylogenetic studies from our group and by others showed that genes in these families have essential functions in DNA replication, repair, and recombination and are broadly distributed, highly conserved and orthologous in representative eukaryotes (Lin et al. 2006, 2007; Surcel et al. 2008; Liu et al. 2009) (fig. 2), suggesting the possibility of these genes being good phylogenetic markers.
Fig. 2.—

Schematic representation of the evolutionary histories of (A) recA/RAD51 family; (B) MSH family; (C) MLH family; (D) SMC family; and (E) MCM family. The topologies are adopted from previous phylogenetic analysis on each gene families. Dotted boxes denote ancient duplication events in early eukaryotes. Prokaryotic outgroups are shown in grey.
To characterize the phylogenetic patterns of the 26 candidate marker genes in eukaryotes, we conducted exhaustive homolog searches in an extensive set of sequenced eukaryotic genomes and performed phylogenetic analysis for each family and subfamily. Most of these genes were found in almost all species examined in this study, while some others showed various degrees of patchy phylogenetic distribution (supplementary table S4, Supplementary Material online); for instance, MCM8 and MCM9 were detected in most eukaryotes except for fungi, while MLH2/3 and MSH1/3/4/5 were missing from most species outside animals, plants and fungi. In addition, these candidate marker genes are single-copy in most organisms that still had them (fig. 3; supplementary table S4, Supplementary Material online). In particular, all these genes remained single-copy following well-documented WGD events in yeast and vertebrates, with the only exception being SMC1. Extra copies of a gene were detected only in a few cases, most of which were likely derived from recent lineage-specific duplications (data not shown). Furthermore, the vast majority of supported bipartitions in single-gene trees was congruent with well-established organismal relationships (e.g., see supplementary fig. S3, Supplementary Material online), suggesting that these genes have most likely maintained orthologous relationship in eukaryotes. Our results suggest that these genes are potentially useful phylogenetic markers across a broad range of eukaryotic diversity.
Fig. 3.—

A matrix showing the distribution of selected marker genes in eukaryotes. The presence/absence of genes is highlighted by color: blank, absence; green, single copy; blue, two copies due to lineage-specific duplication; purple, more than two copies due to lineage-specific duplications; red, more than one copy due to duplications shared by more than one species. The Index of Single Copyness (ISC) is defined as , where n is the total number of species, mi is equal to the gene copy number for species with a single copy of the gene or more than one copies of terminal paralogs (mi is > 0; for species that do not have the gene, 1/mi = 0), k is equal to the total number of species with paralogs shared by two or more species. This matrix includes the 18 genes that were included in both the Euk-S39 and the Fun-S114 datasets, and four commonly used eukaryotic marker genes as comparison.
Case Study I: Deep Relationships Within and Between Eukaryotic Supergroups
To investigate the general utility of the selected marker genes, we first analyzed the most ancient relationships in the eukaryotic tree of life. Considering that MLH2/3 and MSH1/3/4/5 are mostly absent outside animal and fungi lineage, we included the other 20 genes that have broad distribution in eukaryotes, with the hope that they are widely applicable for the study of eukaryotic phylogeny (see supplementary table S3, Supplementary Material online for gene ID and supplementary table S4, Supplementary Material online for gene distribution). Eukaryotic diversity has been very unevenly sampled by genome sequencing projects, with the overwhelming majority of the sequenced genomes belonging to Opisthokonta (animals and fungi). To maximize the representation of eukaryotic diversity in our study, we included a number of sequenced genomes in Amoebozoa, SAR, and Excavata, and selected species from fungi (including Microsporidia), animals, plants and their related protists, with a total of 39 species representing the 5 eukaryotic supergroups (supplementary table S1, Supplementary Material online). The combination of 39 species with 20 genes resulted in dataset Euk-S39G20 (table 1).
Our Bayesian analysis was able to recover a robust phylogeny with strong support for overwhelming majority of nodes in the eukaryotic tree (fig. 4). The well-supported relationships included the monophyly of each of three supergroups, Opisthokonta, Amoebozoa, and Excavata, and most nodes within each supergroup. Many relationships that have often been uncertain in previous single-gene analyses (Parfrey et al. 2006) were robustly resolved in this analysis. For instance, the supergroup Opisthokonta includes a strongly supported close relationship of Microsporidia with fungi (see below for more discussion of Microsporidia). Moreover, the supergroup Amoebozoa was strongly supported despite inclusion of the highly divergent E. histolytica. Strikingly, a moderate number of genes used here was able to successfully recover a monophyletic Excavata, with an internal topology in accordance with recent analyses (Burki et al. 2008; Hampl et al. 2009; Parfrey et al. 2010).
Fig. 4.—

A Bayesian tree of 39 eukaryotes using 20 genes. The topology was estimated from the Euk-S39G20 dataset by Phylobayes using CAT model. The five eukaryotic supergroups are colored as following; red, Amoebozoa; black, Opisthokonta; green, Archaeplastida; blue, SAR; and brown, Excavata. The branch leading to Giardia is shown as a quarter of the original length. Posterior Probability (PP) support values from Bayesian analyses using Euk-S39G20 (first number) and Euk-S39G25 (second number) are shown for each nodes. Black dots indicate 100% PP support from both 20- and 25-gene analyses.
Our analysis also strongly supported the monophyly of stramenopiles and alveolates, two major groups of the supergroup SAR, and the sister relationship between them. In addition, the close relationship between haptophytes and green plants received high support. However, the supergroup Archaeplastida was not recovered; the two red algae were placed sister to the clade uniting green plants, haptophytes, aveolates, and stramenophiles. The same tree topology was recovered from Bayesian analysis using another dataset Euk-S39G25, which included five more widely used marker genes (RPA1, RPB1, RPC1, eIF1A, and eIF5B); however, while most other nodes received higher support, the position of red algae became only marginally supported. We also attempted to include other lineages such as Rhizaria, cryptophytes, and glaucophytes, however their relationships could not be resolved (data not shown) using the small set of 25 genes. These results illustrate the difficulty in resolving the relationships between distant groups of organisms that are photosynthetic (or with photosynthetic ancestry).
Among supergroups, our eukaryotic tree was in agreement with the division of “Unikonta” (with one or no flagellum; Opisthokonta and Amoebozoa) from “Bikonta” (with two flagella; Excavata, Archaeplastida, and SAR), a relationship proposed on the basis of several derived gene fusion events (Stechmann and Cavalier-Smith 2003). In addition, members of Archaeplastida and SAR, as well as haptophytes formed a well-supported clade [“corticates”—Cavalier-Smith 2010; fig. 4], even though photosynthesis in these groups has complex origins. Overall, with 20 or 25 nuclear marker genes, we obtained eukaryotic phylogenies that are largely consistent with both our results based on larger datasets (fig. 1) and previous phylogenomic results (Burki et al. 2008; Hampl et al. 2009; Parfrey et al. 2010).
Case Study II: Relatively Recent Relationships Between Fungal Species
To further assess the utility of the marker genes described here within specific major eukaryotic lineages, we performed an in-depth analysis of the fungal phylogeny. Among the 26 genes in the recA/RAD51, MSH, MLH, SMC, MCM gene families, MLH2/3 and MSH1/3/4/5 are present in fungi, but MCM8 and MCM9 are absent (supplementary table S4, Supplementary Material online). Thus 18 of the 20 genes used in case study I (without MCM8 and MCM9) and 6 more genes (MLH2, MLH3, MSH1/3/4/5) were used as phylogenetic markers for fungi (dataset Fun-S114G24, table 1; see supplementary tables S3 and S5Supplementary Material online for gene and species information). The relationships within fungi have been extensively studied (Fitzpatrick et al. 2006; James et al. 2006; Wang et al. 2009; Medina et al. 2011; Ebersberger et al. 2012; Schoch et al. 2012; James et al. 2013), allowing a good comparison of our results. For example, we analyzed two taxon sets matching previously phylogenomic studies (Fitzpatrick et al. 2006; Wang et al. 2009); our results using only 24 genes (supplementary figs. S4 and S5, Supplementary Material online) were in excellent agreement with the previously reported phylogenies based on 153 genes or whole genome data, indicating that the selected markers have strong resolving power.
We performed both Bayesian and ML analyses on a large dataset (Fun-S114G24) that includes 100 fungal species, covering a large fraction of sequenced fungal genomes. The two approaches resulted in phylogenies that are largely congruent and provided maximum supports for more than 80% of all nodes including both higher-level and recent relationships (fig. 5). In particular, previous studies have generated inconsistent results regarding the relationship of fungi (and other eukaryotes) with Microsporidia (Corradi and Keeling 2009), which are intracellular obligate parasites of major groups of animal and have highly reduced genomes. Recent studies also revealed that Rozella allomycis (Cryptomycota), Mitosporidium daphnia, and Microsporidia together form a monophyletic group as the earliest branching clade of the fungal lineage (Capella-Gutierrez et al. 2012; James et al. 2013; Haag et al. 2014). We found here maximum Bayesian and ML support both for a monophyletic group of R. allomycis, M. daphnia, and two Microsporidia species and for the sister relationship of this group with the rest of the fungi. Further AU test revealed that alternative placements of Microsporidia and R. allomycis could be confidently rejected (supplementary table S6, Supplementary Material online).
Fig. 5.—
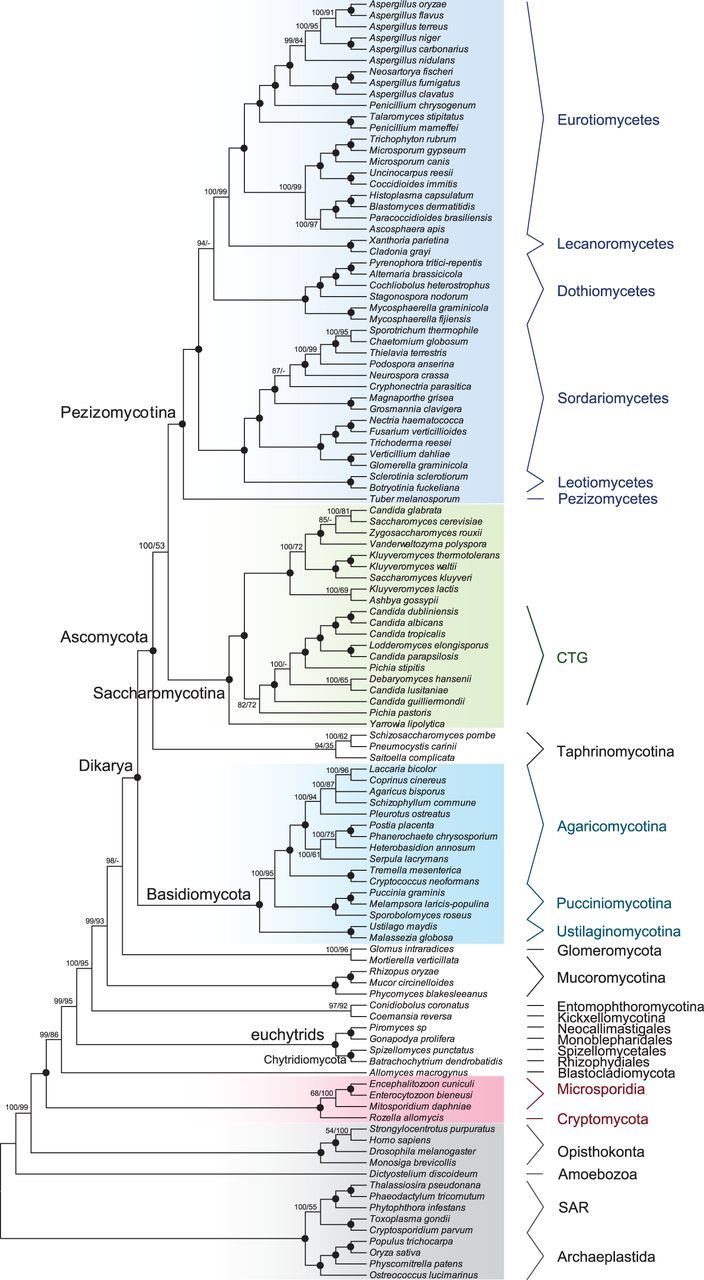
Cladogram of 100 fungal species with 14 other eukaryotic species using 24 genes. The topology was estimated from Fun-S114G24 dataset by Phylobayes using CAT model. Black dots indicate 100% support from both Posterior Probability (PP) and bootstrap (BS) support from ML analysis (based on 100 replicates). Support values are only shown for nodes that do not receive 100% support. Dashes indicate lack of support from the ML analysis.
In addition, both phylogenetic approaches showed strong support for (1) the grouping of Chytridiomycota (fungi that reproduce through flagellated spores) and Neocallimastigomycota (anaerobic inhabitants of herbivore gut); (2) the sister relationship between Entomophthoromycotina and Kickxellomycotina; and (3) successive branching orders of Blastocladiomycota, Chytridiomycota + Neocallimastigomycota, and Entomophthoromycotina + Kickxellomycotina. Both Bayesian and ML analyses showed that Glomeromycota and Mucoromycotina diverged next; however, their relationship to each other was not consistently resolved by Bayesian and ML analyses. While Glomeromycota and Murcoromycotina formed a clade in the ML result, the Bayesian analysis supported a sister relationship between Dikarya and the grouping of Glomeromycota and Mortierella verticillata, a member of Murcoromycotina. The relationship between Glomeromycota and Mucoromycotina is also controversial in previous studies. For example, some results supported the monophyly of the two taxa as shown in our ML results (Capella-Gutierrez et al. 2012; James et al. 2013), but another study supported sister relationship between Dikarya and Mucoromycotina (Ebersberger et al. 2012). Thus, additional genes and/or taxa might be able to place these groups more definitively.
Regarding the other fungal lineages, there was strong support for the monophyly of Dikarya, the subkingdom that includes the phyla Basidiomycota (e.g., mushrooms) and Ascomycota (e.g., baker’s yeast), both of which are important ecologically and economically, as they include many other useful fungi. The three major clades of Basidiomycota all received maximal support from both Bayesian and ML approaches, and the orders of their separation was confidently resolved. Within the phylum Ascomycota, we found strong support for the sister relationship of two major subphyla, Saccharomycotina and Pezizomycotina; in addition, our results support the monophyly of Taphrinomycotina, which include the model organism fission yeast (Schizosaccharomyces pombe) and parasites of animals and plants. We also recovered with maximal support the CTG clade with species that translate CTG as serine instead of leucine. For the relationships among major lineages within Pezizomycotina, Pezizomycetes (represented by Tuber melanosporum) was sister to the rest of Pezizomycotina with high confidence. Also, the monophyly of Leotiomycetes, Sordariomycetes, Dothideomycetes, Eurotiomycetes, and Lecanoromycetes all received maximum Bayesian and ML support. In addition, the Bayesian analysis provided maximum support for the position of Dothideomycetes sister to Eurotiomycetes + Lecanoromycetes. In conclusion, our analyses of fungal phylogeny indicate that the moderate number of markers used here can both recovered well-recognized groups and resolved many of the relationships between groups, with very high support.
Analyses of 943 Genes and a 52-Gene Subset with Additional Taxa Yielded Robust Phylogenies
During the preparation of this manuscript, genome-scale datasets became available for several important lineages such as Apusozoa, Rhizaria, glaucophytes, and haptophytes, whose placements in the eukaryotic phylogeny either have recently been revised or remain uncertain. For example, Rhizaria was originally an independent eukaryotic supergroup but there is increasing support for its clustering with stramenopiles and alveolates, two members of the previously defined supergroup “Chromalveolata” (Adl et al. 2005, 2012). Cryptophytes and haptophytes also previously belong to “Chromalveolata”; however, recent studies have suggested several alternative placements of them such as the association with either SAR or Archaeplastida (Burki et al. 2008, 2009; Parfrey et al. 2010; Burki et al. 2012, 2016; Katz and Grant 2014). We thus expanded our analyses to include nine additional eukaryotic species representing these lineages, to examine whether the 943 genes could be obtained from new genome (for seven of the nine) or transcriptome (for the other two) assemblies and to investigate their challenging relationships using the marker genes identified here.
As a result, orthologs for more than 85% of the 943 genes were found in each of the nine species. We then constructed a number of datasets by adding these orthologs from some of the newly included species to the aforementioned datasets consisting of genes from 33 species (i.e. “Euk-S33” datasets). We first performed analyses on datasets containing the two representatives of Rhizaria (i.e., “Euk-S35” datasets). The monophyly of Rhizaria and the supergroup SAR was recovered by the conserved gene sets Euk-S35G68/138 and Euk-S35G78/139, as well as the majority of sub dataset Euk-S35G478-sub1-7 and Euk-S35G465-sub1-7 (supplementary table S7, Supplementary Material online). With the inclusion of Rhizaria, however, the supergroup Excavata was supported only by the two datasets (Euk-S35G78 and Euk-S35G139) of the Class II conserved genes.
We further expanded the taxa to include the other seven additional eukaryotes, resulting in datasets with a total number of 42 species (i.e., “Euk-S42” datasets). Our Bayesian analyses of the total sets of Class I and Class II genes (Euk-S42G478 and Euk-S42G465) have again resulted in highly similar trees (fig. 6 and supplementary fig. S2B, Supplementary Material online) with strong support for most relationships, although the monophyly of Excavata was still not recovered. Importantly, both trees provided (nearly) maximal support for the sister relationship between Apusozoa and Opisthokonta, the monophyly of the supergroup SAR, and a monophyletic clade including Archaeplastida (green plants, red algae, glaucophytes) plus the grouping of cryptophytes and haptophytes. On the other hand, the positions of these lineages were much less resolved in the analyses using sub-datasets (Euk-S42G478-sub1 to -sub7 and Euk-S42G465-sub1 to -sub7; table 3), suggesting that small sub-datasets do not contain sufficient information to address these difficult relationships, similar to the above results using 25 genes.
Fig. 6.—

Bayesian analyses of eukaryotic phylogeny using 478 Class I marker genes with 42 species. Topologies were estimated from Euk-S42G478 by Phylobayes using CAT model. The five eukaryotic supergroups are colored as following; red, Amoebozoa; black, Opisthokonta; green, Archaeplastida; blue, SAR; and brown, Excavata. Posterior Probability (PP) support values are shown for each nodes. Black dots indicate 100% PP support.
Table 3.
Support for Major Eukaryotes Clades in the Analyses of Subsets of Euk-S42G478 and Euk-S42G465
|
Euk-S42G478
|
Euk-S42G465
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| sub1 | sub2 | sub3 | sub4 | sub5 | sub6 | sub7 | sub1 | sub2 | sub3 | sub4 | sub5 | sub6 | sub7 | |
| Animals | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Fungi | 100 | 100 | nm | nm | 100 | 99 | 84 | 100 | 100 | 100 | 100 | 100 | 100 | 99 |
| Opisthokonta | 100 | 100 | nm | 100 | 100 | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Apusozoa + Opisthokonta | nm | nm | nm | 99 | nm | nm | 59 | nm | nm | 99 | nm | nm | nm | nm |
| Amoebozoa | 99 | 100 | nm | nm | 59 | 79 | 65 | 100 | nm | 100 | 93 | 99 | 99 | nm |
| Archaeplastida | 99 | nm | nm | nm | nm | nm | nm | nm | nm | 91 | nm | nm | nm | nm |
| Excavata | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm | nm |
| Rhizaria | 100 | nm | 100 | nm | nm | 100 | 99 | nm | nm | 100 | 100 | nm | 100 | 100 |
| Alveolates | 100 | 100 | 100 | 100 | nm | 100 | 99 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Stramenopiles | 100 | 100 | 50 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| SAR | 100 | nm | nm | nm | nm | 100 | nm | nm | nm | 99 | Nm | 50 | nm | 100 |
Note.—nm - not monophyletic.
To further test the monophyly of Excavata, a site stripping strategy was adopted for 24 representative supermatrices (described in Materials and Methods), resulting in three classes of datasets including slow1 (without the fastest BIN20), slow2 (without BIN19 and BIN20), and rm_C/S (without constant sites and singltons). Throughout the analysis, similar topologies were consistently supported. Interestingly, improved statistical support values for the monophyly of Excavata were observed for Euk-S42G478-slow1, Euk-S33G465-rm_C/S, Euk-S42G478-slow1 and Euk-S42G478-rm_C/S, as shown in supplementary table S8, Supplementary Material online. However, improvements resulting from site stripping analyses were limited, and should be further considered carefully.
To test the effect of long branches on the monophyly of Excavata, we performed additional analyses without the two most rapidly-evolving excavates (G. lamblia and T. vaginalis), leading to the “Euk-S40” datasets. While the overall results from the conserved genes sets (Euk-S40G68/138 and Euk-S40G78/139) and sub-dataset (Euk-S40G478-sub1-7 and Euk-S40G465-sub1-7) became marginally better, the relationships of Rhizaria, cryptophytes, haptophytes, and others were still not well resolved in most cases (supplementary table S7, Supplementary Material online). Nevertheless, Euk-S40S478-sub1, the sub-dataset consisting of the longest and most conserved 24 genes in Class I, was able to fully resolve the relationships among all 40 species (see supplementary fig. S6, Supplementary Material online).
We described above that the Euk-S39G20 dataset (with members from well-characterized gene families) resulted in the monophyly of Excavata; here the Euk-S40G478-sub1 dataset yields well-supported relationship among all 40 species, especially in Archaeplastida and SAR supergroup. To test whether a combination of these genes could have broad applications, we constructed another gene set termed Euk-G52 as an aggregation of those two datasets (the genes used in the case studies 1 and 2 totalled 31, but MLH2 is not in the orthoMCL database, so not included here; the remaining 30 genes had 2 in common with the 24 genes in G478-sub1, making the final number 52; see supplementary table S3, Supplementary Material online for their orthoMCL ID), hoping that monophyly of Excavata and the relationships between Archaeplastida, cryptophytes and haptophytes could both be solved. To make the evaluation criteria self-consistent, Euk-G52 was tested with four species datasets mentioned in Materials and Methods (Supermatrix datasets for marker gene evaluation), termed Euk-S33G52, Euk-S35G52, Euk-S40G52, and Euk-S42G52. As expected, our analysis revealed a robust phylogeny among most nodes in the eukaryotic phylogeny (supplementary figs. S7–S10, Supplementary Material online), highly congruent with the phylogenetic relationship yielded by Euk-S33G478 and Euk-S42G478. Strikingly, the monophyly of both Excavata and SAR supergroup were well-supported from all four datasets. Notably, the phylogeny of Euk-S33G52 and Euk-S35G52 were identical to figures 1 and 6, except for the placements of T. adhaerens and N. vectensis, which is interestingly in agreement with reference eukaryotic phylogeny shown in supplementary figure S1, Supplementary Material online. For Euk-S40G52 and Euk-S42G52, the monophyly of green plants, Haptophyta (E. Huxley, Pavlovales sp.i) and Cryptophyta (G. theta) was still supported, but the monophyly of Archaeplastida itself was intruded by the latter two taxa (supplementary figs. S9 and S10, Supplementary Material online). Thus we obtained a relatively small subset Euk-G52 that can resolve highly consistent phylogenetic relationships for most taxa among four species datasets, revealing its applicability to resolve deep Eukaryotic relationships.
Discussion
Reporting a Wealth of Eukaryotic Phylogenetic Markers
In this study, we performed a systematic search and identified 943 genes shared among representatives of highly divergent eukaryotic supergroups as candidate phylogenetic markers. The characterization of additional phylogenetic markers is important for eukaryotic phylogeny and has received sustained efforts (e.g., Philippe et al. 2005; Aguileta et al. 2008; Tekle et al. 2010). For example, Tekle et al. (2010) analyzed the KOG database and identified 17 promising marker genes for eukaryotic phylogeny. In addition, a list of 146 genes curated by Philippe et al. (2005) has been widely used in recent phylogenomic analyses, with demonstrated performance in resolving relationships within and among eukaryotic lineages (Hampl et al. 2009; Liu et al. 2009a, 2009b; Philippe et al. 2009, 2011). Almost all the genes identified in these previous studies (15 of 17 genes—Tekle et al. 2010, and 135 of 146 genes—Philippe et al. 2005) were recovered in the study here (supplementary table S2, Supplementary Material online), indicating that the screen performed here was able to capture excellent marker genes for eukaryotic phylogeny. On the other hand, the genes described here had less overlap with those reported by two other phylogenomic studies of more restricted groups of taxa (Aguileta et al. 2008; Dunn et al. 2008) (supplementary table S2, Supplementary Material online). Only 153 of the 246 single-copy genes identified in fungi (Aguileta et al. 2008) and 70 of the 150 single-copy genes identified in animals (Dunn et al. 2008) were recovered by our datasets. Among the 93 fungal single-copy genes and the 80 animal single-copy genes that were excluded in our study, most genes (65 and 64, respectively) failed to meet our criterion that the genes be present in at least 75% of representative organisms, suggesting that they are more likely lost in some eukaryotic lineages and more suitable for phylogeny of specific groups.
In total, 650 out of the 943 eukaryotic marker genes are newly identified in this study, not included in the aforementioned published gene sets (Philippe et al. 2005; Aguileta et al. 2008; Dunn et al. 2008; Tekle et al. 2010) (supplementary table S2, Supplementary Material online). Therefore, the 650 new marker genes are valuable additions to previously available eukaryotic markers. Sequence features of these new marker genes, such as the length of alignable regions and average identity, are similar to those of the previously described 146 genes (Philippe et al. 2005) (supplementary fig. S11, Supplementary Material online). In addition, we provide the phylogenetic informativeness profiles of the new marker genes, as estimates of phylogenetic signals of these genes. The data indicate that these marker genes carry phylogenetic signals even for ancient relationships in eukaryotes (supplementary table S2, Supplementary Material online). At the deepest nodes, most genes have informativeness per site values greater than 0.2 (supplementary table S2, Supplementary Material online), which is as good as the markers identified previously (Tekle et al. 2010). These profiles can also be used to inform the selection of best marker genes for resolving specific phylogenetic relationships. More importantly, we have demonstrated that these genes provide the power to resolve multiple phylogenetic relationships, resulting in a well-supported eukaryotic phylogeny using datasets that moslty consisted of the new marker genes (figs. 1 and 6 and supplementary fig. S2, Supplementary Material online). Therefore, the 650 newly identified marker genes have characteristics and performance that make them excellent markers for eukaryotic phylogeny. In addition, we also examined the copy number and distribution of 943 marker genes and found that most of them are single copy, with 75 of them having homologues in prokaryotes (supplementary table S9, Supplementary Material online). Further studies of these genes might be informative for rooting the eukaryotic tree.
A Smaller Subset of High-Quality Curated Marker Genes
Among the 943 genes, we further characterized 20 markers for broad eukaryotic phylogeny and several additional markers for fungi. For each gene, we conducted exhaustive search in genomic sequences of 231 eukaryotes to characterize its phyletic pattern, and performed careful phylogenetic analysis to assess its orthology. This rigorous procedure ensures the reliability of our results, and distinguishes our approach from other strategies that rely on existing gene annotations or pre-compiled orthologous groups, or that focus on relatively small samplings of species. The procedure is quite time-consuming due to the large number of genomes being screened; however, with potential automation in the future, it will be worth considering applying the procedure in both the identification of new marker genes and the evaluation of existing ones.
The 20 genes we characterized are widely present and single-copy in most eukaryotes analyzed here, and they perform informational functions such as DNA replication, repair, recombination, and chromatin structure maintenance. Both their distribution patterns and functionality suggest that these genes are less prone to HGT (Jain et al. 1999; Lapierre and Gogarten 2009). Unlike the well-appreciated role of bacterial HGT, the prevalence of HGT in eukaryotes is less clear. Recent studies have increasingly revealed an important contribution of HGT in the evolution of eukaryotic gene families, especially for genes with patchy distributions and metabolic functions (Andersson et al. 2006; Andersson 2009, Andersson 2011; Wisecaver and Rokas 2015). Thus, the genes known to be less impacted by HGT, such as the new marker genes reported here, should have a better chance of being orthologous in organisms with uncertain phylogenetic positions.
These desirable features make these genes promising markers for eukaryotic phylogeny. Our case studies of eukaryotic supergroups and fungi have demonstrated that these genes have excellent resolving power at different taxonomic levels; the phylogenies we obtained with this moderate number of genes were largely congruent with previous studies based on larger phylogenomic datasets. Therefore, these newly identified markers can be useful for different branches on the eukaryotic tree of life and will allow easy integration of such separate studies.
Implications for Eukaryotic Phylogeny
Earlier molecular phylogenetic evidence for the relationships within and among eukaryotic supergroups mainly came from similar gene sets (e.g., a few universal markers, see Parfrey et al. 2006); in addition, many recent phylogenomic studies were based on a set of 146 genes (Bapteste et al. 2002; Rodriguez-Ezpeleta et al. 2005, 2007a, b; Burki et al. 2008; Hampl et al. 2009). It is therefore of great interest to compare the results from independent sets of marker genes. With these newly identified markers, our analyses generated well-resolved phylogenies that enabled the test of several important hypotheses, including some that have been hotly debated in recent years, such as the monophyly and relationships of deep eukaryotic supergroups, the origin of Microsporidia, and relationships among fungal lineages.
Eukaryotic Supergroups
The estimation of deep relationships in eukaryotic phylogeny is challenging; the monophyly of some of the supergroups has not been consistently supported (Parfrey et al. 2006), two of the original six supergroups (“Chromalveolata” and “Rhizaria”) were substantially revised (Adl et al. 2005, 2012), and the placements of several “orphan” lineages are still controversial (e.g., cryptophytes and haptophytes) (Burki 2014; Burki et al. 2016). Our analyses using both large (Euk-S33G478/465 and Euk-S42G478/465) and smaller (Euk-S39G20, Euk-S39G25 and Euk-S33/35/40/42/-G52) sets of newly identified markers provided strong support for the monophyly of not only major eukaryotic lineages (such as animals, fungi, and green plants), but also some supergroups, such as Amoebozoa, Opisthkonta, as well as the sisterhood of Amoebozoa and Opisthkonta (figs. 1, 4, and 6; supplementary figs. S2 and S6–S10, Supplementary Material online). Analyses of the smaller datasets also successfully recovered the supergroup Excavata, in agreement with most previous studies (e.g., Burki et al. 2016; Katz and Grant 2014).
Importantly, the large datasets (i.e., the total sets of all Class I or Class II genes) fully corroborated the recently recognized supergroup SAR, and strongly supported the affinity of cryptophytes and haptophytes with members of Archaeplastida (fig. 6 and supplementary fig. S2B, Supplementary Material online). Similarly, both smaller datasets Euk-G20, and Euk-G52 also supported the grouping of haptophytes with green plants (fig. 4, supplementary figs. S7–10, Supplementary Material online). Although the affinity of haptophytes, cryptophytes and Archaeplastida are highly supported throughout our study, it should be noted that an alternative placement of haptophytes as sister group with SAR is supported by Burki et al. (2016), thus further studies are still needed. Altogether, both our phylogenetic results and Burki’s are consistent with the paraphyly of the previous supergroup “Chromalveolata” which was based on the hypothesis that a single ancestral secondary endosymbiotic event contributed to the red plastids in all its members (e.g., cryptophytes, haptophytes, and stramenopiles) (Cavalier-Smith 1999). Thus, our results also lend support to the scenario of two or more separate secondary endosymbiotic events in different lineages of “Chromalveolata” (Archibald 2009; Baurain et al. 2010).
Interestingly, the close relationship between red algae and green plants received maximum support from the datasets containing hundreds of genes (figs. 1 and 6 and supplementary fig. S2, Supplementary Material online) and a relatively small subset Euk-G52 (supplementary figs. S7–10, Supplementary Material online). However, the position of red algae was not confidently resolved in our analysis of eukaryotic phylogeny using 20–25 genes (fig. 4). One possible reason is the limited taxon sampling. The monophyly of Archaeplastida has also been uncertain in recent phylogenetic analyses (Parfrey et al. 2006, 2010; Kim and Graham 2008; Yoon et al. 2008). As suggested by others (Yoon et al. 2008; Parfrey et al. 2010), improved taxon sampling, as well as gene sampling (Katz and Grant 2014), might be critical for understanding the evolutionary history of Archaeplastida. It is therefore possible that, given the limited taxon sampling in Archaeplastida, many more genes are needed to resolve the relationships among these distant photosynthetic lineages. Future studies with more taxa should also be able to address this question.
Microsporidia and Cryptomycota
As mentioned earlier, Microsporidia include rapidly evolving parasites of animals and some other protists, and their phylogenetic placement has been difficult. Earlier phylogenies based on small subunit ribosomal RNA showed an early divergence of Microsporidia in the eukaryotic tree of life (Knoll 1992; Sogin and Silberman 1998), which was likely affected by the extreme long branches of Microsporidia (Fischer and Palmer 2005). Recent analyses of protein coding genes with improved phylogenetic methods suggested that Microsporidia is related to fungi (Hirt et al. 1999; Katinka et al. 2001; Keeling 2003; Gill and Fast 2006; James et al. 2006), yet the specific relationship between Microsporidia and various fungal lineages remains unresolved. Different relationships were proposed, including a position within Zygomycota (Keeling 2003) and a sister relationship to almost all fungi (James et al. 2006), but these hypotheses were not well supported. Our analyses using several taxa and gene selections consistently placed Mircrosporidia sister to all true fungi (figs. 1 and 4–6, supplementary figs. S2 and S6–S10, Supplementary Material online) with high confidence, congruent with recent studies using internal transcribed spacer (ITS) sequences (Schoch et al. 2012) or large phylogenomic datasets (Capella-Gutierrez et al. 2012; Ebersberger et al. 2012; James et al. 2013), thus providing an evolutionary basis for further comparative studies.
Recent studies have also reported additional taxa related to fungi that form a clade with microsporidia; these organisms include M. daphnia, a relative of microsporidia and a more distantly related organism R. allomycis (Cryptomycota) (James et al. 2013; Haag et al. 2014). Microsporidia are generally adapted to intracellular parasitism and mostly only have remnants degenerated from mitochondria, thus lacking the ability to produce ATP, whereas Mitosporidium still possesses a mitochondrial genome. The other endoparasite Rozella shares similar nucleotide transport elements to those of microsporidia to obtain energy from their hosts. Phylogenetic support for these species have only recently been revealed (James et al. 2013; Haag et al. 2014), and not widely accepted. Our phylogenetic and AU test analysis are in strong agreement with that Mitosporidium diverged from other, more typical, microsporidia as the earliest branch, and that both forms of microsporidia form a well-supported sister clade to Rozella, providing strong evidence for their evolutionary relatedness. In addition, the successful resolution of relationships between these fast-evolving and not-well understood lineages also revealed the strong potential of the 24 novel marker genes for deep fungal phylogeny.
Early-Branching Fungal Lineages
Our study of fungal phylogeny included representatives of several early-branching or phylogenetically uncertain fungal lineages that were not present in many other phylogenomic studies (Fitzpatrick et al. 2006; Wang et al. 2009; Medina et al. 2011) (fig. 5). By resolving many of the relationships among these lineages, our study provides important insights into the history of early fungal evolution. First, there is strong support for the monophyly of ‘euchytrids’, including Chytridiales, Monoblepharidales, Neocallimastigales, and Spizellomycetales. In addition, our results strongly supported the grouping of Entomophthoromycotina and Kickxellomycotina, and their more recent divergence in the fungal phylogeny as compared to ‘euchytrids’ and Blastocladiomycota. These relationships provide strong support for the hypothesis that fungi evolved from an aquatic ancestor (James et al. 2006).
However, our analysis showed that Blastocladiomycota split from other fungi before ‘euchytrids’, as opposite to the order revealed in previous studies (James et al. 2006; Liu et al. 2009b). Another alternative topology, the sister relationship between these two early-branching fungal lineages, was found in a recent phylogenomic study using their most appropriate dataset and phylogenetic approach (Ebersberger et al. 2012). Similarly, the position of Glomeromycota and Murcoromycotina remained unresolved; our Bayesian and ML approaches yielded inconsistent topologies, a recent phylogenomic study also showed varying results depending on datasets and methods (Ebersberger et al. 2012). It should be noted that these early-branching fungal lineages are poorly represented in our analysis and other phylogenomic studies; a better understanding of these difficult relationships would likely require improved taxon-sampling which will be greatly facilitated with more phylogenetic markers available (e.g., the hundreds of eukaryotic marker genes we reported in this study).
Ascomycota
Our results also reveal intriguing relationships in Ascomycota (fig. 5). At the base of Ascomycota, previous studies using few genes or phylogenomics datasets showed support for the monophyly of Taphrinomycotina (Sugiyama et al. 2006; Liu et al. 2009a). However, another recent study showed that Taphrinomycotina was not consistently supported as being monophyletic by all datasets (Ebersberger et al. 2012). In this study, we find strong support for the monophyly of Taphrinomycotina in Bayesian analysis, but only weak support in ML analysis. The fact that conflicting results were obtained from different datasets suggests that the position of Saitoella complicata need further investigation. Furthermore, the relationships among major clades in Pezizomycotina have been controversial (Fitzpatrick et al. 2006; Robbertse et al. 2006; Spatafora et al. 2006; Schoch et al. 2009; Wang et al. 2009). We sampled additional representatives from Lecanoromycetes and our Bayesian analysis showed maximal support for placing Dothideomycetes as sister to Eurotiomycetes + Lecanoromycetes. Our results yielded strong support for the relationships from other recent studies (Medina et al. 2011; Ebersberger et al. 2012), suggesting that they likely reflect the true evolutionary history of Pezizomycotina.
The Marker Genes Are Useful for Both Gene-Rich and Taxon-Rich Approaches
Both gene-rich and taxon-rich approaches have greatly contributed to the assembly of the eukaryotic tree of life. The relative importance of more genes or more taxa has been debated for a long time, perhaps largely because of our limited ability of sequence acquisition. It is unlikely that genome-scale data from the majority of eukaryotes will soon become available, although such information is certainly desirable for analyses of eukaryotic phylogeny. Moreover, recent gene-rich and taxon-rich analyses have both emphasized more balanced sampling of genes and taxa. On the one hand, broader taxon sampling has been achieved in gene-rich analyses through EST and transcriptome sequencing projects in targeted organisms, aiming to reduce systematic errors in phylogenomic studies (Philippe et al. 2005; Dunn et al. 2008; Pick et al. 2010; Misof et al. 2014). One the other hand, taxon-rich analyses have expanded the selection of marker genes (Parfrey et al. 2010; Katz and Grant 2014), because a moderate number of genes carry much stronger phylogenetic signals than one or a few genes. At least in the near future both approaches will continue to play important roles in the study of eukaryotic phylogeny.
In this study, we described 943 promising eukaryotic marker genes. They can either be combined with other suitable genes to investigate new relationships or be used as independent dataset to test existing hypotheses. As we have demonstrated, these genes can be sampled through transcriptomic/genomic sequencing projects, and can be valuable tools for phylogenomic studies. We also provided additional information of these genes, such as lengths of alignable regions, average protein sequence identity, and phylogenetic informativeness profiles. Based on such information, researchers can freely decide which genes to include in their analyses.
In addition, using smaller subsets of ∼20 genes (Euk-S39G20, Euk-S39G25, and Fun-S114G24) and 52 genes (Euk-S33/35/40/42-G52) as examples, our results further suggest that many excellent marker genes for taxon-rich analyses can also be developed from the 943 genes reported here. Importantly, we showed that the ∼20 genes have likely maintained single-copy and orthologous relationship in most eukaryotes; thus, they are less likely to have the issue of hidden paralogy, which might be difficult to reveal using transcriptome data (e.g., a paralog is expressed instead of the ortholog). Moreover, this moderate gene set can provide strong resolving power for both very ancient and subsequent eukaryotic relationships. Therefore, these genes, and perhaps others among the 943 genes that show orthology in a wide range of eukaryotes, should be given priority in the selection of phylogenetic markers for phylogenomic studies. Again, the many marker genes we report here are highly useful for both gene-rich and taxon-rich analyses; they can greatly facilitate the study of specific clades, and also have the potential to serve as the common threads to allow for the integration of eukaryotic tree of life studies.
Supplementary Material
Acknowledgments
We thank Profs Blair Hedges and Claude dePamphilis for helpful discussions. We thank Prof Frank Anderson, Dr Allen Collins, Dr Timothy James and the reviewers for very constructive comments and suggestions on the manuscript. This study was supported by a grant from the Chinese National Natural Science Foundation (91531301) to H.M., and by funds from the State Key Laboratory of Genetic Engineering and Fudan University, the Biology Department, the Eberly College of Sciences, and the Huck Institutes of the Life Sciences, the Pennsylvania State University. H.M. was also supported by funds from Fudan University. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Literature Cited
- Adl SM, et al. 2005. The new higher level classification of eukaryotes with emphasis on the taxonomy of protists. J Eukaryot Microbiol. 52:399–451. [DOI] [PubMed] [Google Scholar]
- Adl SM, et al. 2012. The revised classification of eukaryotes. J Eukaryot Microbiol. 59:429–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguileta G, et al. 2008. Assessing the performance of single-copy genes for recovering robust phylogenies. Syst Biol. 57:613–627. [DOI] [PubMed] [Google Scholar]
- Andersson JO. 2009. Gene transfer and diversification of microbial eukaryotes. Annu Rev Microbiol. 63:177–193. [DOI] [PubMed] [Google Scholar]
- Andersson JO. 2011. Evolution of patchily distributed proteins shared between eukaryotes and prokaryotes: dictyostelium as a case study. J Mol Microbiol Biotechnol. 20:83–95. [DOI] [PubMed] [Google Scholar]
- Andersson JO, Hirt RP, Foster PG, Roger AJ. 2006. Evolution of four gene families with patchy phylogenetic distributions: influx of genes into protist genomes. BMC Evol Biol. 6:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archibald JM. 2009. The puzzle of plastid evolution. Curr Biol. 19:R81–R88. [DOI] [PubMed] [Google Scholar]
- Bapteste E, et al. 2002. The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proc Natl Acad Sci U S A. 99:1414–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baurain D, et al. 2010. Phylogenomic evidence for separate acquisition of plastids in cryptophytes, haptophytes, and stramenopiles. Mol Biol Evol. 27:1698–1709. [DOI] [PubMed] [Google Scholar]
- Baurain D, Brinkmann H, Philippe H. 2007. Lack of resolution in the animal phylogeny: closely spaced cladogeneses or undetected systematic errors? Mol Biol Evol. 24:6–9. [DOI] [PubMed] [Google Scholar]
- Burki F. 2014. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb Perspect Biol. 6:a016147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F, et al. 2009. Large-scale phylogenomic analyses reveal that two enigmatic protist lineages, telonemia and centroheliozoa, are related to photosynthetic chromalveolates. Genome Biol Evol. 1:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F, et al. 2016. Untangling the early diversification of eukaryotes: a phylogenomic study of the evolutionary origins of Centrohelida, Haptophyta and Cryptista. Proc R Soc B. 283:20152802.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F, Okamoto N, Pombert JF, Keeling PJ. 2012. The evolutionary history of haptophytes and cryptophytes: phylogenomic evidence for separate origins. Proc Biol Sci. 279:2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burki F, Shalchian-Tabrizi K, Pawlowski J. 2008. Phylogenomics reveals a new 'megagroup' including most photosynthetic eukaryotes. Biol Lett. 4:366–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella-Gutierrez S, Marcet-Houben M, Gabaldon T. 2012. Phylogenomics supports microsporidia as the earliest diverging clade of sequenced fungi. BMC Biol. 10:47.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. 2000. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 17:540–552. [DOI] [PubMed] [Google Scholar]
- Cavalier-Smith T. 2010. Kingdoms Protozoa and Chromista and the eozoan root of the eukaryotic tree. Biol Lett. 6:342–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. 1999. Principles of protein and lipid targeting in secondary symbiogenesis: euglenoid, dinoflagellate, and sporozoan plastid origins and the eukaryote family tree. J Eukaryot Microbiol. 46:347–366. [DOI] [PubMed] [Google Scholar]
- Chen F, Mackey AJ, Stoeckert CJ, Jr, Roos DS. 2006. OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res. 34:D363–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli FD, et al. 2006. Toward automatic reconstruction of a highly resolved tree of life. Science 311:1283–1287. [DOI] [PubMed] [Google Scholar]
- Corradi N, Keeling PJ. 2009. Microsporidia: a journey through radical taxonomical revisions. Fungal Biol Rev. 23:1–8. [Google Scholar]
- Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. 2008. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A. 105:20356–20361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins CA, McInerney JO. 2011. A method for inferring the rate of evolution of homologous characters that can potentially improve phylogenetic inference, resolve deep divergence and correct systematic biases. Syst Biol. 60:833–844. [DOI] [PubMed] [Google Scholar]
- Darriba D, Taboada GL, Doallo R, Posada D. 2011. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27:1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn CW, et al. 2008. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature 452:745–749. [DOI] [PubMed] [Google Scholar]
- Ebersberger I, et al. 2012. A consistent phylogenetic backbone for the fungi. Mol Biol Evol. 29:1319–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebersberger I, Strauss S, Von Haeseler A. 2009. HaMStR: profile hidden markov model based search for orthologs in ESTs. BMC Evol Biol. 9:157.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer WM, Palmer JD. 2005. Evidence from small-subunit ribosomal RNA sequences for a fungal origin of Microsporidia. Mol Phylogen Evol. 36:606–622. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DA, Logue ME, Stajich JE, Butler G. 2006. A fungal phylogeny based on 42 complete genomes derived from supertree and combined gene analysis. BMC Evol Biol. 6:99.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill EE, Fast NM. 2006. Assessing the microsporidia-fungi relationship: combined phylogenetic analysis of eight genes. Gene 375:103–109. [DOI] [PubMed] [Google Scholar]
- Haag KL, et al. 2014. Evolution of a morphological novelty occurred before genome compaction in a lineage of extreme parasites. Proc Natl Acad Sci U S A. 111:15480–15485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampl V, et al. 2009. Phylogenomic analyses support the monophyly of Excavata and resolve relationships among eukaryotic “supergroups”. Proc Natl Acad Sci U S A. 106:3859–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He D, et al. 2014. An alternative root for the eukaryote tree of life. Curr Biol. 24:465–470. [DOI] [PubMed] [Google Scholar]
- Hedtke SM, Townsend TM, Hillis DM. 2006. Resolution of phylogenetic conflict in large data sets by increased taxon sampling. Syst Biol. 55:522–529. [DOI] [PubMed] [Google Scholar]
- Hirt RP, et al. 1999. Microsporidia are related to Fungi: evidence from the largest subunit of RNA polymerase II and other proteins. Proc Natl Acad Sci U S A. 96:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Rivera MC, Lake JA. 1999. Horizontal gene transfer among genomes: the complexity hypothesis. Proc Natl Acad Sci U S A. 96:3801–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James TY, et al. 2006. Reconstructing the early evolution of Fungi using a six-gene phylogeny. Nature 443:818–822. [DOI] [PubMed] [Google Scholar]
- James TY, et al. 2013. Shared signatures of parasitism and phylogenomics unite Cryptomycota and microsporidia. Curr Biol. 23:1548–1553. [DOI] [PubMed] [Google Scholar]
- Jeffroy O, Brinkmann H, Delsuc F, Philippe H. 2006. Phylogenomics: the beginning of incongruence? Trends Genet. 22:225–231. [DOI] [PubMed] [Google Scholar]
- Katinka MD, et al. 2001. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature 414:450–453. [DOI] [PubMed] [Google Scholar]
- Katz LA. 2012. Origin and diversification of eukaryotes. Annu Rev Microbiol. 66:411–427. [DOI] [PubMed] [Google Scholar]
- Katz LA, Grant JR. 2014. Taxon-rich phylogenomic analyses resolve the eukaryotic tree of life and reveal the power of subsampling by sites. Syst Biol. 64:406–415. [DOI] [PubMed] [Google Scholar]
- Keeling PJ. 2003. Congruent evidence from alpha-tubulin and beta-tubulin gene phylogenies for a zygomycete origin of microsporidia. Fungal Genet Biol. 38:298–309. [DOI] [PubMed] [Google Scholar]
- Keeling PJ, Inagaki Y. 2004. A class of eukaryotic GTPase with a punctate distribution suggesting multiple functional replacements of translation elongation factor 1alpha. Proc Natl Acad Sci U S A. 101:15380–15385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Graham LE. 2008. EEF2 analysis challenges the monophyly of Archaeplastida and Chromalveolata. PLoS One 3:e2621.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AH. 1992. The early evolution of eukaryotes: a geological perspective. Science 256:622–627. [DOI] [PubMed] [Google Scholar]
- Lapierre P, Gogarten JP. 2009. Estimating the size of the bacterial pan-genome. Trends Genet. 25:107–110. [DOI] [PubMed] [Google Scholar]
- Lartillot N, Lepage T, Blanquart S. 2009. PhyloBayes 3: a Bayesian software package for phylogenetic reconstruction and molecular dating. Bioinformatics 25:2286–2288. [DOI] [PubMed] [Google Scholar]
- Le SQ, Gascuel O. 2008. An improved general amino acid replacement matrix. Mol Biol Evol. 25:1307–1320. [DOI] [PubMed] [Google Scholar]
- Lin Z, Kong H, Nei M, Ma H. 2006. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci U S A. 103:10328–10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Nei M, Ma H. 2007. The origins and early evolution of DNA mismatch repair genes—multiple horizontal gene transfers and co-evolution. Nucleic Acids Res. 35:7591–7603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. 2009a. Phylogenomic analyses support the monophyly of Taphrinomycotina, including Schizosaccharomyces fission yeasts. Mol Biol Evol. 26:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. 2009b. Phylogenomic analyses predict sistergroup relationship of nucleariids and fungi and paraphyly of zygomycetes with significant support. BMC Evol Biol. 9:272.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Richards TA, Aves SJ. 2009. Ancient diversification of eukaryotic MCM DNA replication proteins. BMC Evol Biol. 9:60.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayrose I, Graur D, Ben-Tal N, Pupko T. 2004. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol Biol Evol. 21:1781–1791. [DOI] [PubMed] [Google Scholar]
- Medina EM, Jones GW, Fitzpatrick DA. 2011. Reconstructing the fungal tree of life using phylogenomics and a preliminary investigation of the distribution of yeast prion-like proteins in the fungal kingdom. J Mol Evol. 73:116–133. [DOI] [PubMed] [Google Scholar]
- Misof B, et al. 2014. Phylogenomics resolves the timing and pattern of insect evolution. Science 346:763–767. [DOI] [PubMed] [Google Scholar]
- Parfrey LW, et al. 2006. Evaluating support for the current classification of eukaryotic diversity. PLoS Genet. 2:e220.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parfrey LW, et al. 2010. Broadly sampled multigene analyses yield a well-resolved eukaryotic tree of life. Syst Biol. 59:518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, et al. 2011. Acoelomorph flatworms are deuterostomes related to Xenoturbella. Nature 470:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, et al. 2004. Phylogenomics of eukaryotes: impact of missing data on large alignments. Mol Biol Evol. 21:1740–1752. [DOI] [PubMed] [Google Scholar]
- Philippe H, et al. 2009. Phylogenomics revives traditional views on deep animal relationships. Curr Biol. 19:706–712. [DOI] [PubMed] [Google Scholar]
- Philippe H, et al. 2011. Resolving difficult phylogenetic questions: why more sequences are not enough. PLoS Biol. 9:e1000602.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippe H, Lartillot N, Brinkmann H. 2005. Multigene analyses of bilaterian animals corroborate the monophyly of Ecdysozoa, Lophotrochozoa, and Protostomia. Mol Biol Evol. 22:1246–1253. [DOI] [PubMed] [Google Scholar]
- Pick KS, et al. 2010. Improved phylogenomic taxon sampling noticeably affects non-bilaterian relationships. Mol Biol Evol. 27:1983–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quang LS, Gascuel O, Lartillot N. 2008. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics 24:2317–2323. [DOI] [PubMed] [Google Scholar]
- Robbertse B, Reeves JB, Schoch CL, Spatafora JW. 2006. A phylogenomic analysis of the Ascomycota. Fungal Genet Biol. 43:715–725. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ezpeleta N, et al. 2005. Monophyly of primary photosynthetic eukaryotes: green plants, red algae, and glaucophytes. Curr Biol. 15:1325–1330. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ezpeleta N, et al. 2007a. Toward resolving the eukaryotic tree: the phylogenetic positions of jakobids and cercozoans. Curr Biol. 17:1420–1425. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Ezpeleta N, et al. 2007b. Detecting and overcoming systematic errors in genome-scale phylogenies. Syst Biol. 56:389–399. [DOI] [PubMed] [Google Scholar]
- Rokas A, Carroll SB. 2005. More genes or more taxa? The relative contribution of gene number and taxon number to phylogenetic accuracy. Mol Biol Evol. 22:1337–1344. [DOI] [PubMed] [Google Scholar]
- Rokas A, Williams BL, King N, Carroll SB. 2003. Genome-scale approaches to resolving incongruence in molecular phylogenies. Nature 425:798–804. [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. 2003. R8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19:301–302. [DOI] [PubMed] [Google Scholar]
- Schoch CL, et al. 2012. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. 109:6241–6246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoch CL, et al. 2009. The Ascomycota tree of life: a phylum-wide phylogeny clarifies the origin and evolution of fundamental reproductive and ecological traits. Syst Biol. 58:224–239. [DOI] [PubMed] [Google Scholar]
- Shimodaira H, Hasegawa M. 2001. CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247. [DOI] [PubMed] [Google Scholar]
- Simpson AG, Inagaki Y, Roger AJ. 2006. Comprehensive multigene phylogenies of excavate protists reveal the evolutionary positions of “primitive” eukaryotes. Mol Biol Evol. 23:615–625. [DOI] [PubMed] [Google Scholar]
- Simpson AG, Perley TA, Lara E. 2008. Lateral transfer of the gene for a widely used marker, alpha-tubulin, indicated by a multi-protein study of the phylogenetic position of Andalucia (Excavata). Mol Phylogen Evol. 47:366–377. [DOI] [PubMed] [Google Scholar]
- Sogin ML, Silberman JD. 1998. Evolution of the protists and protistan parasites from the perspective of molecular systematics. Int J Parasitol. 28:11–20. [DOI] [PubMed] [Google Scholar]
- Spatafora JW, et al. 2006. A five-gene phylogeny of Pezizomycotina. Mycologia 98:1018–1028. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. [DOI] [PubMed] [Google Scholar]
- Stechmann A, Cavalier-Smith T. 2003. The root of the eukaryote tree pinpointed. Curr Biol. 13:R665–R666. [DOI] [PubMed] [Google Scholar]
- Sugiyama J, Hosaka K, Suh SO. 2006. Early diverging Ascomycota: phylogenetic divergence and related evolutionary enigmas. Mycologia 98:996–1005. [DOI] [PubMed] [Google Scholar]
- Surcel A, Zhou X, Quan L, Ma H. 2008. Long-term maintenance of stable copy number in the eukaryotic SMC family: origin of a vertebrate meiotic SMC1 and fate of recent segmental duplicates. J Syst Evol. 46:405–423. [Google Scholar]
- Tekle YI, Grant JR, Kovner AM, Townsend JP, Katz LA. 2010. Identification of new molecular markers for assembling the eukaryotic tree of life. Mol Phylogen Evol. 55:1177–1182. [DOI] [PubMed] [Google Scholar]
- Townsend JP. 2007. Profiling phylogenetic informativeness. Syst Biol. 56:222–231. [DOI] [PubMed] [Google Scholar]
- Townsend JP, Lopez-Giraldez F. 2010. Optimal selection of gene and ingroup taxon sampling for resolving phylogenetic relationships. Syst Biol. 59:446–457. [DOI] [PubMed] [Google Scholar]
- Wang H, Xu Z, Gao L, Hao B. 2009. A fungal phylogeny based on 82 complete genomes using the composition vector method. BMC Evol Biol. 9:195.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisecaver JH, Rokas A. 2015. Fungal metabolic gene clusters-caravans traveling across genomes and environments. Front Microbiol. 6:161.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HS, et al. 2008. Broadly sampled multigene trees of eukaryotes. BMC Evol Biol. 8:14.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.