

Abstract
A catalytic, enantioselective decarboxylative allylic alkylation of 4-thiopyranones is reported. The α-quaternary 4-thiopyranones produced are challenging to access by standard enolate alkylation owing to facile ring-opening β-sulfur elimination. In addition, reduction of the carbon-sulfur bonds provides access to elusive acyclic α-quaternary ketones. The alkylated products are obtained in up to 92% yield and 94% enantiomeric excess.
Graphical Abstract
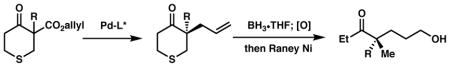
The enantioselective construction of all-carbon quaternary stereogenic centers, particularly acyclic ones, is a significant challenge in organic synthesis.1 Among the most reliable methods available for forming all-carbon quaternary stereocenters, such as conjugate addition and sigmatropic rearrangements, palladium-catalyzed decarboxylative allylic alkylation has become a widely adopted and studied transformation owing to its mild nature, broad scope, and versatility of the introduced allyl group for further synthetic manipulations.2 The mild nature of this transformation was hypothesized to allow access to α-quaternary 4-thiopyranones, which are troublesome to access through standard enolate chemistry owing to the β-disposed sulfur and the propensity for forming ring-opened sulfur alkylation products.3 Despite these disadvantages, such heterocyclic ketones have been shown to undergo aldol4 and Mannich5 reactions, although degradation of the products by retro-aldol processes during purification is common.6 The thiopyranone motif has also been shown to tolerate amine, thiourea dual catalysis for enantioselective conjugate addition forming an α-quaternary 4-thiopyranone in an enantioenriched fashion.7 The value of this motif has been demonstrated by its use as a building block in medicinal chemistry applications,8 therefore new methods for constructing chiral, enantioenriched thiopyranone derivatives would allow access to new chemical space.
Our approach would entail a palladium-catalyzed decarboxylative allylic alkylation of a thiopyranone allyl β-ketoester (1a, Table 1), which would allow access to a diverse range of α-quaternary 4-thiopyranones in an enantioselective fashion. Indeed, a general strategy that has been reported to access alkylated 4-thiopyranones is by first α–functionalization of the corresponding β-ketoester followed by decarboxylation9 or tin hydride mediated addition of alkyl and aryl halides.10
Table 1.
Optimization of Alkylation Conditions.a
| |||||
|---|---|---|---|---|---|
| entry | Pd source | ligand | solvent | yieldb | eec,d |
| 1 | Pd(dba)2 | (S)-L1 | THF | NR | - |
| 2 | Pd(dba)2 | (S)-L2 | THF | 24 | −78 |
| 3 | Pd(dba)2 | (R,R)-L3 | THF | <5 | 24 |
| 4 | Pd(dba)2 | (R,R)-L4 | THF | <5 | 66 |
| 5 | Pd(dba)2 | (R,R)-L5 | THF | 85 | 69 |
| 6 | Pd(dba)2 | (R,R)-L5 | dioxane | –e | 54 |
| 7 | Pd(dba)2 | (R,R)-L5 | EtOAc | –e | 73 |
| 8 | Pd(dba)2 | (R,R)-L5 | toluene | –e | 83 |
| 9 | Pd(dba)2 | (R,R)-L5 | benzene | –e | 78 |
| 10 | Pd(dba)2 | (R,R)-L5 | hexane/toluene (2:1) | –e | 80 |
| 11 | Pd(dba)2 | (R,R)-L5 | TBME | 78 | 87 |
| 12 | [PdCl(allyl)]2 | (R,R)-L5 | TBME | –e | 90 |
| 13 | Pd(dmdba)2 | (R,R)-L5 | TBME | –e | 91 |
| 14 | Pd2(pmdba)3 | (R,R)-L5 | TBME | 90 | 93 |
| 15f | Pd2(pmdba)3 | (R,R)-L5 | TBME | 92 | 94 |
Conditions: 0.1 mmol 1a, 2.5 mol % Pd2(dba)3, 6 mol % ligand, 1 mL solvent.
Yield of isolated product.
Determined by chiral SFC.
Absolute configuration determined by vibrational circular dichroism.
Conversion not monitored.
Reaction performed with 0.2 mmol 1a, 1.0 mol % Pd2(pmdba)3, 2.4 mol % ligand and 2 mL solvent.
In addition to accessing a synthetically challenging quaternary stereocenter, we also envisioned utilizing the sulfur as a removable handle to access acyclic, all-carbon quaternary stereocenters,1c by treatment with Raney nickel. Exploiting a rigid, cyclic thioether to access stereodefined acyclic systems is a valuable strategy utilized in organic synthesis to access polypropionates, most notably Woodward’s synthesis of Erythromycin,11 with this chemistry later expanded upon by other research groups.12 By using a cyclic system, the issue of controlling tri- and tetrasubstituted enolate geometry, a difficult task in acyclic molecules, is circumvented. While certain acyclic tri-substituted enolates can be formed with a high degree of stereochemical fidelity (e.g. oxizolidinones), the methodology to produce tetra-substituted enolates en route to all-carbon quaternary stereocenters is less developed. A notable example comes from the Marek lab, in which a stereoselective carbometalation of alkynes is exploited en route to a number of different motifs including homoallylic alcohols13 and aldol adducts.14 In fact, only a couple reports of forming enantioenriched acyclic α-quaternary ketones exist in the literature.15
We initiated our investigations with α-benzyl β-ketoester 1a and phosphinooxazoline (PHOX) ligands L1 and the electronically deficient L2, used previously by our group for asymmetric allylic alkylation (Table 1).2a Initial reactions employed 5 mol% Pd(dba)2 and 6 mol% ligand in THF. The presence of the Lewis basic sulfur was envisioned to be trivial owing to prior examples of such functionalities (i.e. thioethers) being tolerated under analogous conditions.2a Unfortunately, in this case both PHOX ligands performed poorly, producing either no reaction with L1 (entry 1) or low conversion with L2 (entry 2), albeit in moderate enantiomeric excess.
Bisphosphine ligands L3 and L42b were found to perform equally poorly, providing very low conversion to product (>5%) and enantiomeric excesses of 24 and 66%, respectively (entries 3 and 4). On the other hand, anthracene diamine deriviced bisphosphine L5 was found to affect the desired transformation in 85% isolated yield and 69% enantiomeric excess (entry 5). Despite obtaining a lower enantiomeric excess with L5 compared to L2, the improved reactivity was promising and further optimization efforts focused on L5 (36 additional ligands were examined, see SI for details).
With the ideal ligand identified, the remaining reaction parameters were examined. More polar solvents such as dioxane and EtOAc were found to give decreased enantiomeric excesses of 54% and 73%, respectively (entries 6 and 7). The use of toluene resulted in an increased enantiomeric excess of 83% (entry 8), while benzene and a 2:1 mixture of hexane/toluene were found to give slightly lower levels of selectivity (entries 9 and 10). TBME provided an elevated enantiomeric excess of 87% (entry 11) and was advanced further as the solvent of choice. Additional improvements in enantioselectivity were achieved with different palladium sources (entries 12–14), with Pd2(pmdba)3 providing the best result and furnishing the α-quaternary thiopyranone in 90% isolated yield and 93% enantiomeric excess (entry 14). Lastly, it was found that lowering the loading of catalyst and ligand by 2.5 times resulted in no loss of either yield or enantiomeric excess, with only a modest increase in reaction time from 3 h to 9 h (entry 15), therefore this lowered loading was used for the remainder of the experiments. The absolute stereochemistry of 2a was determined via by vibrational circular dichroism (see SI for details).
With the optimized conditions identified, a variety of different α-substituents were examined in order to evaluate the scope of the chemistry (Scheme 1).16 Simple alkyl groups,17 such as methyl (2b) and ethyl (2c), provided the desired product in moderate yields and moderate to excellent enantiomeric excess. Substituted olefins such as vinyl bromide 2d and prenylated ketone 2e were tolerated, however only moderate enantiomeric excess was obtained. An ester (2f) and an α,β-unsaturated ethyl ester (2g) provided the desired products, again with only moderate selectivity. Alkyl substitution on the 2-position of the allyl was not well tolerated in the reaction, providing the product (2h) in low yield. Lastly, a variety of different benzyl derivatives performed well in the reaction, furnishing the corresponding alkylated products with good to excellent yields and excellent enantioselectivities (2i–2l).
Scheme 1. Substrate Scopea.
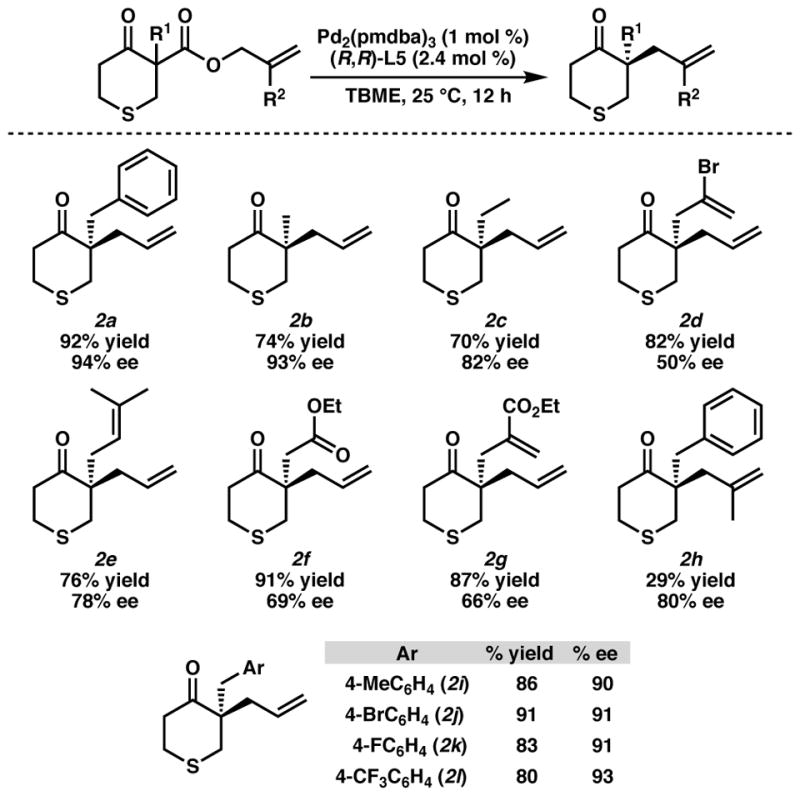
aConditions: 0.2 mmol β-ketoester, 1.0 mol % Pd2(pmdba)3, 2.4 mol % ligand, 2 mL TBME.
With the scope established, we sought to examine our hypothesis pertaining to the formation of acyclic all-carbon quaternary stereocenters through reductive cleavage of the thioether (Scheme 2). When alkylation product 2a was treated directly with Raney nickel in methanol at reflux, the desired desulfurization occurred, however concomitant olefin hydrogenation was also observed. All attempts to prevent olefin hydrogenation while still achieving the desired desulfurization failed. To circumvent this issue, the allyl group can be functionalized prior to the desulfurization step, exemplified by the hydroboration-oxidation of 2a prior to treatment with Raney nickel, providing acyclic ketone 3 with a synthetic handle for further manipulations.
Scheme 2.
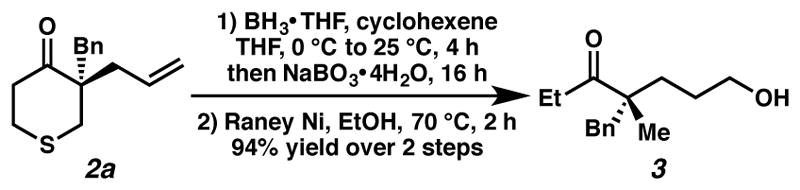
Transformation of Alkylation Product to an Acyclic Ketone.a
In summary, we have developed an enantioselective Pd-catalyzed decarboxylative allylic alkylation of thiopyranones. Conditions were identified that tolerate the presence of the Lewis basic thioether, providing alkylated products in moderate to excellent levels of selectivity, with no issues of ring-opening β-elimination. While the cyclic products formed have multiple handles for further derivatization, the thioether can be reduced to afford desulfurized molecules containing an acyclic, all-carbon quaternary stereocenter. We envision that such acyclic α-quaternary ketones, as well as the parent thiopyranones, will find use in synthesis and medicinal chemistry.
Supplementary Material
Figure 1.
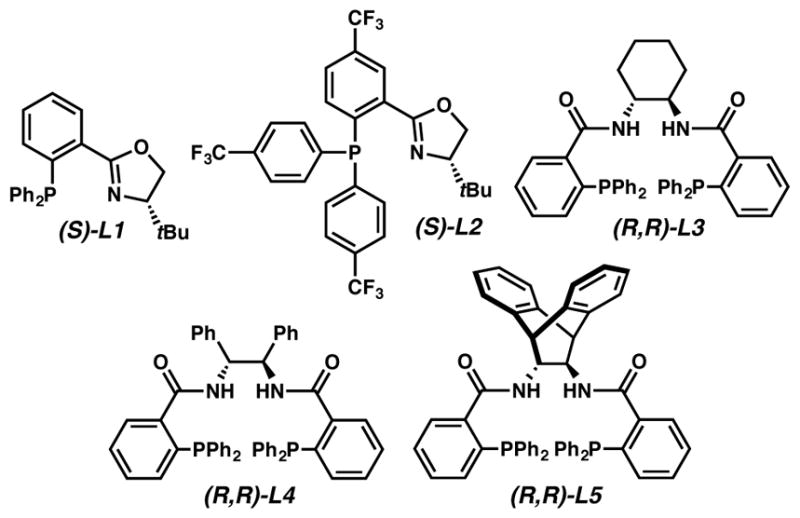
Representative Ligands Examined.
Acknowledgments
We thank NIH-NIGMS (R01GM080269), Amgen, the Gordon and Betty Moore Foundation, and Caltech for financial support. E.J.A. is grateful to the National Science Foundation for a predoctoral fellowship. We thank Dr. David VanderVelde (Caltech) for NMR expertise, and Dr. Mona Shahgholi (Caltech) and Naseem Torian (Caltech) for mass spectrometry assistance.
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, characterization data, vibrational circular dichroism determination of absolute stereochemistry. (PDF)
References
- 1.Selected reviews: Martin SF. Tetrahedron. 1980;36:419–460.Douglas CJ, Overman LE. Proc Natl Acad Sci U S A. 2004;101:5363–5367. doi: 10.1073/pnas.0307113101.Das JP, Marek I. Chem Commun. 2011;111:1846–1913.Liu Y, Han SJ, Liu WB, Stoltz BM. Acc Chem Res. 2015;48:740–751. doi: 10.1021/ar5004658.
- 2.(a) Behenna DC, Mohr JT, Sherden NH, Marunescu SC, Harned AM, Tani K, Seto M, Ma S, Novak Z, Krout MR, McFadden RM, Roizen JL, Enquist JA, Jr, White DE, Levine SR, Petrova KV, Iwashita A, Virgil SC, Stoltz BM. Chem - Eur J. 2011;17:14199–14223. doi: 10.1002/chem.201003383. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343–18357. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Batten RJ, Coyle JD, Taylor RJK, Vassiliou S. J Chem Soc, Perkin Trans 1. 1982:1177–1182. [Google Scholar]
- 4.Jheengut V, Ward DE. J Org Chen. 2007;72:7805–7808. doi: 10.1021/jo701546f. [DOI] [PubMed] [Google Scholar]
- 5.Hatano M, Ishihara K. Synthesis. 2010;22:3785–3801. [Google Scholar]
- 6.Hayashi T. Tetrahedron Lett. 1991;32:5369–5372. [Google Scholar]
- 7.Kang JY, Carter RG. Org Lett. 2012;14:3178–3181. doi: 10.1021/ol301272r. [DOI] [PubMed] [Google Scholar]
- 8.(a) Rovnyak GC, Millonig RC, Schwartz J, Shu V. J Med Chem. 1982;25:1482–1488. doi: 10.1021/jm00354a018. [DOI] [PubMed] [Google Scholar]; (b) Shankaran K, Donnelly KL, Shah SK, Caldwell CG, Chen P, Hagmann WK, MacCoss M, Humes JL, Pacholok SG, Kelly TM, Grant SK, Wong KK. Bioorg Med Chem Lett. 2004;14:5907–5911. doi: 10.1016/j.bmcl.2004.09.019. [DOI] [PubMed] [Google Scholar]; (c) Tan KL, Ali A, Du Y, Fu H, Jin HX, Chin TM, Khan M, Go ML. J Med Chem. 2014;57:5904–5918. doi: 10.1021/jm401352a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cersosimo U, Sgorbissa A, Foti C, Drioli S, Angelica R, Tomasella A, Picco R, Semrau MS, Storici P, Benedetti F, Berti F, Brancolini C. J Med Chem. 2015;58:1691–1704. doi: 10.1021/jm501336h. [DOI] [PubMed] [Google Scholar]; (e) Pan J, Xu T, Xu F, Zhang Y, Liu Z, Chen W, Fu W, Dai Y, Zhao Y, Feng J, Liang G. Eur J Med Chem. 2017;125:478–491. doi: 10.1016/j.ejmech.2016.09.033. [DOI] [PubMed] [Google Scholar]; (f) Zhao X, Lin K, Ma Z, Chui WK, Zhou W. Eur J Med Chem. 2017;125:1279–1288. doi: 10.1016/j.ejmech.2016.11.010. [DOI] [PubMed] [Google Scholar]
- 9.(a) Takemura T, Jones JB. J Org Chem. 1983;48:791–796. [Google Scholar]; (b) Lane S, Quick SJ, Taylor RJK. J Chem Soc, Perkin Trans. 1985;1:893–898. [Google Scholar]; (c) Casy G, Lane S, Taylor RJK. J Chem Soc, Perkin Trans. 1986;1:1397–1404. [Google Scholar]; (d) Casy G, Sutherland AG, Taylor RJK, Urben PG. Synthesis. 1989;10:767–769. [Google Scholar]
- 10.Ward DE, Gai Y, Lai Y. Synlett. 1996;3:261–262. [Google Scholar]
- 11.(a) Woodward RB, Logusch E, Nambiar KP, Sakan K, Ward DE, Au-Yeung B-W, Balaram P, Browne LJ, Card PJ, Chen CH, Chênevert RB, Fliri A, Frobel K, Gais H-J, Garratt DG, Hayakawa K, Heggie W, Hesson DP, Hoppe D, Hoppe I, Hyatt JA, Ikeda D, Jacobi PA, Kim KS, Kobuke Y, Kojima K, Krowicki K, Lee VJ, Leutert T, Malchenko S, Martens J, Matthews RS, Ong BS, Press JB, Rajan Babu TV, Rousseau G, Sauter HM, Suzuki M, Tatsuta K, Tolbert LM, Truesdale EA, Uchida I, Ueda Y, Uyehara T, Vasella AT, Vladuchick WC, Wade PA, Williams RM, Wong HN-C. J Am Chem Soc. 1981;103:3210–3213. [Google Scholar]; (b) J Am Chem Soc. 1981;103:3213–3215. [Google Scholar]; (c) J Am Chem Soc. 1981;103:3215–3217. [Google Scholar]
- 12.Ward DE. Chem Commun. 2011;47:11375–11393. doi: 10.1039/c1cc13323c. [DOI] [PubMed] [Google Scholar]
- 13.Sklute G, Marek I. J Am Chem Soc. 2006;128:4642–4649. doi: 10.1021/ja060498q. [DOI] [PubMed] [Google Scholar]
- 14.Minko Y, Pasco M, Lercher L, Botoshansky M, Marek I. Nature. 2012;490:522–526. doi: 10.1038/nature11569. [DOI] [PubMed] [Google Scholar]
- 15.(a) Meyers AI, Lefker BA. Tetrahedron Lett. 1987;28:1745–1748. [Google Scholar]; (b) Meyers AI, Westrum LJ. Tetrahedron Lett. 1993;34:7701–7704. [Google Scholar]; (c) Doyle AG, Jacobsen EN. Angew Chem Int Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901. [DOI] [PubMed] [Google Scholar]
- 16.Substrate synthesis required the use of a Dieckmann cyclization to form the cyclic β-ketoester, with other strategies such as α-acylation of the parent thiopyranone being unsuccessful. Additionally, attempts to prepare thiochromanone derivatives also failed.
- 17.Bulkier alkyl groups were poorly tolerated in the reaction, providing low levels of enantioselectivity.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.