

Abstract
Monoclonal antibodies (mAbs) are essential reagents for deciphering gene or protein function and have been a fruitful source of therapeutic and diagnostic agents. However, developing anticarbohydrate antibodies to target glycans for those purposes has been less successful because the molecular basis for glycan-mAb interactions is poorly understood relative to protein- or peptide-binding mAbs. Here, we report our investigation on glycan-mAb interactions by using the unique architectural scaffold of 2G12, an antibody that targets oligomannoses on the HIV-1 glycoprotein gp120, as the template for engineering highly specifc mAbs to target glycans. We first analyzed 24 different X-ray structures of antiglycan mAbs from the Protein Data Bank to determine side chain amino acid distributions in of glycan-mAb interactions. We identified Tyr, Arg, Asn, Ser, Asp, and His as the six most prevalent residues in the glycan-mAb contacts. We then utilized this information to construct two phage display libraries in which positions on the heavy chain variable domains of 2G12 were allowed to vary in restricted manner among Tyr, Asp, Ser, His, Asn, Thr, Ala and Pro to interrogate the minimal physicochemical requirements for oligomannose recognition. We characterized 39 variants from Lib1 and 14 variants from Lib2 following selection against gp120, the results showed that there is a high degree of malleability within the 2G12 for glycan recognitions. We further characterized five unique phage clones from both libraries that exhibited a gp120-specific binding profile. Expression of two of these variants as soluble mAbs indicated that, while specificity of gp120-binding was retained, the affinity of these mutants was significantly reduced relative to WT 2G12. Nonetheiess, the results indicate these is some malleability in the identity of contact residues and provide a novel insight into the nature of glycan-antibody interactions and how they may differ from protein-protein binding interactions.
Graphical abstract
1. Introduction
Glycans (oligosaccharides) are critical information carriers in biology but their precise roles are poorly defined in many cases1–2. Subtle changes in glycan composition of cellular surfaces can signal significant biological transitions, for example tumor metastasis3. However, detection and therapeutic targeting of such biologically important glycan changes is largely limited by a lack of specific and high-affinity binding reagents. Glycan-binding agents such lectins and monoclonal antibodies (mAb) are available and can be used to decipher glycosylation status of cell surfaces or proteins. However, most lectins and mAbs target terminal saccharides in low affinity, and thus are of limited utility for detecting more complex glycosylation patterns4. Furthermore, individual glycan-protein interactions are typically lower affinity (KD values in the micromolar range) than protein-protein or protein-nucleic acid interactions, because glycans have lower overall hydrophobic functionality available to participate in extensive intermolecular associations. As a result, applications of glycan-binding domains typically require high-density multimerization of the lectin to achieve the required affinity5.
The high-affinity HIV-1 antibody 2G12 represents a unique immunological solution to carbohydrate recognition6. 2G12 is a human antibody from an HIV-1 patient that contains an unusual domain exchange architecture. Instead of the canonical IgG1, where the two antigen-binding fragments (Fabs) independently engage antigens in an overall bivalent fashion, the two heavy chain variable domains of 2G12 (VH and VH’, variable domains from one Fab chain denoted with a prime throughout) participate in a domain exchange across the two Fabs to create an extended overall recognition surface (Fig. 1)6. Somatic hypermutations at the VH-VH’ interface, as well as those at VH-VL’ and VH’-VL interfaces, give rise to four overall potential sites for antigen recognition. Two of these binding sites lie at VH-VL’ or VH’-VL interfaces on both “domain exchanged” Fabs, akin to the canonical combining sites found on regular antibodies. The other two potential binding sites are located diametrically opposed at the VH-VH’ interface. Cryoelectron microscopy studies suggest that the 2G12 IgG1 can engage up to four oligomannose glycans simultaneously on its heavily glycosylated target, gp120, which consists of ~50% oligomannose by weight7. Furthermore, 2G12 has been found to dimerize, forming more complex interactions with multiple gp120 subunits on the viral particle8–9. Because of the extended and multivalent nature of the 2G12 oligosaccharide binding surface, 2G12 binds to gp120 with high-affinity (reported KD of 5.6–16.1 nM for binding to gp120)6, 10–11, despite the fact that it does not require any protein binding components unlike other HIV-1 mAbs that target oligomannose as well as protein epitopes within gp12012. Furthermore, glycan array analysis has demonstrated that 2G12 has a relatively high level of specificity, able to differentiate among similar oligomannose glycans11. This specificity likely arises from the fact that the extended interaction surface that results from the VH-VH’ domain exchange provides contacts with terminal sugars from two of the three arms of the triantennaray Man9GlcNAc2 carbohydrate that is present on gp120. For these reasons, 2G12 is an attractive scaffold to utilize for structure-based engineering of glycan-binding antibodies, and serves as an excellent model for studying the basis of high-affinity antibody-glycan interactions.
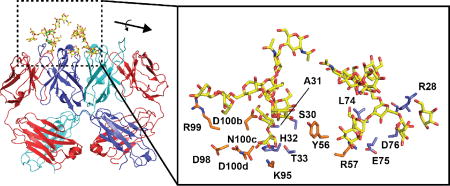
Fig. 1. Domain-exchange architecture of 2G12.

(A) Schematic of domain architecture for canonical IgG molecules (top) and 2G12 (bottom). Canonical IgG architecture results in two antigen binding sites at the interface of light and heavy chain variable domains (VL-VH and VL’-VH’). The VH-VH’ domain exchange in 2G12 creates an extended antigen binding surface that contains up to two additional binding sites at the VH-VH’ interface. (B) Crystal structure of the 2G12 Fab dimer in complex with Man9GlcNAc2 (orange) reported by Calarese et al. (PDB ID 1OP5)6. Domain colors as shown in panel A. (C) Fab monomer of 2G12 showing features that stabilize the domain exchange.
Here, we utilize phage display to explore recognition requirements on 2G12 for binding to gp120. We employ high-throughput mutagenesis techniques to interrogate importance of particular residues to the interaction. We undertook to understand glycan-antibody interactions from a global perspective, and thus performed an analysis of 24 antibody-glycan X-ray structures in the protein data bank. We identified biases within glycan-interacting residues toward certain side chains which, when compared to overall prevalence of side chains in functional antibodies, suggest unique physicochemical properties are required for glycan recognition relative to other biopolymers. We generated and screened limited diversity libraries using the “glycan recognition privileged” 2G12 scaffold to dissect functional requirements for binding to gp120. These results provide novel insights into glycan recognition and the potential for harnessing the 2G12 scaffold for engineering naïve glycan-specific antibody libraries.
2. Results and Discussion
2.1 Optimization of functional expression of domain-exchanged 2G12 (Fab)2 on phage
The basis of the VH-VH’ exchange on 2G12 is a P113 somatic mutation in the region between the variable domain and first constant domain on the heavy chain, that redirects the polypeptide toward the adjacent Fab (Fig. 1)6, 13–15. Additional somatic mutations in regions that would normally correspond to the surface-exposed side of the VH domains result in a novel VH-VH’ interface that stabilizes the overall architecture. Thus, all of the mutations that promote the domain-exchange architecture can be contained within the 2G12 Fab fragment. Previously, we described an approach for functional display of 2G12 (Fab)2 on the surface of M13 bacteriophage using the pAPIII6 phagemid that encoded the 2G12 Fab upstream of the IgG hinge region and a GCN4 leucine zipper to promote on-phage dimerization16. This construct provided functional display of 2G12 (Fab)2 and was amenable to mock selections. However, when complex libraries were generated and subjected to selections, we repeatedly isolated truncation mutants of the 2G12 (Fab)2. These results suggested that there was some inherent instability of the 2G12 (Fab)2 in this format, and thus the selections favored elimination of the display element rather than enrichment of functional clones.
To overcome these limitations, we engineered 2G12 (Fab)2 display sequences into the pHP153 phagemid vector that has been utilized for successful display and library screening of numerous protein and antibody fragments17–18. Furthermore, since all required mutations for the 2G12 domain exchange architecture was contained in the (Fab)2, we examined both a GCN4 leucine zipper-contaning construct (“pHP153-2G12z”) as well as one containing only the IgG hinge region (“pHP153-2G12h”) (Fig. 2a). Although the presence of the GCN4 region presumably provides a driving force for ensuring dimerization of the (Fab)2 fragments on phage, the presence of the stabilizing VH-VH’ interactions in combination with the homodimeric disulfide bonds in the IgG1 hinge region may be sufficient for promoting dimerization of two Fab fragments on phage. However, pHP153-2G12h displayed assembly is predicted to be 64 residues (32 per GCN monomer) shorter than the pHP153-2G12z assembly and thus there may be less inherent bias against display of this element. Both construct contained a FLAG epitope at the C-terminus of the light chain for detection purposes. Secretion of both heavy and light chains into the E. coli periplasm was mediated by an StII signal sequence.
Fig. 2. Phage display of functional 2G12.

(A) Coding regions of pHP153-2G12z and pHP153-2G12h. In both cases, the light chain and heavy chain VH-CH fragment were coexpressed in the periplasm using an stII signal sequence. A FLAG epitope was included at the light chain C-terminus. (B and C) Phage ELISA of particles produced from pHP153-2G12z (B) or pHP153-2G12h (C). The anti-FLAG antibody M2 was used to confirm expression of the construct.
Phage particles derived from both pHP153-2G12z and pHP153-2G12h could be generated readily to titers of 8×1011 and 3×1012 infectious units (iu)/mL, respectively. Phage ELISA of both clones against both gp120 and M2, which recognizes the FLAG epitope that was included at the C-terminus of the light chain, suggested that pHP153-2G12h had higher levels of functional 2G12 (Fab)2 display (Fig. 2b). To test whether pHP153-2G12h was dependent on the domain exchange architecture for gp120 recognition, two variants of pHP153-2G12h were generated containing substitutions that are known to destroy the VH-VH’ interface. Both pHP153-2G12h(I19A/F77A/Y79A), a triple mutant, and pHP153-2G12h(I19R) exhibited no reactivity toward gp120 despite binding strongly to M2 (Supplementary Figure 1), indicating these mutants were expressed on phage but did not assemble functionally into the domain-exchanged architecture that is required for gp120 recognition6, 13–16. Together, these results indicate that functional expression of domain-exchanged 2G12 (Fab)2 can be achieved by inclusion of the IgG1 hinge region at the C-terminus of the first constant domain.
2.2 Surveys of glycan-antibody residue-specific interactions
The goal of this work is to examine the requirements for glycan recognition within the context of the “privileged” 2G12 scaffold. Combined bioinformatics and empirical work has demonstrated that protein recognition, within the context of the 4D5 antibody (Herceptin®) scaffold, can be reduced to a small subset of the 20 naturally occurring amino acids19–22. In the most extreme cases, phage antibody libraries built on the 4D5 scaffold containing binomial diversity (Tyr or Ser) in the CDR regions affords high affinity and specific recognition19–20. More recent antibody libraries based on this scaffold employ greater but still restricted diversity in the CDR-L3 and CDR-H3 regions18, 23–25. The design of such restricted diversity libraries is guided by observations that natural combining sites are enriched in residues such as Tyr, Ser, Ala, and Asp26.
We sought to perform a bioinformatics analysis of glycan-specific antibodies to determine if any side chain-specific biases were present, and whether this information could be utilized to generate glycan-specific restricted diversity antibody libraries. We combed the Protein Data Bank (PDB) for available glycan-antibody complex structures and identified a total of 24 complexes (Supplementary Table 1). Antibodies in these complexes target five distinct classes of carbohydrate antigens. Within these complexes, we examined side chains that were within van der Waals radius (<2.5 Å) of a glycan atom within the antibody combining site. In total, there were 325 side chain-glycan interactions. Interestingly, the distribution of these interactions involved Tyr, Arg, Asn, Ser, Asp, and His as the six most prevalent residues (Fig. 3a). Overall, the distribution of side chains involved in glycan-antibody interactions contains similarities to overall biases in antibody combining sites in that Tyr, Ser, and Asp were prevalent. However, the inclusion of Arg, Asn and His appears to be glycan specific as these residues are not abundantly represented in global surveys of CDR-H3 segments from functional antibodies26.
Fig. 3. Frequency of specific amino acid-glycan interactions.

Observed frequency of direct interactions with glycan antigens for all antigens in the dataset (A) or classified into type of antigen (B–F).
When residue-specific glycan interactions were separated into subsets of four different antigen classes, the distributions were found to vary by antigen composition (Figs. 3b–3f). The number of specific interactions that comprised the antigen-specific datasets ranged from 33 total contacts for sialic acid to 110 for known HIV-1 gp120 antigens. Overall there appeared to be clear preferences toward different subsets of amino acids for each antigen. For example, HIV-1-specific antibodies, which target the oligomannoses on gp120, as well as tumor-specific antibodies, which target GlcNAc, fucose, and galactose-containing saccharides, and the sialic acid interactions all favored Asp more strongly than did interactions toward internal and external lipopolysaccharides (LPSs). The LPS antigens contain ketodeoxyoctulosonic acid and rhamnose. Furthermore, in the two antigen classes that contained a sampling of interactions > n = 100, there were clear distinctions among the top four observed contacting side chains. In all antigen classes, however, Tyr was the dominant residue, observed in more than 15% of the interactions in all four antigen classes. Pro and Met were not observed with high frequency in any of the antigen classes. These results suggest that chemically distinct glycan antigens have different preferences for side chain recognition by antibodies.
2.3 Residue-specific biases for oligomannose recognition explored by limited diversity 2G12-based libraries
The structural surveys above suggest that side chain biases may exist for recognition of glycans by antibodies. In order to further explore these potential biases within the specific context of the 2G12 scaffold, we created limited diversity phage display libraries in which heavy chain contact residues were allowed to vary with the NMC degenerate codon (N = A/T/C/G, M = A/C) which encodes for the eight amino acids Tyr, Asp, Ser, His, Asn, Thr, Ala, and Pro. This codon was chosen because it contains Tyr, Asn, Ser, and Asp which were highly represented in the structural survey, as well as His which provides some positive charge character that may be important as both Arg and His were among the top six most frequently observed side chains. Two phage libraries (Lib1 and Lib2) based upon the pHP153-2G12h clone were produced where residues in CDR-H1 and FR3 (Lib1) and CDR-H2 and CDR-H3 (Lib2) were allowed to vary (Fig. 4a). These regions were targeted for mutagenesis because they made direct contacts with oligomannose in the primary (“canonical”) combining site (CDR-H3) or at the secondary binding sites formed by the VH-VH’ interface (CDR-H1, FR3, and CDR-H2). Residues D98 and R99 do not make direct contacts with the glycan antigen but may serve to orient the proper CDR-H3 topology and were thus included in the analysis. The libraries were subjected to selection against gp120, the native antigen to gain insight into “functional” requirements, or to M2 in order to control for potential display biases. Clones that exhibited a phage ELISA signal of greater than 3-fold for wells coated with gp120 over BSA (“functional selection”) or M2 over BSA (“display selection”) were sequenced. A total of 39 and 94 clones were analyzed from Lib1 against gp120 and M2, respectively; and 13 and 40 for Lib2 against gp120 and M2, respectively. Notably, fewer positive clones were obtained from the Lib2 selection against gp120, suggesting the presence of fewer functional clones in this library.
Fig. 4. Limited diversity 2G12 library design and results from selection.

(A) Positions that were targeted for mutagenesis involve direct contact residues for glycans at both the primary and secondary sites; blue side chains in the close-up were for Lib1 and orange side chains were for Lib 2. All of the residues were allowed to vary among eight residues (Y/D/S/H/N/T/A/P) using the NMC degenerate codon. For clarity, only one antigen for each is shown. (B and C) Percent occurrence of the eight possible residues following selection against gp120 at each position for Lib1 (B) and Lib2 (C). For reference and to control for expression biases, a parallel selection was performed against M2, an anti-FLAG antibody, and the percent occurrence listed in brackets.
Figs. 4b and 4c show the distribution of amino acids at each of the randomized positions within the ELISA-positive pools. In theory, any specific positional preferences for particular side chains should become apparent from this analysis as highly favored in the gp120 (functional) selection but not in the M2 (display) selection. A purely random distribution would predict the population contains 12.5% of each of the eight possibilities at each position. Although not distributed equally among the gp120 clones, it is apparent that none of the positions harbors a strong preference for any one of the eight residues. Notably, Ala was disfavored at positions H32 and T33 in CDR-H1 (observed only once out of 43 clones, or 2%, in each instance), and Pro was not observed at all at H32. Although some preferences against these residues were observed in the M2 selection, they were not as strong suggesting that such substitutions are deleterious for glycan recognition. Both H32 and T33 make direct contacts with the terminal mannose on the D1 arm, which is buried deep within the primary combining sites. Thus, mutation of H32 and T33 to Ala, or of H32 to Pro is likely to be deleterious to binding. It is interesting, however, that Tyr is tolerated at both positions, indicating some malleability in packing arrangements. There were fewer selected functional clones from Lib2 (only 13 sequences could be recovered), thus the analysis not as complete, but nonetheless among this set, strong preferences for the WT Asp residue at position D98 in CDR-H3 and Tyr at position Y56 in CDR-H2 were apparent (6/13 and 5/13 contained this identity, respectively). These preferences were also observed in the M2 selection suggesting that these residues participate overall importantly to structure, folding and expression. Notably, several substitutions were not observed at all in the functional selection despite being present in the display selection: R57T, K95P, D98S, R99Y, N100cH, D100dN, D100dA, D100dY. Furthermore, the native Asn was not observed at position 100c in the functional selection despite the fact that it was present 6/40 times in the display selection. Residue K95 makes direct contacts with the D1 arm in the primary site, and thus mutation to Pro likely disrupts both direct interactions as well as conformation of the surrounding side chain. Residues D98, and R99, do make direct contacts with the oligomannose and thus these preferences may contribute to promoting proper CDR conformations for productive interactions. R57 lies at the VH-VH’ interface, and is proximal to the glycans located in the secondary sites, and thus the preference against Thr may be due to conformational effects, VH-VH’ packing effects, direct contacts with the glycan or some combination of these three. N100c forms peripheral interactions with the terminal D2 mannose, and D100d lines the primary combining site pocket that interacts with the D1 arm) and thus the preferences observed at these positions may reflect the requirements for recognition of the oligomannose.
Taken together, these results suggest that there is a high degree of malleability within the 2G12 combining site for gp120 recognition. While there are some clear preferences within CDR H3 at positions N100c and D100d, for WT residue, and some potential disfavored substitutions at H32 and T33 in CDR-H2, overall a number of residues from the limited codon set could be well tolerated in both the functional and display selections.
2.4 Characterization of individual clones from focused libraries
Given that results from the restricted diversity libraries suggested that substitutions were among the restricted diversity set permitted at varied positions, we sought to identify and characterize representative single clones for their reactivity toward gp120. From Lib1, we identified five unique clones (L1–14, L1–42, L1–43, L1–50, and L1–74) that contained 5–8 substitutions relative to WT 2G12 (Fig. 5a). Monoclonal phage ELISA with these clones indicated that these clones exhibited specific reactivity toward gp120, albeit at higher phage titers than WT, with minimal or no cross-reactivity toward BSA (Fig. 5b). Similarly, five clones from Lib2 (L2–13, L2–20, L2–23, L2–67, and L2–83) containing 5 to 8 substitutions were found to have similar specific reactivity profiles against gp120, again requiring higher phage titers than WT 2G12 (Figs. 5a and 5b). These results further suggest that binding specificity can be maintained with high levels of substitutions. The requirement for higher phage titers for reactivity against gp120 for the selected clones relative to WT 2G12 suggests that, while specificity toward the gp120 epitope is maintained, the affinity may be decreased by these substitutions. We chose L1–42 and L2–83 for further analysis as purified antibodies to explore affinity. The IgG1 molecules for these mAbs, along with WT 2G12 were purified from HEK293 cells. The reactivity of these mAbs for binding to gp120 was tested using ELISA (Fig. 5c). As reported previously, 2G12 bound to gp120 in high affinity with a half-maximal binding titer (EC50) of 2.3 × 10−6 µM6, 10–11. By contrast, L1–42 and L2–83 had EC50 values ~6-orders of magnitude higher (1.0 and 1.6 µM), suggesting much lower binding affinity. Nonetheless, the binding remained specific to gp120 as neither L1–42 nor L2–83 had strong reactivity toward BSA. Furthermore, L1–42 and L2–83 did not exhibit any binding reactivity toward commercial preparations of human erythropoietin (hEPO) and human chorionic gonadotropin (hCG), which both contain N- and O-linked carbohydrates that differ from Man9GlcNAc2, further validating the glycan specificity of 2G12 and variants (Supplementary Fig. 2). These results suggest that recognition of oligomannoses on gp120 is possible in the domain-exchanged scaffold with 7–8 substitutions in critical residues, but that this can result in drastic decreases in binding affinity
Fig. 5. Characterization of individual clones from limited diversity libraries.

(A) Sequences of mutants from Lib1 and Lib2 that were further studied. The WT residue identity at each position is indicated, those positions that were identical to WT are shaded. (B) ELISA of phage displayed 2G12 and mutants against gp120, M2 (anti-FLAG), and BSA. The WT 2G12 phage were tested at a phage titer of ~1010 iu/mL whereas the variants were tested at ~1011–1012 iu/mL. (C) ELISA of purified 2G12 mAb, and variants L1–42 and L1–83 against gp120 and BSA. The EC50s for gp120 were 2.3 pM for 2G12, and 1.0 and 1.6 µM for L1–42 and L2–83, respectively.
3. Conclusions
Here we have used phage display and structure-guided design to explore functional requirements for oligomannose recognition within the domain-exchanged architecture of the 2G12 scaffold. While a number of previous structure-activity relationship studies have been performed on 2G12, these have generally focused on understanding minimal requirements for induction of 2G12-like antibodies with carbohydrate immunogens or on deciphering the salient features required for potent virus neutralization. Currently, we have sought to explore physicochemical requirements for recognition within the “privileged” domain-exchanged scaffold. Ultimately, such studies can provide insight for development of naïve antibody libraries that can be used to target a broad array of glycan targets. A robust and generalizable strategy for generation of specific carbohydrate antibodies would be a significant advance. Glycans play important roles in biology, and cell surface glycan changes can signal major transitions (e.g. tumor metathesis)1. However, glycans themselves tend to be poorly immunogenic, and thus routine methods for generation of glycan-specific antibodies have been met with limited success. This represents a major roadblock to the development of glycan-specific antibodies for diagnostic or therapeutic uses. Alternative approaches, such as the use of glycan-binding domains (e.g., lectins) have been employed, but typically require multimerization of the carbohydrate-bonding moiety to achieve the required affinity4–5. Furthermore, mAbs have a number of advantages relative to other protein-based agents as potential therapeutics such as long in vivo half-life and high general safety profile.
In comparison to protein-binding antibodies, there are far fewer available X-ray structures of glycan-binding antibodies. Nonetheless, our structural survey of 24 carbohydrate-antibody structures indicated that intrinsic side chain biases may exist for glycan recognition. While some of the side chain preferences echo those that have previously been determined from structural and empirical studies for protein-binding antibodies21, 24–26, such as the prevalence of Tyr, Asp, and Ser, there were other features that were unique to the glycan-specific set. These include positively charged residues such as His and Arg, as well as Asn. From a physicochemical perspective, the prevalence of Tyr, Asp, and Ser can be straightforwardly rationalized. Tyr contains both hydrophobic character via the aromatic ring, as well as the capacity to participate in hydrogen bonding at the phenol. Furthermore, host-guest studies and broader structural surveys of carbohydrate-aromatic amino acid side chain interactions have illustrated the importance of the “CH-π” interaction27–28. In this model, the axial hydroxyl groups of monosaccharides serve to induce an overall dipole that runs perpendicular to the plane of the carbohydrate ring, providing an electropositive character to the glycan C-H bond that can participate in interactions with the π cloud of aromatic residues. In one study, specific CH-π interactions on opposing β-strands of a β-hairpin peptide were found to account for as much 0.8 kcal/mol in folding energy for the peptide28. A more recent structural survey has revealed biases in the specific orientation of the C-H bond relative to the plane of the aromatic group across structures, providing support that this specific type of interaction is important in glycan recognition27. In our antibody study, the preferential use of Tyr as a primary interacting residues may well provide the energetic driving force required for recognition. Notably, however, both host-guest as well as structural surveys have found that Trp is the most favored for individual carbohydrate-aromatic side chain interactions27–28. While Trp was generally represented in our survey, it was not among the most frequently observed side chains involved in specific interactions for any of the antigen classes. The prevalence of Ser and Asp may be due to the fact that these side chains are capable of participating in hydrogen bonding interactions, which may be important for specificity. While the chemical composition of different sugar groups is relatively invariant (a central sugar ring with hydroxyl or amine groups extending from the carbons), there is diversity in the relative regio- and stereochemical positioning of the heteroatoms. Therefore, positional requirements for hydrogen bond donor and/or acceptor groups on the antibody may be necessary for differentiating among carbohydrates. While the specific context of the interactions varies from protein-antibody interactions, the general physicochemical features of the Tyr, Ser, and Asp residues that favors their abundance at intermolecular interfaces is likely similar for both groups of antigens.
The preference for positively charged residues may be specific to glycan recognition. A possibility is that such residues have the capacity to participate in dipole interactions induced by axial hydroxyl groups on the sugar, somewhat akin to cation-π interactions that are known to be important in protein core structures29. Furthermore, it seems apparent that the relative distribution of side chain preferences is somewhat dependent on the specific glycan class, as the different antigens had unique distributions of preferred side chains.
Investigation of the predicted side chain preferences within the context of the 2G12 scaffold for oligomannose recognition revealed that substitution of many of the glycan-interacting residues with a limited diversity subset of side chains was generally tolerated while maintaining the oligomannose binding potential, but that such substitutions are deleterious to affinity. The purified L1–42 and L2–83 mAbs had drastically lower reactivity toward gp120 by ELISA; while not a precise KD measurement, such reactivity studies can often be used to gauge relative affinity, particularly in cases where there are orders of magnitude in difference. Nonetheless, it is notable that many of the characterized Lib1 and Lib2 clones exhibited specific binding to gp120 despite the fact that anywhere from 5 to 8 contact residues were replaced. This number of substitutions, if deleterious to binding, would be predicted to result in the complete abolishment of activity. Thus, our results suggest that there is a high degree of tolerance within the 2G12 combining site for substitutions within this subset, albeit with a precipitous decline in binding affinity. While specificity toward the gp120 antigen relative to BSA was examined in these cases, which provides information about general “stickiness”, we did not explore effects of these substitutions on specificity toward oligomannose over other large oligosaccharides. It is possible that such substitutions decrease the overall specificity toward the Man9GlcNAc2 glycan.
Overall our results suggest that the 2G12 scaffold has the potential for development of generalizable glycan-binding antibody libraries. A number of substitutions were tolerated within the combining site, but with significant effects on binding activity. A challenge moving forward will be to identify mutagenesis strategies that allow alteration of the specificity toward oligomannose while maintaining high affinity. The results presented here provide novel insight into the nature of glycan-antibody interactions and how they may differ from protein-binding interactions.
4. Experimental
4.1 Phage Display
The pHP153 vector was used to display 2G12 (Fab)2 on the surface of filamentous bacteriophage17. The Escherichia coli codon-optimized synthetic genes for 2G12 (light chain with a C-terminal FLAG tag, heavy chain with and without a GCN4 leucine zipper) were obtained from Genewiz (South Plainfield, NJ). The 2G12z and 2G12h constructs were cloned into phagemid pHP153 using NsiI and FseI restriction sites to form pHP153-2G12z and pHP153-2G12h. This cloning strategy results the functional display of 2G12 (Fab)2 on the surface of phage as a heavy chain fusion to the minor phage coat protein pIII and as a C-terminal FLAG epitope on the light chain for detection by the monoclonal antibody M2. In addition, the constructs are translated with stII signal peptide and under the control of the pho A promoter. Phage particles expressing 2G12 (Fab)2 were produced by separately electroporating each of the phagemids, pHP153-2G12z and pHP153-2G12h, into E. coli XL1-Blue cells. Transformed cells were grown for 5 h at 37 °C in 2×YT media containing 10 µg/mL tetracycline and 50 µg/mL carbenicillin. This culture media was then coinfected with 1010 plaque forming units (PFU) of M13K07 helper phage for 1 h at 37 °C followed with adding 50 µg/mL of kanamycin. The culture was further incubated overnight at 37°C. Subsequently, the phage particles were precipitated by addition of 4% (w/v) polyethylene glycol (PEG) 8000 and 3% (w/v) NaCl after removal of cells by centrifugation. Precipitated phage were centrifuged and then resuspended in PBS containing 1% (w/v) BSA.
Functional display of 2G12 (Fab)2 on the surface of phage were confirmed using a phage ELISA. gp120 and M2 were coated on Costar EIA/RIA high-binding plates (Fisher Scientific, Nepean, ON, Canada) at 0.9 µg per well in PBS pH=8.0 overnight at 4 °C. All further incubations were completed at 37°C. Plates were blocked with 1% (w/v) BSA in PBS (pH=7.4) for 2 h. Wells were washed with PBS containing 0.05% (v/v) Tween-20 (PBS-T) and 2G12 (Fab)2 -expressing phage were then added to the wells for 1 h. Wells were washed 5 times with PBS-T. An anti-M13 HRP-conjugated antibody was then allowed to bind for 1 h. The wells were washed with PBS-T and the anti-M13-HRP conjugate was detected using 3,3′,5,5′-tetramethylbenzidine (TMB) (Sigma-Aldrich, St. Louis, MO). The data was graphed using GraphPad Prism (GraphPad Software, La Jolla, CA).
4.2 Structural survey of glycan-antibody interactions
The structure information of the selected antibodies were obtained from Protein Data Bank (http://www.rcsb.org/pdb) and then analyzed by using PyMol (http://www.pymol.org/). The glygan-contact residues were determined as those within van der Waals radius (1.4~2.5 Å, radius=distance/2) of a glycan atom, and the list of residues manually collated.
4.3 Generation and screening of 2G12 phage libraries
Limited diversity phage display libraries were constructed in which contact residues were allowed to vary with the NMC degenerate codon (N = A/T/C/G, M = A or C) which encodes for the eight amino acids Tyr, Asp, Ser, His, Asn, Thr, Ala, and Pro. Oligonucleotide-directed Kunkel mutagenesis was used to incorporate diversity elements using the following primers: Lib1, TGCGGTGTTTCTAACTTCNMCATCNMCNMCNMCNMCATGAACTGGGTTCGTCG and ACCGTTTCTCGTGACGACNMCNMCNMCTTCGTTTACCTGCAGATG; Lib2 TGGGTTGCTTCTATCTCTACCTCTTCCACCNMCNMCGACTATGCTGACGCTGTTAAAG and GCTATCTACTACTGCGCTCGTNMCGGTTCTNMCNMCCTGTCTNMCNMCNMCCCGTTCGACGCTTG17. The library DNA was electroporated into SS320 E. coli and library phage amplified according to standard protocols17. Briefly, each library was subjected to one round of selection against the anti-FLAG antibody M2 (display selection), or one round against M2 then three rounds against gp120 (functional selection). For the functional selections, each round consisted of coating seven wells with 0.8 µg of gp120 overnight at 4°C. Wells were blocked with 1% (w/v) BSA in PBS for 2 h and library phage were then allowed to bind for 1 h before washing 5 times with PBS-T. Bound phage were eluted by incubating with 100 mM glycine pH=2.0 for 5 min at room temperature (RT), and neutralizing with 2 M Tris pH=7.5. M2 selections were performed similarly, except seven wells were coated with a 1:500 dilution of M2 in PBS pH=8.0 overnight. After each round of selection, eluted phage were amplified by infecting E. coli XL1-Blue cells, coinfecting with M13K07 helper phage and precipitated as described above for further rounds of selection.
To analyze individual clones for binding to gp120, individual clones were grown in 1 mL 2×YT media supplemented with 1010 PFU M13K07 helper phage, and 100 µg/mL carbenicillin in 96 deep-well plates overnight at 37 °C. Cells were removed by centrifugation and the phage-containing supernatants were applied to plates containing gp120 or BSA. Wells were washed 5 times with PBS-T and were incubated with an anti-M13-HRP conjugate for 1 h. Wells were washed with PBS-T and bound clones were detected using TMB substrate. Clones that demonstrated an ELISA-positive signal 3-fold higher for the gp120 plates compared with the BSA control plates were sequenced. The M2 display sorts were performed similarly, except the wells were coated with a 1:500 dilution of M2.
4.4 Expression of recombinant 2G12 mAb and analogues
The recombinant 2G12 mAb and analogues were amplified from phage particles by PCR and subcloned into pMAZ-IgL and pMAZ-IgH vectors30. Vectors for the heavy and light chain were transfected into HEK293F cells (Invitrogen, Grand Island, NY) using 2 µg/mL linear polyethylenimine (PEI) molecular weight 25,000 Daltons according to the manufacturer’s instructions (Polysciences, Warrington, PA). Cell cultures were incubated at 37 °C and 8% CO2 for 5–6 days post-transfection. The cell cultures were centrifuged and the supernatants were applied to a protein-A affinity column (~1 mL packed beads per 600 mL culture) (Pierce, ThermoScientific, Rockford, IL). Antibodies were purified using the Gentle Antibody Elution System (Pierce, ThermoScientific, Rockford, IL) as per manufacturer’s protocol. Antibodies were desalted by PD-10 desalting column into 150 mM HEPES and 200 mM NaCl pH 7.4 buffer, and the final protein level was assayed using A280 with an extinction coefficient of 1.4. The product was stored at − 20 or 2–8 °C.
4.5 ELISA for 2G12 mAb and analogues
To analyze the binding of 2G12 and its variants for different glycans/glycoproteins, ELISA plates were coated with 0.5ug gp120 (NIH reagent program), hEPO (Millipore EMD 329871), hCG (Sigma C1063), or BSA overnight in 4 °C. Wells were washed 5 times with PBS-T and incubated with 2G12 and its variants for 1 hr. Subsequently, wells were washed with PBS-T again and incubated with protein A-HRP. The signals were detected using TMB substrate as before.
Supplementary Material
Supplementary Figure 1: Phage ELISA of pHP153-2G12h(I19A/F77A/Y79A) and pHP153-2G12h(I19R).
Supplement Table 1: List of antibodies that were included in the structural survey.
Supplementary Figure 2: ELISA of 2G12, L1–42, and L2–83 against hEPO and hCG.
Acknowledgments
This work was funded by the National Institutes of Health (R01-AI125462-01A1). J.R.L. acknowledges an Irma T. Hirschl/Monique Weill-Caulier Career Scientist Award from the Irma T. Hirschl Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Bertozzi CR, Kiessling LL. Chemical glycobiology. Science (New York, N.Y.) 2001;291(5512):2357–64. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 2.Prescher JA, Bertozzi CR. Chemical technologies for probing glycans. Cell. 2006;126(5):851–4. doi: 10.1016/j.cell.2006.08.017. [DOI] [PubMed] [Google Scholar]
- 3.Lee HY, Chen CY, Tsai TI, Li ST, Lin KH, Cheng YY, Ren CT, Cheng TJ, Wu CY, Wong CH. Immunogenicity study of Globo H analogues with modification at the reducing or nonreducing end of the tumor antigen. Journal of the American Chemical Society. 2014;136(48):16844–53. doi: 10.1021/ja508040d. [DOI] [PubMed] [Google Scholar]
- 4.Collins BE, Paulson JC. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Current opinion in chemical biology. 2004;8(6):617–25. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Blixt O, Collins BE, van den Nieuwenhof IM, Crocker PR, Paulson JC. Sialoside specificity of the siglec family assessed using novel multivalent probes: identification of potent inhibitors of myelin-associated glycoprotein. The Journal of biological chemistry. 2003;278(33):31007–19. doi: 10.1074/jbc.M304331200. [DOI] [PubMed] [Google Scholar]
- 6.Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. Antibody domain exchange is an immunological solution to carbohydrate cluster recognition. Science (New York, N.Y.) 2003;300(5628):2065–71. doi: 10.1126/science.1083182. [DOI] [PubMed] [Google Scholar]
- 7.Murin CD, Julien JP, Sok D, Stanfield RL, Khayat R, Cupo A, Moore JP, Burton DR, Wilson IA, Ward AB. Structure of 2G12 Fab2 in complex with soluble and fully glycosylated HIV-1 Env by negative-stain single-particle electron microscopy. Journal of virology. 2014;88(17):10177–88. doi: 10.1128/JVI.01229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein JS, Webster A, Gnanapragasam PN, Galimidi RP, Bjorkman PJ. A dimeric form of the HIV-1 antibody 2G12 elicits potent antibody-dependent cellular cytotoxicity. AIDS (London, England) 2010;24(11):1633–40. doi: 10.1097/qad.0b013e32833ad8c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.West AP, Jr, Galimidi RP, Foglesong CP, Gnanapragasam PN, Huey-Tubman KE, Klein JS, Suzuki MD, Tiangco NE, Vielmetter J, Bjorkman PJ. Design and expression of a dimeric form of human immunodeficiency virus type 1 antibody 2G12 with increased neutralization potency. Journal of virology. 2009;83(1):98–104. doi: 10.1128/JVI.01564-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoorelbeke B, van Montfort T, Xue J, LiWang PJ, Tanaka H, Igarashi Y, Van Damme EJ, Sanders RW, Balzarini J. HIV-1 envelope trimer has similar binding characteristics for carbohydrate-binding agents as monomeric gp120. FEBS letters. 2013;587(7):860–6. doi: 10.1016/j.febslet.2013.02.037. [DOI] [PubMed] [Google Scholar]
- 11.Calarese DA, Lee HK, Huang CY, Best MD, Astronomo RD, Stanfield RL, Katinger H, Burton DR, Wong CH, Wilson IA. Dissection of the carbohydrate specificity of the broadly neutralizing anti-HIV-1 antibody 2G12. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(38):13372–7. doi: 10.1073/pnas.0505763102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pejchal R, Doores KJ, Walker LM, Khayat R, Huang PS, Wang SK, Stanfield RL, Julien JP, Ramos A, Crispin M, Depetris R, Katpally U, Marozsan A, Cupo A, Maloveste S, Liu Y, McBride R, Ito Y, Sanders RW, Ogohara C, Paulson JC, Feizi T, Scanlan CN, Wong CH, Moore JP, Olson WC, Ward AB, Poignard P, Schief WR, Burton DR, Wilson IA. A potent and broad neutralizing antibody recognizes and penetrates the HIV glycan shield. Science (New York, N.Y.) 2011;334(6059):1097–103. doi: 10.1126/science.1213256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doores KJ, Fulton Z, Huber M, Wilson IA, Burton DR. Antibody 2G12 recognizes di-mannose equivalently in domain- and nondomain-exchanged forms but only binds the HIV-1 glycan shield if domain exchanged. Journal of virology. 2010;84(20):10690–9. doi: 10.1128/JVI.01110-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gach JS, Furtmuller PG, Quendler H, Messner P, Wagner R, Katinger H, Kunert R. Proline is not uniquely capable of providing the pivot point for domain swapping in 2G12, a broadly neutralizing antibody against HIV-1. The Journal of biological chemistry. 2010;285(2):1122–7. doi: 10.1074/jbc.M109.058792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huber M, Le KM, Doores KJ, Fulton Z, Stanfield RL, Wilson IA, Burton DR. Very few substitutions in a germ line antibody are required to initiate significant domain exchange. Journal of virology. 2010;84(20):10700–7. doi: 10.1128/JVI.01111-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stewart A, Liu Y, Lai JR. A strategy for phage display selection of functional domain-exchanged immunoglobulin scaffolds with high affinity for glycan targets. Journal of immunological methods. 2012;376(1–2):150–155. doi: 10.1016/j.jim.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frei JC, Lai JR. Protein and Antibody Engineering by Phage Display. Methods in enzymology. 2016;580:45–87. doi: 10.1016/bs.mie.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koellhoffer JF, Chen G, Sandesara RG, Bale S, Ollmann Saphire E, Chandran K, Sidhu SS, Lai JR. Two synthetic antibodies that recognize and neutralize distinct proteolytic forms of the Ebola virus envelope glycoprotein. Chembiochem. 2012;13(17):2549–2557. doi: 10.1002/cbic.201200493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Regula LK, Stewart A, Lai JR. Synthetic Fab fragments that bind the HIV-1 gp41 heptad repeat regions. Biochemical and biophysical research communications. 2011;413(4):611–615. doi: 10.1016/j.bbrc.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fellouse FA, Li B, Compaan DM, Peden AA, Hymowitz SG, Sidhu SS. Molecular recognition by a binary code. Journal of molecular biology. 2005;348(5):1153–62. doi: 10.1016/j.jmb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- 21.Fellouse FA, Wiesmann C, Sidhu SS. Synthetic antibodies from a four-amino-acid code: a dominant role for tyrosine in antigen recognition. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(34):12467–72. doi: 10.1073/pnas.0401786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sidhu SS, Fellouse FA. Synthetic therapeutic antibodies. Nature chemical biology. 2006;2(12):682–688. doi: 10.1038/nchembio843. [DOI] [PubMed] [Google Scholar]
- 23.Uchime O, Dai Z, Biris N, Lee D, Sidhu SS, Li S, Lai JR, Gavathiotis E. Synthetic Antibodies Inhibit Bcl-2-associated X Protein (BAX) through Blockade of the N-terminal Activation Site. The Journal of biological chemistry. 2016;291(1):89–102. doi: 10.1074/jbc.M115.680918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, Koide S, Sidhu SS. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. Journal of molecular biology. 2007;373(4):924–40. doi: 10.1016/j.jmb.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 25.Persson H, Ye W, Wernimont A, Adams JJ, Koide A, Koide S, Lam R, Sidhu SS. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. Journal of molecular biology. 2013;425(4):803–11. doi: 10.1016/j.jmb.2012.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birtalan S, Zhang Y, Fellouse FA, Shao L, Schaefer G, Sidhu SS. The intrinsic contributions of tyrosine, serine, glycine and arginine to the affinity and specificity of antibodies. Journal of molecular biology. 2008;377(5):1518–28. doi: 10.1016/j.jmb.2008.01.093. [DOI] [PubMed] [Google Scholar]
- 27.Hudson KL, Bartlett GJ, Diehl RC, Agirre J, Gallagher T, Kiessling LL, Woolfson DN. Carbohydrate-Aromatic Interactions in Proteins. Journal of the American Chemical Society. 2015;137(48):15152–60. doi: 10.1021/jacs.5b08424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laughrey ZR, Kiehna SE, Riemen AJ, Waters ML. Carbohydrate-pi interactions: what are they worth? Journal of the American Chemical Society. 2008;130(44):14625–33. doi: 10.1021/ja803960x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gallivan JP, Dougherty DA. Cation-pi interactions in structural biology. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(17):9459–64. doi: 10.1073/pnas.96.17.9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mazor Y, Barnea I, Keydar I, Benhar I. Antibody internalization studied using a novel IgG binding toxin fusion. Journal of immunological methods. 2007;321(1–2):41–59. doi: 10.1016/j.jim.2007.01.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Phage ELISA of pHP153-2G12h(I19A/F77A/Y79A) and pHP153-2G12h(I19R).
Supplement Table 1: List of antibodies that were included in the structural survey.
Supplementary Figure 2: ELISA of 2G12, L1–42, and L2–83 against hEPO and hCG.