

Abstract
Background
Rare DNA breakage-repair disorders predispose to infection and lympho-reticular malignancies. Hematopoietic cell transplantation (HCT) is curative but co-administered chemo- or radio-therapy is damaging due to systemic radio-sensitivity. We collected HCT outcome data for Nijmegen Breakage syndrome (NBS), DNA ligase IV deficiency (LIG4), Cernunnos-XLF deficiency and ataxia-telangiectasia.
Methods
Data from 38 centres worldwide, including indication, donor, conditioning regimen, graft-versus-host disease (GvHD) and outcome were analyzed. Conditioning was classified as myeloablative (MAC) if it contained radiotherapy or alkylators and reduced intensity (RIC) if no alkylators and/or fludarabine ≤150 mg/m2 and cyclophosphamide ≤ 40 mg/kg were used.
Results
55 new, 14 updated and 18 previously published patients were analyzed. Median age at HCT was 48 (range 1.5 – 552) months. 29 were transplanted for infection, 21 malignancy, 13 bone marrow failure, 13 pre-emptively, 5 had multiple indications, and 6 had no information. 22 received MAC, 59 RIC, 4 were infused;-information unavailable for 2. 73/77 patients with LIG4, Cernunnos-XLF deficiency or NBS received conditioning. Survival was 53/77 (69%), worse for MAC than RIC (p=0.006). Most deaths occurred early post-transplant suggesting poor tolerance of conditioning. Survival in ataxia-telangiectasia patients was 25%. 41/83 patients experienced aGvHD (49%): less in RIC compared to MAC, 26/56 (46%) vs 12/21 (57%) (p=0.45). Median follow-up was 35 (range 2-168) months. No secondary malignancies were reported during 15 years follow-up. Growth and developmental delay remained post-HCT; immune-mediated complications resolved.
Conclusion
RIC-HCT resolves DNA repair disorder-associated immunodeficiency. Long-term follow-up is required for secondary malignancy surveillance. Routine HCT for ataxia-telangiectasia is not recommended.
Keywords: Ataxia-Telangiectasia, Cernunnos-XLF deficiency, DNA repair disorders, DNA Ligase 4 deficiency, Hematopoietic stem cell transplantation, Nijmegen Breakage syndrome
Introduction
Maintenance of genomic stability requires repair of DNA, damaged through endogenous processes such as meiotic and mitotic replication errors, and exogenous processes including exposure to oxidising radicals, DNA-damaging chemicals, ultra-violet and ionizing radiation. Several repair pathways regulate the cell cycle, and recognize and repair DNA damage. One of the most serious events to threaten genomic stability, DNA-double strand breaks (DNA-dsb), if unchecked, lead to loss of genomic material, mutagenesis and oncogenesis or cell death1. Two pathways are employed to repair such damage: homologous recombination, which functions primarily in dividing cells and S phase, and requires a homologous template to maintain replication accuracy, and non-template dependent non-homologous end joining (NHEJ), which is particularly employed during phases of the cell cycle when a homologous template is not present. The latter is an especially error-prone process with some loss of DNA information at the site of the DNA-dsb2.
Development of normal adaptive immunity requires generation of a wide range of T-and B-lymphocyte receptors to recognise unique antigen/MHC combinations and provide effective defence against a broad repertoire of pathogens. Many genetically diverse receptors are generated in the thymus and bone marrow, by breaking, stochastically rearranging and re-joining DNA sequences coding for antigen receptors, a process known as VDJ recombination. Additional diversity is created in B-lymphocytes during immunoglobulin class switch recombination, and somatic hypermutation. The DNA repair mechanisms required to maintain somatic genomic stability are also utilized during lymphocyte VDJ recombination to repair intermediate DNA hairpins and physiological DNA-dsb created following activation of recombination activating gene 1 and 2 (RAG1/2)3. Seven ubiquitously-expressed proteins are associated with NHEJ – Ku70/80 and DNA-PKcs, which stabilize the DNA break, the DNA endo/exonuclease Artemis, important for processing RAG-induced hairpin intermediate joins and the DNA ligase 4, Cernunnos-XLF and XRCC4 complex, which together are responsible for the ligation step. Additionally, ataxia-telangiectasia-mutated and nibrin proteins are involved in the initial cell cycle arrest and recruitment of NHEJ proteins to the breakage site4 (Supplementary Figure 1).
Defects in the lymphoid-specific RAG1/2 proteins lead to T-lymphocyte negative, B-lymphocyte negative, natural killer lymphocyte positive (T-B-NK+) severe combined immunodeficiency (SCID) (5). Defects in Artemis, DNA-PKcs, DNA Ligase 4 and Cernunnos-XLF proteins also lead to T-B-NK+ SCID, and combined immunodeficiencies, often associated with other developmental anomalies, particularly microcephaly in patients with DNA Ligase 4 and Cernunnos-XLF deficiency, as a result of the ubiquitous expression of these proteins6-14. Hematopoietic cell transplantation (HCT) is curative for T-B-NK+ SCID, but best results with donor myeloid chimerism and long-term immune reconstitution are obtained if preparative chemotherapy is administered prior to transplantation15. However, in Artemis-deficient radiosensitive SCID patients, although overall survival is equivalent to patients with RAG-deficient SCID, significant long-term sequelae result from the administration of alkylating agents, which are required to gain donor stem cell engraftment with sustained, long-term thymopoiesis. The use of alkylating chemotherapy does not result in increased short-term toxicities or increased transplant-related mortality, but long-term effects on growth and development are observed, due to the effect of chemotherapy on other somatic cells that harbor the genetic defect15. Similar significant effects of chemotherapy are seen in patients with Fanconi anaemia (OMIM 227650) and dyskeratosis congenita (OMIM 127550) – both DNA fragility syndromes16,17. Given the systemic nature of the DNA-dsb defect in other DNA-dsb repair disorders and the finding that the radiosensitivity is generally more severe than in Artemis-deficiency, it is possible that pre-administration of DNA-damaging chemotherapy prior to transplantation will lead to significant systemic morbidity and possible increased mortality.
Due to the primary immunodeficiency phenotype and the frequent occurrence of malignancy, a number of patients with DNA-dsb repair disorders have undergone HCT10-13,18-28. To assess outcome of HCT for DNA-dsb repair disorders, we surveyed patients transplanted for DNA ligase 4 deficiency (LIG4), Cernunnos-XLF deficiency (XLF or NHEJ1), Nijmegen Breakage Syndrome (NBS) and Ataxia-Telangiectasia (AT), using base line data from Stem CEll Transplant for primary Immune Deficiencies in Europe (SCETIDE), Inborn Errors Working Party (IEWP) of the European Society for Blood and Marrow Transplantation (EBMT) registry, the Center for International Blood and Marrow Transplant Research (CIBMTR) and the North American Primary Immune Deficiency Treatment Consortium (PIDTC) and supplemented with additional information from individual centers where available. Patients with mutations in RAG1/2 and DCLRE1C (encoding Artemis) were excluded from the study, as HCT outcomes for these conditions have recently been reported15.
Methods
Data collection
Data on patients with defined mutations in LIG4 (OMIM 606593), NBN (OMIM 602667), NHEJ1 (OMIM 611290) and ATM (OMIM 607585), who had undergone HCT were gathered from the IEWP of EBMT, SCETIDE, CIBMTR and the North American PIDTC. Further patients were identified from previously published data and case reports. Centers with identified patients completed a proforma to gather data on genetic diagnosis, patient demographic, reason for HCT, type and source of HCT, conditioning regimen employed, rates and severity of graft-versus-host disease (GvHD) and survival post-HCT.
Inclusion criteria were any patient having a confirmed genetic diagnosis and having undergone HCT.
The reason to offer HCT was defined as any category or combination of:
infection, (defined as any listed severe infection or recurrent infections)
malignancy
bone marrow failure, (defined as leukopenia, anemia or thrombocytopeniawithout the presence of infection or malignancy)
autoimmunity
pre-emptive.
Conditioning was categorized as either myeloablative conditioning or reduced intensity conditioning. Myeloablative conditioning (MAC) was defined as any regimen using high dose alkylating agents, typically melphalan or busulphan, thiotepa, or total body irradiation at any dose. Although a low dose 200-400cGy regimen can normally be considered non-myeloablative, we reasoned that radiation-sensitive cells were best not exposed to ionising radiation. If the regimen did not use alkylating agents and/or had doses of fludarabine ≤150 mg/m2 and cyclophosphamide ≤ 40 mg/kg it was defined as reduced intensity conditioning (RIC)29. A modified Fanconi-regimen was based on fludarabine 120-150 mg/m2 (30 mg/m2/day in 4-5 divided doses), cyclophosphamide 20-40 mg/kg (in 4 divided doses) with or without anti-thymocyte globulin (ATG) or alemtuzumab serotherapy30,31 or fludarabine 180/m2 (in 6 divided doses), busulphan 1.6mg/kg (in 2 divided doses) and cyclophosphamide 40mg/kg (in 2 divided doses)32. The use of targeted agents such as antibodies, for example alemtuzumab, did not affect the classification of the conditioning.
The primary outcome that was measured was survival. Secondary outcome measures sought were presence, severity and outcome of GvHD, other transplant-related complications and survival.
Analysis
Significance of results was determined by use of Fisher's exact test, utilising 2×2 contingency tables. A two-tailed p value of ≤ 0·05 was considered significant. Kaplan-Meier curves were created based on last known status at time of proforma received, cases where survival was not listed have been excluded from the survival analysis. All statistics were calculated using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, California).
Results
Data were collected from 38 centers worldwide, culminating in 55 newly identified patients, and 14 previously published patients with updated new information, giving new information on 69 patients. Available data from 18 previously published cases10-14,18-28 were included where possible, totalling 87 cases. The median age of patients at HCT was 48 months (range 1.5 - 552 months), 47 were male (54%).
Mutations in LIG4 were most commonly represented, (36 patients, 32 unpublished or with new information) (Table S1), 26 with NBN mutations (17 unpublished or with new information) (Table S2), 17 with NHEJ1 mutations (12 unpublished or with new information) (Table S3), and 8 with ATM mutations (all with new information, 2 previously published, updated in this report) (Table S4). All patients received allogeneic hematopoietic stem cells, except two published cases who died immediately prior to HCT, whilst receiving MAC, but whose data were included in the study.
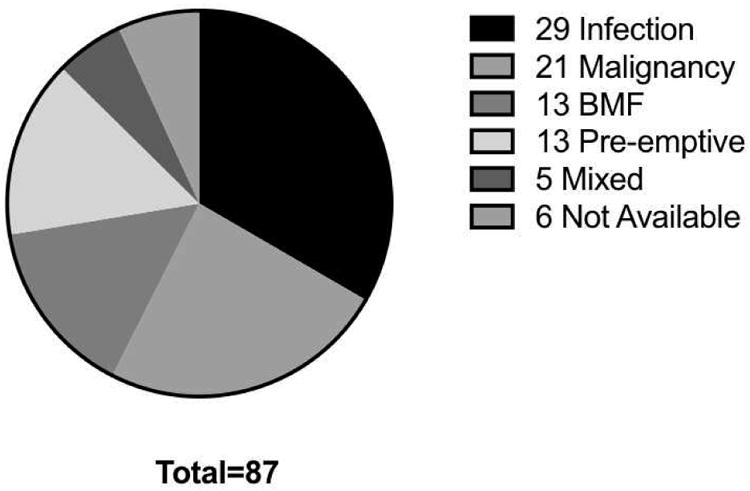
Information was provided on the primary reason for HCT in 83 patients (figure 1). Significant or repeated infections were the most commonly cited reason (29 patients, 35% - 12 with LIG4 mutations, 11 with NHEJ1 mutations), 13 patients were transplanted for bone marrow failure (15%) and 21 patients (24%) for malignancy (17 with NBN mutations). Thirteen patients were transplanted pre-emptively on the basis of a SCID-like diagnosis (15%), 10 with LIG4 mutations. Five patients had a mixture of the above indications, and in 6 patients, the reason for HCT was not available.
Figure 1. Indication for hematopoietic cell transplantation.
Twenty two patients received MAC, and 59 RIC, of which 30 were based on a modified Fanconi anaemia conditioning regimen. Four patients received a stem cell infusion without prior conditioning, data were unavailable for 2 patients. Two received radiotherapy (5Gy, 2Gy) as part of the conditioning regimen.
Survival
Of patients with DNA ligase 4, Cernunnos-XLF deficiency and NBS, there were survival data for 77, of whom 73 received conditioning. Overall survival was 53/77 (69%) (Figure 2A), of whom 2 died from relapse of malignancy giving a transplant-related survival of 71%. One patient with NBS rejected the graft, and is alive with disease. One rejected and succumbed to malignancy. Survival among those receiving myeloablative conditioning was significantly worse at 41% (7/17) compared with 79% (44/56) for those receiving reduced intensity conditioning, (p=0.006) (Figure 2B), describing 2 patients who died of malignancy relapse as survivors. There was no significant difference in transplanted-related mortality between those who received a modified Fanconi or other reduced intensity conditioning regimens (p=0.13). The Kaplan Meier curve demonstrates that the majority of deaths occur early in the course of transplant, particularly in those receiving myeloablative conditioning, suggesting poor tolerance of the conditioning regimen.
Figure 2.
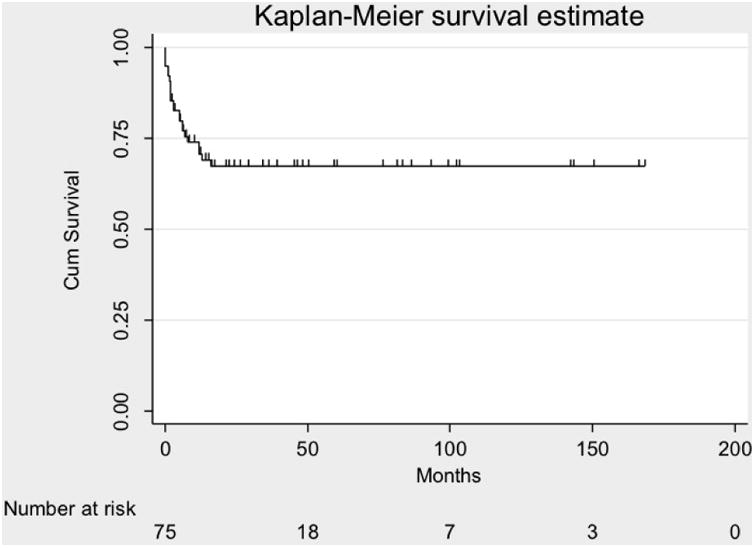
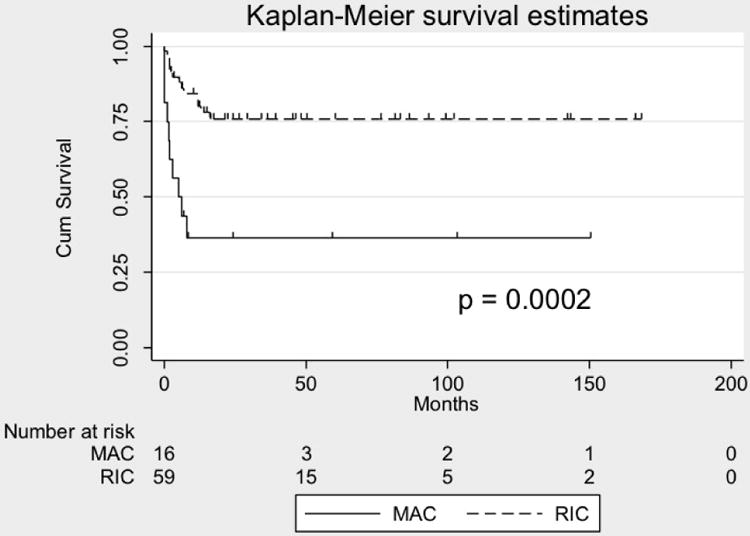
Probabilities of overall survival.
A. Kaplan Meier curve showing overall survival of 74 patients with with DNA ligase 4, Cernunnos-XLF deficiency and Nijmegen Breakage Syndrome
B. Kaplan Meier curve demonstrating differences in survival of 74 patients with DNA ligase 4, Cernunnos-XLF deficiency and Nijmegen Breakage Syndrome transplanted using a reduced intensity or myeloablative conditioning regimens.
In patients with Ataxia-Telangiectasia, overall survival was 25%. Of the 2 patients who survived, both received a modified Fanconi conditioning regimen and neither experienced GvHD, unlike all patients who received myeloablative conditioning. The 6 patients who died experienced GvHD grade 2-3 (67%%), despite well-matched donors. Death was due to multi-organ failure, viral activation or post-transplant lymphoproliferative disorder (PTLD).
Transplant-related survival in the entire cohort for whom data were available was 66% (56/85), with a survival of 75% (45/60) following reduced intensity conditioning and 32% (7/22) following myeloablative conditioning (p= 0·0006). There was no significant difference in outcome between those who underwent HCT for malignancy (12/22 survivors) or for other indications (37/57 survivors, p=0.44). There was no significant difference in survivors for those receiving RIC (11/17) or MAC (1/5) conditioning when malignancy was the reason for HSCT (p=0.14). There was also no significant difference in survivors for those receiving RIC (5/25) or MAC (4/9) conditioning when infection was the reason for HSCT (p=0.09). There were too few cases who were transplanted for bone marrow failure to make a similar comparison.
There were no differences in survival between donor sources whether matched sibling, matched unrelated or mis-matched unrelated donors were used (18/25, 20/27, 5/8 respectively).
Graft versus host disease
Data on presence or absence of acute (a) GvHD was available for 83 patients; in 41 of these, aGvHD was present (49%). Of the reported patients with aGvHD, 24 (59%) had mild (grade 1- 2), and 15 (37%) had severe (grade 3-4) aGvHD (a grade was unavailable for 2 patients). Rates of aGvHD were lower in the RIC group, 26/56 cases (46%) for which data were available, compared to the MAC group, in whom 12/21 cases (57%) experienced aGvHD although this was not statistically significant (p=0.45). Three of 4 patients who received infused stem cells with no preconditioning experienced aGvHD grade 1, 3 and 4 respectively. There was no significant difference in survival between those experiencing grades 0-1 compared with grades 2-4 aGvHD (p=0.22).
Mortality
Overall mortality was 29/85 (34%) - information was unavailable for 2 previously published patients24. Two patients died of multi-organ failure during the conditioning process – both received full myeloablative conditioning. Eleven others died of multi-organ failure post-transplantation, making multi-organ failure the most common cause of death (45%). Eleven deaths were predominantly infectious (38%)
Other Complications
The most common non-aGvHD complication was viremia due to adenovirus, cytomegalovirus (CMV), Epstein-Barr virus (EBV) or a combination, reported in 24/79 patients (30%), of whom 6 died. There were 6 cases (8%) of EBV-related PTLD. Six patients (8%) experienced severe mucositis, 14 developed chronic (c) GvHD (18%). Seven patients rejected the graft – 2 after stem cell infusion (1 with serotherapy), 2 after T-lymphocyte depleted transplants (1 myeloablative, 1 reduced intensity conditioning) and 3 after MAC or RIC transplants. Patients receiving RIC were less likely to develop severe mucositis, veno-occlusive disease or PTLD than those who received MAC (7/59 vs 8/22, p=0.0215).
Follow up
Given the retrospective and multi-institutional nature of the study, detailed information regarding long term (> 5years) follow up was scarce. Median length of follow up was 35 months (range 2-168 months). No secondary malignancies were reported during the follow up period, which although short overall, does include patients with almost 15 years follow up. Pre-existing growth retardation and developmental issues appear to remain post- HCT: more detailed examination would be required to determine whether HCT ameliorates these features. A predisposition to infection or hematological cytopenia pre-existing before HCT appears to have been abolished.
Discussion
Many patients with DNA-dsb repair defects exhibit immunodeficiency, ranging from mild to severe combined immunodeficiency. They are at increased risk of developing lymphoid malignancy. Allogeneic HCT is curative for many immunodeficiencies33. Establishment of effective DNA repair mechanisms in lymphoid progenitors leading to restoration of functional adaptive immunity may prevent the future development of lymphoid malignancy in this cohort of patients. Lymphoid malignancy is difficult to treat effectively when established because of the aggressive nature of the tumours and poor tolerance of patients to cytotoxic radio- and chemotherapy34. It is therefore a reasonable strategy to consider HCT in these patients. However, as most patients have some residual immunity, and even in the SCID phenotype, natural killer cells are present, rejection and poor stem cell engraftment are likely without some preparative cytoreductive pre-conditioning. The systemic nature of the genetic defect, however, increases the risk of substantial morbidity or mortality from chemotherapy or ionising radiation administered prior to transplantation. Only a few small case series of patients with DNA-dsb repair defects undergoing HCT have been published. To date there has been no formal large case series from which to gauge experience.
We now report a multi-institutional retrospective survey on outcome of HCT for 55 previously unpublished patients and update information for 18 previously reported patients with DNA-dsb repair defects. We have demonstrated that HCT can correct the hematopoietic defect and underlying immunodeficiency. Furthermore we have demonstrated that survival is significantly superior when reduced intensity conditioning is used. It is likely that chemotherapy agents, especially alkylating agents, induce systemic double strand breaks, which are not readily repaired because of the underlying genetic defect. These systemic double strand breaks may contribute to the early mortality seen following myeloablative therapy. This intolerance, clinically manifest as severe toxicity, sometimes followed by higher grade GvHD, suggests that when considering HCT, a reduced intensity conditioning regimen should be used in patients with known ionising radiation sensitivity and/or proven diagnosis of a DNA-dsb repair defect, and that radiotherapy should be omitted. Given the equivalence of outcome results when comparing modified Fanconi anemia-based regimens with other reduced intensity regimens, the former may be preferred. Longer term-follow up is required to determine impact of HCT on future prevention of lymphoid malignancy.
The rate of aGvHD overall was 49%, of which 37% was grades 3-4. The rate of cGvHD was 18%. The incidence of severe (grade 3-4) aGvHD and cGvHD is higher than that reported for transplantation of patients with other primary immunodeficiencies35-38. It is not clear whether this is due to the greater use of matched unrelated donors, rather than matched sibling donors (although, in the modern era, outcome of HCT using matched siblings or unrelated donors approaches equivalence), particularly in those receiving reduced intensity conditioning. Significant co-morbidities may also have contributed to the increased incidence of GvHD. However, it may be that the underlying molecular defect causing impaired DNA repair and reduced cellular repair capability predisposes to GvHD following cellular damage, as is found in Fanconi anemia or dyskeratosis congenita17,39.
Patients showed a range of other early post-HCT complications in addition to GvHD. Most common were viral reactivations, which in the case of EBV led to PTLD in 6 patients. Severe mucositis and veno-occlusive disease were commonly encountered.
Three patients experienced veno-occlusive disease and two who had undergone transplant for malignancy, experienced relapse of the primary malignancy. Three patients developed autoimmune thyroid disease, and autoimmune cytopenias were also manifest.
Within this patient cohort there are few data on long-term follow up. Transplantation, unsurprisingly given the systemic nature of the defect, appears not to improve the effects of the primary disease on growth or neurological development. It may be, as in patients with Artemis-SCID, that use of any alkylating agent leads to long-term sequalae15. It will be difficult to predict whether growth or development has been improved or deteriorated as a result of chemotherapy, given the scarce data available on the natural history of these diseases, and the variability of phenotype already reported. However, determining the long-term and late beneficial and adverse effects of HCT in DNA-dsb defects will be important to inform about the utility of this treatment approach. A recent report on a cohort of patients with mutations in NBN documented poor survival in those developing malignancy24. Given the good survival outcome in this cohort amongst those who received reduced intensity conditioning regimens – a pre-emptive approach to transplant may be considered. Of particular importance, therefore, will be long-term follow up to determine frequency of secondary malignancies – not reported so far in other primary immunodeficiency transplant series, but a well-recognised complication in patients transplanted for Fanconi anemia39.
Whilst the outcome of HCT in patients with mutations in LIG4, NBN and NHEJ1 is favourable, particularly when reduced intensity conditioning regimens are employed, the data for patients with Ataxia-Telangiectasia undergoing HCT are disappointing. Whether this is specifically due to the use of myeloablative conditioning regimens, or the presence of malignancy, precipitating transplantation as a therapeutic option, is not clear. With current results, it is difficult to recommend HCT as a treatment option for patients with Ataxia-Telangiectasia, except in clinical trials. In contrast, patients with the other conditions described have transplant outcomes similar to other primary immunodeficiencies when choosing a reduced intensity conditioning. Therefore, transplantation could be considered more favourably as a pre-emptive therapeutic approach, particularly if radiotherapy is omitted from the conditioning regimen, and low intensity conditioning regimens are employed. The high rate of post-transplant complications, including GvHD, remains a concern however, and should drive the development of alternative low or non-toxic conditioning approaches that relieve these patients of the deleterious effects of alkylating therapy but enable full T- and B-lymphocyte reconstitution. In the meantime, careful follow up is required to observe further systemic benefits from transplantation, if any, and importantly to monitor for long-term adverse events. In the future, gene therapy may be an acceptable alternative treatment strategy for this group of patients.
Supplementary Material
Figure S1. V(D)J Recombination
Figure 1A. A. DNA is uncoiled at transcription “factories” within the cell, where the associated recombination and repair proteins co-localize.
B. The lymphoid specific recombinase activating gene 1 and 2 (RAG1/2) proteins recognize and bind the recombination signal sequences (RSS) that flank the V(D)J gene segments, and introduce site-specific DNA-DSBs.
C. The phosphorylated blunt signal ends and the covalently sealed hairpin intermediate of the coding end are held together by the RAG complex.
Figure 1B. D. The MRN complex binds the broken DNA ends and activates ATM which initiates cell cycle arrest and attraction of the repair proteins. H2AX, 53BP1 and RNF168, and with other proteins stabilize the damaged chromatin.
Ei. Ku70/Ku80 heterodimer binds the coding ends and recruits DNA-PKcs and Artemis, which is required to open the hairpin intermediates. The covalently sealed hairpin intermediate is randomly nicked by the DNA-Pkcs/Artemis complex, which generates a single stranded break with 3′ or 5′ overhangs.
Eii. XRCC4, DNA ligase 4 and cernunnos-XLF (C-XLF) co-associate and are recruited to the ends. The signal ends are directly ligated by the XRCC4/DNA-LIG4/C-XLF complex. The opened hairpin intermediate is modified by polymerases, exonucleases and the lymphoid-specific terminal deoxynucleotidyl transferase (TdT), before
Eiii. being repaired and ligated by the XRCC4/DNA-LIG4/C-XLF complex (Reproduced from reference 43 with permission)
Table S1. Characteristics of patients with DNA ligase 4.
Table S2. Characteristics of patients with defects in NBN.
Table S3. Characteristics of patients with defects in NHEJ1.
Table S4. Characteristics of patients with Ataxia Telangiectasia.
Clinical implications.
Hematopoietic cell transplant cures DNA breakage-repair disorders. Cernunnos-XLF deficiency, LIG4 and Nijmegen breakage syndrome patients receiving alkylator or radiotherapy pre-conditioning have worse survival than those receiving reduced intensity conditioning.
Acknowledgments
Declarations: M. J. Cowan was supported by: the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases; the Intramural Research Program of the National Institute of Allergy and Infectious Diseases; and the Office of Rare Diseases Research, National Center for Advancing Translational Sciences, National Institutes of Health, Bethesda, MD, USA; U54-AI082973
Mary Eapen was funded in part by Public Health Service grant U24-CA76518 from the National Cancer Institute, National Heart Lung and Blood Institute and National Institute of Allergy and Infectious Diseases.
Luis Gonzalez-Granado was supported by a Grant from Fondo de Investigación Sanitaria (FIS-PI16/2053).
Abbreviations
- AT
Ataxia-Telangiectasia
- ATG
anti-thymocyte globulin
- ATM
Ataxia-Telangiectasia mutated
- Cernunnos-XLF
Cernunnos –XRCC4 like factor
- CIBMTR
Center for International Blood and Marrow Transplant Research
- CMC
cytomegalovirus
- DNA-dsb
DNA double strand breaks
- DNA-PKcs
DNA protein kinase catalytic subunit
- EBMT
European Society for Blood and Marrow Transplantation
- EBV
Epstein-Barr virus
- GvHD
graft-versus-host disease
- Gy
Gray
- HCT
Hematopoietic cell transplantation
- IEWP
Inborn Errors Working Party
- LIG4
DNA ligase 4 deficiency
- MAC
Myeloablative conditioning
- NBS
Nijmegen Breakage Syndrome
- NHEJ
non-homologous end joining
- NHEJ1
Non-Homologous End Joining Factor 1
- PIDTC
North American Primary Immunodeficiency Treatment Consortium
- PTLD
post-transplant lymphoproliferative disorder
- RAG 1/2
recombination activating gene 1/2
- RIC
reduced intensity conditioning
- SCETIDE
Stem CEll Transplant for primary Immune Deficiencies in Europe
- SCID
severe combined immunodeficiency
- XRCC4
X-ray repair cross-complementing protein 4
Footnotes
Author contributions: ARG conceived and designed the study, interpreted the data and wrote the manuscript, JS and MAS collated and helped interpret the data and write the manuscript, all other authors provided and assisted in analysis of data, and helped write and revise the manuscript. All authors have seen and approved the final version for submission.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang C, Lees-Miller SP. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys. 2013;86:440–9. doi: 10.1016/j.ijrobp.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters CA, Strande NT, Wyatt DW, Pryor JM, Ramsden DA. Nonhomologous end joining: A good solution for bad ends. DNA Repair (Amst) 2014;17:39–51. doi: 10.1016/j.dnarep.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. Adv Genet. 2013;82:1–45. doi: 10.1016/B978-0-12-407676-1.00001-9. [DOI] [PubMed] [Google Scholar]
- 4.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 2014;17:9–20. doi: 10.1016/j.dnarep.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz K, Gauss GH, Ludwig L, Pannicke U, Li Z, Lindner D, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–9. doi: 10.1126/science.274.5284.97. [DOI] [PubMed] [Google Scholar]
- 6.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, et al. Artemis, a novel DNA double-strand break repair/V(D)J recombination protein, is mutated in human severe combined immune deficiency. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 7.Moshous D, Pannetier C, de Chasseval R, le Deist F, Cavazzana-Calvo M, Romana S, et al. Partial T and B lymphocyte immunodeficiency and predisposition to lymphoma in patients with hypomorphic mutations in Artemis. J Clin Invest. 2003;111:381–387. doi: 10.1172/JCI16774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Burg M, Ijspeert H, Verkaik NS, Turul T, Wiegant WW, Morotomi-Yano K, et al. A DNA-PKcs mutation in a radiosensitive T-B- SCID patient inhibits Artemis activation and nonhomologous end-joining. J Clin Invest. 2009;119:91–98. doi: 10.1172/JCI37141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woodbine L, Neal JA, Sasi NK, Shimada M, Deem K, Coleman H, et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. J Clin Invest. 2013;123:2969–2980. doi: 10.1172/JCI67349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O'Driscoll M, Cerosaletti KM, Girard PM, Dai Y, Stumm M, Kysela B, et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol Cell. 2001;8:1175–1185. doi: 10.1016/s1097-2765(01)00408-7. [DOI] [PubMed] [Google Scholar]
- 11.Buck D, Moshous D, de Chasseval R, Ma Y, le Deist F, Cavazzana-Calvo M, et al. Severe combined immunodeficiency and microcephaly in siblings with hypomorphic mutations in DNA ligase IV. Eur J Immunol. 2006;36:224–235. doi: 10.1002/eji.200535401. [DOI] [PubMed] [Google Scholar]
- 12.Enders A, Fisch P, Schwarz K, Duffner U, Pannicke U, Nikolopoulos E, et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J Immunol. 2006;176:5060–5068. doi: 10.4049/jimmunol.176.8.5060. [DOI] [PubMed] [Google Scholar]
- 13.van der Burg M, van Veelen LR, Verkaik NS, Wiegant WW, Hartwig NG, Barendregt BH, et al. A new type of radiosensitive T-B-NK+ severe combined immunodeficiency caused by a LIG4 mutation. J Clin Invest. 2006;116:137–145. doi: 10.1172/JCI26121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buck D, Malivert L, de Chasseval R, Barraud A, Fondanèche MC, Sanal O, et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 15.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014;123:281–289. doi: 10.1182/blood-2013-01-476432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deeg HJ, Socié G, Schoch G, Henry-Amar M, Witherspoon RP, Devergie A, et al. Malignancies after marrow transplantation for aplastic anemia and Fanconi anemia: a joint Seattle and Paris analysis of results in 700 patients. Blood. 1996;87:386–392. [PubMed] [Google Scholar]
- 17.Gadalla SM, Sales-Bonfim C, Carreras J, Alter BP, Antin JH, Ayas M, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013;19:1238–1243. doi: 10.1016/j.bbmt.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gruhn B, Seidel J, Zintl F, Varon R, Tönnies H, Neitzel H, et al. Successful bone marrow transplantation in a patient with DNA ligase IV deficiency and bone marrow failure. Orphanet J Rare Dis. 2007;2:5. doi: 10.1186/1750-1172-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunebaum E, Bates A, Roifman CM. Omenn syndrome is associated with mutations in DNA ligase IV. J Allergy Clin Immunol. 2008;122:1219–1220. doi: 10.1016/j.jaci.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 20.Straathof KC, Rao K, Eyrich M, Hale G, Bird P, Berrie E, et al. Haemopoietic stem cell transplantation with antibody-based minimal intensity conditioning. Lancet. 2009;374:912–920. doi: 10.1016/S0140-6736(09)60945-4. [DOI] [PubMed] [Google Scholar]
- 21.Albert MH, Gennery AR, Greil J, Cale CM, Kalwak K, Kondratenko I, et al. Successful Stem cell transplantation for Nijmegen breakage syndrome. Bone Marrow Transplant. 2010;45:622–626. doi: 10.1038/bmt.2009.207. [DOI] [PubMed] [Google Scholar]
- 22.Woźniak M, Krzywoń M, Hołda MK, Goździk J. Reduced-intensity conditioning umbilical cord blood transplantation in Nijmegen breakage syndrome. Pediatr Transplant. 2015;19:E51–5. doi: 10.1111/petr.12420. [DOI] [PubMed] [Google Scholar]
- 23.Stajner T, Vasiljević Z, Vujić D, Marković M, Ristić G, Mićić D, et al. Atypical strain of Toxoplasma gondii causing fatal reactivation after hematopoietic stem cell transplantion in a patient with an underlying immunological deficiency. J Clin Microbiol. 2013;51:2686–2690. doi: 10.1128/JCM.01077-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dai Y, Kysela B, Hanakahi LA, Manolis K, Riballo E, Stumm M, et al. Nonhomologous end joining and V(D)J recombination require an additional factor. Proc Natl Acad Sci U S A. 2003;100:2462–2467. doi: 10.1073/pnas.0437964100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faraci M, Lanino E, Micalizzi C, Morreale G, Di Martino D, Banov L, et al. Unrelated hematopoietic stem cell transplantation for Cernunnos-XLF deficiency. Pediatric Transplantation. 2009;13:785–789. doi: 10.1111/j.1399-3046.2008.01028.x. [DOI] [PubMed] [Google Scholar]
- 26.Çağdas D, Özgür TT, Asal GT, Revy P, De Villartay JP, van der Burg M, et al. Two SCID cases with Cernunnos-XLF deficiency successfully treated by hematopoietic stem cell transplantation. Pediatr Transplant. 2012;16:E167–71. doi: 10.1111/j.1399-3046.2011.01491.x. [DOI] [PubMed] [Google Scholar]
- 27.Ghosh S, Schuster FR, Binder V, Niehues T, Baldus SE, Seiffert P, et al. Fatal outcome despite full lympho-hematopoietic reconstitution after allogeneic stem cell transplantation in atypical ataxia telangiectasia. J Clin Immunol. 2012;32:438–440. doi: 10.1007/s10875-012-9654-7. [DOI] [PubMed] [Google Scholar]
- 28.Ussowicz M, Musiał J, Duszeńko E, Haus O, Kałwak K. Long-term survival after allogeneic-matched sibling PBSC transplantation with conditioning consisting of low-dose busilvex and fludarabine in a 3-year-old boy with ataxia-telangiectasia syndrome and ALL. Bone Marrow Transplant. 2013;48:740–1. doi: 10.1038/bmt.2012.207. [DOI] [PubMed] [Google Scholar]
- 29.Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the Intensity of Conditioning Regimens: Working Definitions Biol Blood Marrow Transplant. 2009;15:1628–1633. doi: 10.1016/j.bbmt.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de la Fuente J, Reiss S, McCloy M, Vulliamy T, Roberts IA, Rahemtulla A, et al. Non-TBI stem cell transplantation protocol for Fanconi anaemia using HLA-compatible sibling and unrelated donors. Bone Marrow Transplant. 2003;32:653–656. doi: 10.1038/sj.bmt.1704219. [DOI] [PubMed] [Google Scholar]
- 31.https://www.ebmt.org/Contents/About-EBMT/Who-We-Are/ScientificCouncil/Documents/EBMT_ESID_GUIDELINES_FOR_INBORN_ERRORS_ FINAL 2011.pdf (accessed 16th October 2016, 22.00)
- 32.Chao MM, Ebell W, Bader P, Beier R, Burkhardt B, Feuchtinger T, et al. Consensus of German transplant centers on hematopoietic stem cell transplantation in Fanconi anemia. Klin Padiatr. 2015;227(3):157–165. doi: 10.1055/s-0035-1548841. [DOI] [PubMed] [Google Scholar]
- 33.Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of Haematopoietic Stem Cells and Long Term Survival for Primary Immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol. 2010;26:602–610.e1-11. doi: 10.1016/j.jaci.2010.06.015. [DOI] [PubMed] [Google Scholar]
- 34.Wolska-Kusnierz B, Gregorek H, Chrzanowska K, Piątosa B, Pietrucha B, Heropolitańska-Pliszka E, et al. Nijmegen Breakage Syndrome: clinical and immunological features, long-term outcome and treatment options. J Clin Immunol. 2015;35:538–549. doi: 10.1007/s10875-015-0186-9. [DOI] [PubMed] [Google Scholar]
- 35.Güngör T, Albert MH, Teira P, Slatter M, Stüssi G, Stepensky P, et al. Reduced-intensity conditioning and HLA-matched haemopoietic stem-cell transplantation in patients with chronic granulomatous disease: a prospective multicentre study. Lancet. 2014;838:436–448. doi: 10.1016/S0140-6736(13)62069-3. [DOI] [PubMed] [Google Scholar]
- 36.Booth C, Gilmour KC, Veys P, Gennery AR, Slatter MA, Chapel H, et al. X-linked Lymphoproliferative Disease due to SAP/SH2D1A deficiency: A Multicentre Study on the manifestations, management and outcome of the disease. Blood. 2011;117:53–62. doi: 10.1182/blood-2010-06-284935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mahlaoui N, Pellier I, Mignot C, Jais JP, Bilhou-Nabéra C, Moshous D, et al. Characteristics and outcome of early-onset, severe forms of Wiskott-Aldrich syndrome. Blood. 2013;121:1510–6. doi: 10.1182/blood-2012-08-448118. [DOI] [PubMed] [Google Scholar]
- 38.Ouachee-Chardin M, Elie C, de Saint Basile G, Le Deist F, Mahlaoui N, Picard C, et al. Hematopoietic stem cell transplantation in hemophagocytic lymphohistiocytosis: a single-center report of 48 patients. Pediatrics. 2006;117:e743–e750. doi: 10.1542/peds.2005-1789. [DOI] [PubMed] [Google Scholar]
- 39.Peffault de Latour R, Porcher R, Dalle JH, Aljurf M, Korthof ET, Svahn J, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279–4286. doi: 10.1182/blood-2013-01-479733. [DOI] [PubMed] [Google Scholar]
- 40.Tamura S, Higuchi K, Tamaki M, Inoue C, Awazawa R, Mitsuki N, et al. Novel compound heterozygous DNA ligase IV mutations in an adolescent with a slowly-progressing radiosensitive-severe combined immunodeficiency. Clin Immunol. 2015;160:255–60. doi: 10.1016/j.clim.2015.07.004. [DOI] [PubMed] [Google Scholar]
- 41.Ussowicz M, Musiał J, Duszeńko E, Haus O, Kałwak K. Long-term survival after allogeneic-matched sibling PBSC transplantation with conditioning consisting of low-dose busilvex and fludarabine in a 3-year-old boy with ataxia-telangiectasia syndrome and ALL. Bone Marrow Transplant. 2013;48:740–1. doi: 10.1038/bmt.2012.207. [DOI] [PubMed] [Google Scholar]
- 42.Cowan MJ, Gennery AR. Radiation-sensitive severe combined immunodeficiency: The arguments for and against conditioning before hematopoietic cell transplantation--what to do? J Allergy Clin Immunol. 2015;136:1178–85. doi: 10.1016/j.jaci.2015.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. V(D)J Recombination
Figure 1A. A. DNA is uncoiled at transcription “factories” within the cell, where the associated recombination and repair proteins co-localize.
B. The lymphoid specific recombinase activating gene 1 and 2 (RAG1/2) proteins recognize and bind the recombination signal sequences (RSS) that flank the V(D)J gene segments, and introduce site-specific DNA-DSBs.
C. The phosphorylated blunt signal ends and the covalently sealed hairpin intermediate of the coding end are held together by the RAG complex.
Figure 1B. D. The MRN complex binds the broken DNA ends and activates ATM which initiates cell cycle arrest and attraction of the repair proteins. H2AX, 53BP1 and RNF168, and with other proteins stabilize the damaged chromatin.
Ei. Ku70/Ku80 heterodimer binds the coding ends and recruits DNA-PKcs and Artemis, which is required to open the hairpin intermediates. The covalently sealed hairpin intermediate is randomly nicked by the DNA-Pkcs/Artemis complex, which generates a single stranded break with 3′ or 5′ overhangs.
Eii. XRCC4, DNA ligase 4 and cernunnos-XLF (C-XLF) co-associate and are recruited to the ends. The signal ends are directly ligated by the XRCC4/DNA-LIG4/C-XLF complex. The opened hairpin intermediate is modified by polymerases, exonucleases and the lymphoid-specific terminal deoxynucleotidyl transferase (TdT), before
Eiii. being repaired and ligated by the XRCC4/DNA-LIG4/C-XLF complex (Reproduced from reference 43 with permission)
Table S1. Characteristics of patients with DNA ligase 4.
Table S2. Characteristics of patients with defects in NBN.
Table S3. Characteristics of patients with defects in NHEJ1.
Table S4. Characteristics of patients with Ataxia Telangiectasia.