

Abstract
DNA damaging chemotherapy and radiation are widely used standard-of-care modalities for the treatment of cancer. Nevertheless, the outcome for many patients remains poor and this may be attributed, at least in part, to highly effective DNA repair mechanisms. Ataxia-telangiectasia mutated and Rad3-related (ATR) is a key regulator of the DNA-damage response (DDR) that orchestrates the repair of damaged replication forks. ATR is a serine/threonine protein kinase and ATR kinase inhibitors potentiate chemotherapy and radiation. The ATR kinase inhibitor VX-970 (NSC 780162) is in clinical development in combination with primary cytotoxic agents and as a monotherapy for tumors harboring specific mutations. We have developed and validated an LC–MS/MS assay for the sensitive, accurate and precise quantitation of VX-970 in human plasma. A dilute-and-shoot method was used to precipitate proteins followed by chromatographic separation with a Phenomenex Polar-RP 80 Å (4 μm, 50 × 2 mm) column and a gradient acetonitrile-water mobile phase containing 0.1% formic acid from a 50 μL sample volume. Detection was achieved using an API 4000 mass spectrometer using electrospray positive ionization mode. The assay was linear from 3–5,000 ng/mL, proved to be accurate (94.6–104.2%) and precise (<8.4% CV), and fulfilled criteria from the FDA guidance for bioanalytical method validation. This LC-MS/MS assay will be a crucial tool in defining the clinical pharmacokinetics and pharmacology of VX-970 as it progresses through clinical development.
Keywords: VX-970, tandem mass spectrometry, clinical
1 Introduction
Cytotoxic chemotherapies and ionizing radiation that induce DNA damage are widely used standard-of-care modalities for solid malignancies. For many patients the benefit of these therapies is limited, at least in part, by the DNA damage response (DDR), the signaling that orchestrates a wide array of mechanisms that detect and repair DNA damage in cancer cells as well as normal cells [1]. Cancer cells frequently acquire mutations that inactivate specific DNA repair mechanisms and these changes that are not present in normal cells, may be exploited to achieve significant therapeutic index with small molecules such as PARP inhibitors. Furthermore, the DDR may be a barrier to oncogenic transformation that may be elevated in cancer cells and therefore targeted to achieve significant therapeutic index [2].
Ataxia telangiectasia mutated (ATM) and ataxia telangiectasia mutated and Rad3-related (ATR) are two serine/threonine kinases that have been identified as primary activators and coordinators of DDR [3, 4]. ATM recognizes DNA double-strand breaks (DSBs) and ATR recognizes single-strand DNA at damaged replication forks and resected (DSBs) [1]. ATM and ATR initiate complex and overlapping signaling cascades [5]. Up to 70% of tumors display inactivating mutations in either ATM or tumor suppressor protein p53 and it is hypothesized that cells harboring defects in this signaling axis acquire a selective advantage during carcinogenesis that is associated with loss of the G1 cell cycle checkpoint and reduced DNA repair [6]. Cancer cells that have lost the ATM/p53 axis have an increased dependence on the ATR signaling pathway and correspondingly both ATM- and p53-deficient cancer cells are selectively killed by ATR kinase inhibitors in vitro an in vivo [7–9].
VX-970 has recently entered clinical development as a potent and selective inhibitor of ATR [10]. VX-970 has anti-proliferative qualities as a single agent and it potentiates the effects of DNA damaging therapies in ATM-deficient xenograft models [11–13]. Radiation and chemotherapy are typically administered at or near their maximum tolerable dose and potentiating these standard-of-care therapies with VX-970 is a rational approach to increase the efficacy of the primary DNA damaging modality without creating additional toxicity. As VX-970 progresses through clinical development, an accurate description of its pharmacokinetics and potential alterations by primary chemotherapies is crucial. To support these endeavors we developed a sensitive and accurate LC-MS/MS assay to quantify VX-970 in human plasma. The calibration curve ranges from 3–5000 ng/mL to accommodate expected clinical concentrations.
2 Experimental
2.1 Chemicals and reagents
VX-970 (NSC 780162, VRT-0768079, VE-822) (M/(M + M+7) > 99.99%) was obtained from Vertex Pharmaceuticals (Boston, MA) as the free base. Because the drug substance used clinically, and therefore the reported doses, reflect the hydrochloride salt, all concentrations described in this report are expressed as hydrochloride salt. The internal standard, [d7]-VX-970 (VRT-1135046) (M+7/(M + M+7) > 99.66%), was obtained from Vertex Pharmaceuticals (Boston, MA). Bile from VX-970 treated rat was provided by Vertex Pharmaceuticals, London, UK. Acetonitrile, water (both HPLC grade) and dimethyl sulfoxide were purchased from Fisher Scientific (Fairlawn, NJ). Formic acid was purchased from Sigma-Aldrich (St. Louis, MO). Control human plasma was purchased from Valley Biomedical (Winchester, VA) and Lampire (Everett, PA). Bovine serum albumin (BSA), pooled human liver microsomes, pooled female rat microsomes, UDPGA, MgCl2, alamethicin and formic acid were purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Chromatography
The LC system consisted of an Agilent (Palo Alto, CA, USA) 1200 SL autosampler and binary pump and a Phenomenex (Torrence, CA USA) Synergi Polar-RP 80Å (4 μm, 50 × 2.0 mm) column using a gradient style elution program. Mobile phase solvent A consisted of 0.1% formic acid in acetonitrile and mobile phase solvent B consisted of 0.1% formic acid in water. The initial mobile phase composition was 30% solvent A and 70% solvent B. From 0 to 2.2 min, at a 0.4 mL/min flowrate, solvent A was increased to 80% and conditions held until 2.3 min. Between 2.3 and 3.0 min, solvent A was maintained at 80% and the flowrate increased to 0.8 mL/min and conditions held until 3.1 min. At 3.1 min he mobile phase composition was then returned to 30% solvent A until 4 min at a flowrate of 1.0 mL/min. The total run time was 4 min with an injection volume of 5 μL.Mass spectrometry
Mass spectrometric detection was carried out using a SCIEX (Framingham, MA) 4000 hybrid linear ion trap tandem mass spectrometer utilizing electrospray ionization in positive-ion multiple reaction monitoring (MRM) mode. The settings of the mass spectrometer in positive mode scanning parameters were as follows: curtain gas 40, IS voltage 5000 V, probe temperature 500°C, GS1 65, GS2 65, DP 50 V, CE of 25 eV, and an exit potential of 10 V. The temperature of the autosampler was 4 °C. The MRM m/z transitions monitored were: 464.3>433.5 for VX-970 and 471.3>440.3 for [d7]-VX-970. Control of the LC system and mass spectrometer as well as data collection was accomplished with Analyst software (version 1.4.2).
2.3 Preparation of calibration standards and quality control samples
Stock solutions of VX-970 and internal standard [d7]-VX-970 (see Fig. 1 for structures) were prepared independently at 1.0 mg/mL in dimethyl sulfoxide (DMSO) and stored at −80 °C. Working stock solutions of 0.1 mg/mL VX-970 and 0.1 mg/mL [d7]-VX-970 were prepared in DMSO. On the days of analysis, the working stock solution of VX-970 was serially diluted in 10 fold steps using DMSO, and the internal standard was diluted 200 fold to a final concentration of 0.5 μg/mL. VX-970 calibration working stock solutions were then diluted in human plasma to produce the following analyte concentrations: 3, 10, 30, 100, 300, 1,000, 3,000 and 5000 ng/mL. For each calibration series zero and blank samples were also prepared from 50 μL of control plasma.
Fig. 1.
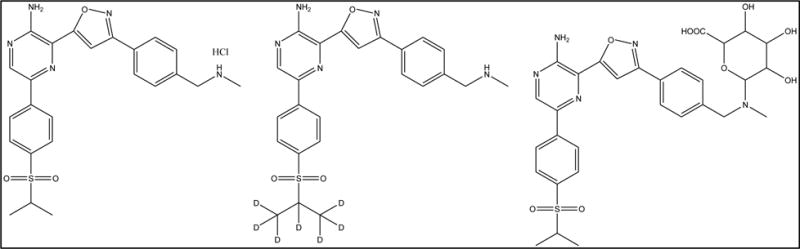
Structures of VX-970 (HCl salt), [d7]-VX-970 internal standard, and VX-970 glucuronide metabolite.
Quality control (QC) stock solutions were stored at −80 °C. These solutions were diluted in human plasma to produce the following QC samples of either: Lower Limit of Quantification (LLOQ) 3 ng/mL, QC Low (QCL) 5 ng/mL; QC Mid (QCM) 150 ng/mL, and QC High (QCH) 4000 ng/mL.
2.4 Sample preparation
Ten μL of 0.5 μg/mL [d7]-VX-970 was added to each microcentrifuge tube except blank plasma samples. This was followed by addition 50 μL plasma (standard, QC or sample plasma). Protein precipitation was sequentially accomplished using 200 μL of acetonitrile (aqueous to organic ratio of 1:4 v/v). Samples were vortexed for 1 min on a Vortex Genie-2 set at 9 (Model G-560 Scientific Industries, Bohemia, NY) and then centrifuged at 13,500 × g at room temperature for 5 min. A 100 μL aliquot of the resulting supernatant was transferred to an autosampler vial followed by addition of 150 μL of water (final organic composition 32%), vial capping and brief vortexing. A 5 μL volume of each sample was then injected into the LC-MS/MS system.
2.5 Validation procedures
2.5.1 Calibration curve and lower limit of quantitation (LLOQ)
Calibration standards and blanks were prepared and analyzed (see paragraph 2.4 and 2.5) in triplicate to establish a calibration range with acceptable accuracy and precision, as previously described [14].
2.5.2 Accuracy and precision
The accuracy and precision of the assay were determined by analyzing samples at the LLOQ, QCL, QCM, and QCH concentrations, as previously described [14].
2.5.3 Selectivity and specificity
Six individual batches of control drug-free human plasma were processed and analyzed (see 2.5) to investigate potential interference of endogenous matrix components with the assay. Responses of VX-970 at the LLOQ concentration were compared with the response of the blank samples.
Cross-talk of VX-970 and [d7]-VX-970 was characterized by detection of competing analyte signal into their respective MRM channels.
2.5.4 Extraction recovery and matrix effect
We determined the extraction recovery of VX-970 from plasma by comparing the absolute response of a neat solution of VX-970 in an extracted control plasma sample to the absolute response of a plasma extract with an equal amount of VX-970 added prior to extraction.
Regions of potential matrix effects on analyte signal by endogenous phospholipids in human plasma were identified by characterizing the chromatographic profiles of phospholipids. This was accomplished using previously reported phospholipid MRM channels and MS settings while maintaining the optimized LC components of the VX-970 assay [Little, 2006, 16497566][15, 16]. The following MRM channels were monitored in addition to VX-970: m/z 184>184, 496>184, 522>184, 524>184, 704>184, 758>184, 760>184, 784>184, 806>184. Trimethylammonium-ethyl phosphate (m/z 184) is a common phospholipid product ion found in human plasma.
The matrix effect caused by endogenous plasma matrix components on VX-970 response was defined as the change in the absolute response of a control plasma extract to which analyte had been added after the extraction relative to the absolute response of reconstitution solvent to which the same amount of VX-970 had been added. Experiments were performed at the three QC concentrations in quadruplicate.
2.5.5 Stability
Long-term stability experiments were performed both in stock solution and plasma after storage at −80 °C. Stability in the stock solution was expressed as the percentage recovery from a freshly prepared solution relative to the stored solution (8 months). The stability of VX-970 in plasma at −80 °C was determined by comparison of plasma samples before and after storage (8 months) against freshly prepared standard curves.
The room temperature stability of VX-970 in stock solution for 6 h was determined by comparison to stock solution stored at −80 °C. All stability testing in plasma was performed in triplicate at the QCL, QCM and QCH concentrations. The effect of 3 freeze/thaw cycles on VX-970 concentrations in plasma was evaluated by analysis of sample after they had been frozen (−80 °C) and thawed on 3 separate days and comparing the results with those of freshly prepared samples. The stability of VX-970 in plasma during sample preparation was evaluated by assaying samples before and after 4 h of storage at room temperature. To evaluate the stability of VX-970 after sample processing, we re-injected QC samples and calibration curves approximately 72 h after the first injection. The concentrations and absolute analyte areas derived from the second injection were then compared with those derived from the first injection. The results were expressed as a percentage of the values of the second run to their respective values in the first run.
2.5.6 Dilution integrity
To demonstrate dilution integrity, plasma samples containing VX-970 concentrations of 50,000 ng/mL were diluted 20-fold (to 2,500 ng/mL) with control plasma and analyzed.
2.5.7 Anti-coagulantia cross validation
To demonstrate the ability of our EDTA plasma-based assay to quantitate heparinized plasma samples, we quantitated and compared our EDTA QCL, QCM, and QCH samples in quadruplicate against both an EDTA and a heparinized plasma duplicate calibration curve.
2.5.1 Glucuronide Metabolite
This method was developed for the quantitation of VX-970 in samples obtained from multiple clinical trials. In preclinical animal testing, the major metabolite of VX-970 was identified as a direct glucuronide conjugate of VX-970 (mass 638.5 amu). In anticipation of possible glucuronide metabolites in plasma of treated with VX-970, we examined bile from a rat that was administered VX-970 (M. Howard, Vertex Pharmaceuticals London UK, estimated concentrations based on HPLC-UV: 9.0 μg/mL VX-970, 31.9 μg/mL VX-970 glucuronide).
We first determined the retention time of the glucuronide metabolite in our assay. To accomplish this we first spiked bile from VX-970 treated rats (10 mg/kg IV infusion) to control plasma and processed these samples as described previously. A full mass scan of this sample was analysed to identify the glucuronide metabolite (+176 m/z). The sample was then re-injected using a product ion scan to determine the fragmentation pattern of the metabolite. This revealed the metabolite fragmented into both VX-970 parent mass (464.3 m/z) as well as the previously selected VX-970 product ion (433.5 m/z). Based on signal intensity, the 433.5 m/z product ion was selected and a 639.5>433.5 m/z MRM channel was added to the method to monitor the metabolite.
Chemical degradation in matrix and in-source fragmentation can cause cleavage of direct glucuronide metabolites, producing parent drug, causing false positive parent compound signal. Potential VX-970 interference from in-source fragmentation of the glucuronide metabolite was assessed by comparison of parent and metabolite retention times. The stability of glucuronide metabolite in human plasma was assessed by taking bile from VX-970 treated mice and spiking it to human plasma. These samples were then analysed before and after a 4 h incubation at room temperature. The stability of the metabolite as a function of pH was also assessed by spiking the bile to phosphate buffers at various pH values (5, 6, 7, 8 and 9). VX-970 was quantitated before and after a 4 h incubation at room temperature in these buffers.
To further elucidate the role of VX-970 glucuronidation as a pathway in human metabolism, separate incubations of VX-970 in permeabilized human and rat microsomes were conducted in triplicate. The incubation for glucuronidation was adapted from a previously published method to allow for a 0.5 mL incubation volume [17]. The final incubation mixture contained 0.1 M phosphate buffer (pH 7.4), 0.25 mg/mL microsomal protein concentration, 25 μg/mL alamethicin, 50 mM Tris-HCl, 10 mM MgCl2, and 5,000 ng/mL of VX-970. Determination of microsome protein content was accomplished using a protein assay kit from BioRad (Hercules, CA) with BSA as the standard. Absorbance readings were recorded at 630 nm using an Infinite M100 Pro plate reader from Tecan (Männedorf, Switzerland). The reaction was initiated with the addition of 5 μM UPDGA. Aliquots (50 μL) were sampled from the reaction mixture at 0 and 60 min with 200 μL of acetonitrile added to terminate the reaction. Samples were stored at −80 °C until the day of analysis upon which 10 μL of internal standard was added and samples processed as described previously. Samples were quantitated with a standard curve produced in phosphate buffer matrix (pH 7.4) with 0.25 mg/mL BSA along with QCs prepared in human plasma. The validity of the assay in this matrix was first validated by preparing a triplicate curve in the matrix and comparing it to QCs prepared in plasma. A negative control was included that lacked UPDGA in a rat microsome incubation. Positive controls were included using SN-38 (active metabolite of irinotecan) in separate human and rat microsome incubations. SN-38 and SN-38 glucuronide were analysed by LC-MS/MS based on a previously published assay [18].
2.6 Application of Assay
To demonstrate the applicability of the assay, we analyzed samples from a patient enrolled in the study of VX-970 in combination with irinotecan (NCT02595931). This patient had signed informed consent on an institutional review board approved protocol. The patient received an IV infusion of VX-970 and blood samples were collected between 0 and 72 hours. The pharmacokinetic parameters from this patient were determined non-compartmentally using PK Solutions 2.0 (Summit Research Services, Montrose, CO). Additionally, the glucuronide metabolite channel was also monitored during sample analysis.
3 Results and Discussion
3.1 Assay Validation
3.1.1 Calibration curve and LLOQ
The selected assay range of 3–5,000 ng/mL fulfilled the FDA criteria for the LLOQ concentration and the calibration curve [19]. Accuracies and precisions at the different concentrations were determined from triplicate calibration curves on 5 separate days and are reported in Table S 1. At most concentrations, the mean square of the within runs was greater than the mean square of the between runs, indicating that there was no significant additional variability due to the performance of the assay in different runs [20]. Representative calibration curves and corresponding correlation and regression coefficients are shown in Fig. S. 1.
3.1.2 Accuracy and precision
The accuracies and intra- and inter-assay precisions for the tested concentrations (LLOQ, QCL, QCM, and QCH) were all within the defined acceptance criteria (Table S 2) [19].
3.1.3 Selectivity and specificity
Chromatograms of six individual control plasma samples contained no co-eluting peaks >20% of the analyte areas at the LLOQ concentration.
Cross-talk calculations were performed and revealed that VX-970 cross-talks with the internal standard channel at approximately 0.0015% with identical retention times. The internal standard is added at approximately 50 ng/mL, which would not be expected to result in meaningful interference with the VX-970 signal at the LLOQ at 3 ng/mL.
3.1.4 Extraction recovery and matrix effect
The recovery of VX-970 was approximately 94% with CVs between 2.7 and 11.8%.
Analysis of phospholipid MRM channels demonstrated no phospholipids co-elute with VX-970 (Fig. S. 3). Matrix effect was less than −7% (i.e. ionization suppression), with CVs between 2.3 and 6.6% (Table S 3).
3.1.5 Stability
The stability of VX-970 stock solution at room temperature for 6 h was 96.7% (Table 1). Stability in stock solution for 8 months at −80 °C was 95.7%. The stability of the analyte after 3 freeze thaw cycles (−80 °C to RT) ranged between 98.1 and 99.6%. Long-term stability (8 months) of the analyte in plasma at −80 °C was adequate with recovery between 92.2 and 100.1%. The absolute responses of plasma extracts of VX-970 at the QC concentrations, when processed and kept in the autosampler for 72 h, were 102.7 to 114.4% of the initial responses (CV 2.6–3.9%), while the response of VX-970 relative to the internal standard signal ranged from 97.0 to 99.3% (CV 1.8–4.2%). Importantly, the reinjection run met the criteria set by the FDA [7].
Table 1.
Stability of VX-970 under varying conditions.
| Storage condition | Concentration (ng/mL) |
Stability (%) |
CV (%) |
Replicates | |
|---|---|---|---|---|---|
| VX-970 | |||||
|
| |||||
| Stock solution 6 h | |||||
| Ambient temp | 100,000 | 96.7 | 10.6 | 3 | |
| Stock solution 8 months | |||||
| −80 °C | 100,000 | 95.7 | 4.0 | 3 | |
| Plasma 4 h | |||||
| Ambient temp. | QCL | 5 | 96.2 | 6.2 | 3 |
| QCM | 150 | 96.0 | 4.3 | 3 | |
| QCH | 4000 | 100.2 | 0.5 | 3 | |
| Plasma 3 freeze-thaw cycles | |||||
| −80 °C | QCL | 5 | 98.1 | 3.9 | 4 |
| QCM | 150 | 99.6 | 5.0 | 4 | |
| QCH | 4000 | 98.1 | 3.4 | 4 | |
| Plasma 8 months | |||||
| −80 °C | QCL | 5 | 92.2 | 4.6 | 4 |
| QCM | 150 | 100.1 | 3.8 | 4 | |
| QCH | 4000 | 96.2 | 4.0 | 4 | |
3.1.6 Dilution integrity
The samples diluted from 50,000 ng/mL to 2500 ng/mL (diluted 20 times) displayed an accuracy of 91.3% with a CV of 4.0%, indicating the dilution integrity of VX-970 in plasma.
3.1.1 Anti-coagulant cross validation
Accuracy and precision of back-calculated concentrations were within 15% at QCL, QCM, and QCH concentrations (Table S 4).
3.2 Development
3.2.1 Mass spectrometry
The mass spectrometric parameters were optimized in order to obtain the largest parent ion and subsequent product ion from fragmentation for both VX-970 and IS. Both compounds were infused separately into the mass spectrometer with mobile phase containing either 50% methanol and 50% water with 0.1% formic acid or 50% acetonitrile and 50% water with 0.1 % formic acid. There was no remarkable difference in the ionization effects between the chosen organic solvents. Acetonitrile was chosen as the organic solvent due to its greater ability to precipitate proteins and create a cleaner final matrix for our desired dilute and shoot sample preparation. Optimized mass spectrometric parameters are listed in section 2.3.
3.2.1 Chromatography
We evaluated the following three columns: Synergi Polar RP 80Å (50 × 2.0 mm, 4 μm), Synergi Hydro RP 80Å (50 × 2.0 mm, 4 μm), and Luna phenylhexyl (50 × 2.0 mm, 3 μm). VX-970 exhibited significant peak-tailing on the Luna column, which was therefore eliminated from the selection. The Polar RP and Hydro RP columns each produced acceptable VX-970 peak shapes but analysis of rat bile containing the glucuronide metabolite demonstrated that the Polar RP column provided better separation of parent and metabolite and was therefore selected for the method.
It was observed that greater than 75% acetontrile in the mobile phase caused rapid elution of VX-970. We found that a gradient style elution gave adequate time to separate endogenous matrix components from VX-970 and still achieve a short run time. The final mobile phase composition elutes VX-970 during the terminal phase of the 0–2.2 min step where mobile phase A is increased from 30 to 80%.
Despite the short elution time, phospholipids from the human plasma matrix did not co-elute with VX-970 indicating that a potential signal interference was minimal.
The approximate retention time of both VX-970 and [d7]-VX-970 was 1.9 min. VX-970 had a capacity factor of 4.6 based on the observed void time of 0.34 min. Representative chromatograms of VX-970 (at the LLOQ), and internal standard in plasma are displayed in Fig. S. 2.
Carry-over was assessed by injecting 50,000 ng/mL of VX-970 and 5,000 ng/mL of IS, followed by injection of 4 control plasma samples. Carry-over amounted to less than 0.02% for VX-970 and 0.2% for IS.
3.2.2 Extraction and sample preparation
To expedite sample preparation, we chose to pursue a method of dilute-and-shoot sample preparation. This process used a 1:4 ratio of sample to acetonitrile (v/v) and demonstrated the ability to obtain our desired sensitivity of the 3 ng/mL LLOQ. Despite achieving our desired sensitivity, the final sample extract produced peak fronting and general irregular chromatography, which was most likely due to the incompatibility of the organic to aqueous ratio between sample extract (80% organic) and initial mobile phase composition (30% organic). Resolving this discrepancy without alteration of our developed chromatographic method would require a decrease in the sample extract organic-to-aqueous ratio. Decreasing the amount of acetonitrile for extraction could compromise protein precipitation so we added an additional step of adding 150 μL of water to 100 μL supernatant of processed samples. This decreased the final organic composition of the sample to approximately 32% and thus closely replicated the initial mobile phase composition of 30% acetonitrile. Initial experimentation with this processing method revealed a crucial step of thoroughly vortexing autosampler vials after capping. This minor addition to sample preparation restored peak shape, maintained LLOQ sensitivity and did not compromise efficiency of protein precipitation.
3.2.3 Glucuronide metabolite
Analysis of bile from rats treated with VX-970 revealed it contained both unconjugated VX-970 (retention 1.9 min) and the direct glucuronide of VX-970 metabolite (retention 0.83 min, see Fig. S. 2C). In-source fragmentation of the metabolite back to the VX-970 parent m/z was also observed. The latter observation is not considered a potential bias for the quantitation of VX-970 as the analytes were appropriately separated. We incorporated the corresponding MRM transition (m/z 639.3>433.5) into the quantitation method to ensure that any metabolite in patient samples would be detected.
Incubation of bile at the tested pH values demonstrated no increase in VX-970 concentrations, indicating stability of the glucuronide metabolite as both a function of pH (5–9) and time (up to 4 h) (Table S 5). Bile spiked plasma incubated at room temperature for 4 h showed VX-970 concentrations were 96.5% stable (CV 4.7%), indicating that the glucuronide metabolite did not hydrolyse to produce parent VX-970.
VX-970 incubations in permeabilized human microsomes revealed no glucuronidation with 99.9% (CV 4.7%) of drug remaining in the incubation mixture after 60 min compared to 87.5% (CV 1.8%) in rat microsomes. No metabolite was observed in the 639.3>433.5 m/z glucuronide channel in rat the microsome incubation, most likely due to being below the limit of sensitivity. These experiments lend further evidence that the glucuronidation pathway for VX-970 is species specific and does not occur in humans. Positive control experiments confirmed glucuronidation of SN-38 in both rat and human microsomes both measured as SN-38 depletion and SN-38 glucuronide generation. These experiments demonstrate that the UGT isoforms responsible for VX-970 glucuronidation in rats are either not present in humans or have a very low activity.
3.3 Application of Assay
All patient plasma samples analysed had VX-970 concentrations within the calibration range. Visual inspection of the time-concentration plot revealed a distinct biphasic elimination profile as seen in Fig. 2. A Cmax of 343 ng/mL was observed near the end of the 1 h infusion. Non-compartmental analysis of this data resulted in the following PK parameter values: T1/2 of 14 h, AUC0-72 2.43 μg•h/mL, AUC0-∞ 2.50 μg•h/mL, Vss 386 L/m2, and Cl 24.0 L/h/m2. Additionally, monitoring the glucuronide metabolite MRM channel revealed no glucuronide metabolite in any sample analyzed.
Fig. 2.
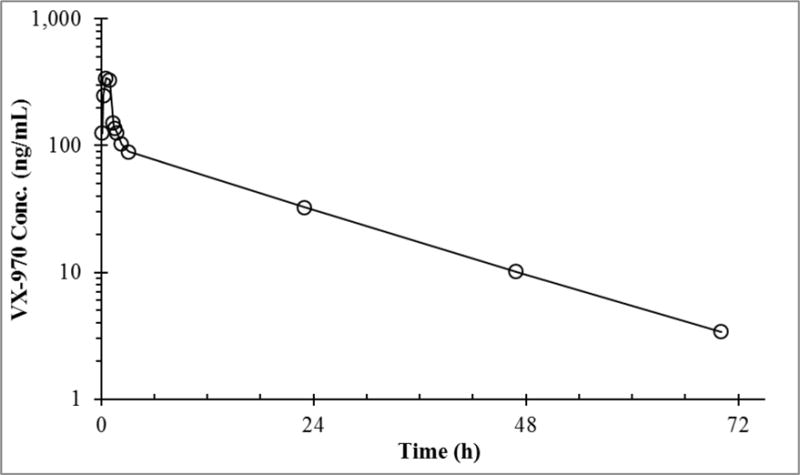
VX-970 (○) plasma concentrations in a patient administered a 1 h intravenous infusion of 60 mg/m2 VX-970.
4 Conclusion
With the clinical development of ATR inhibitors such as VX-970, a thorough description of their clinical pharmacokinetics is needed. To support these investigations, our aim in the present study was to develop and validate an analytical method for VX-970 in human plasma that ensures accurate and reliable quantification. This method will be used to quantitate VX-970 in plasma samples being obtained from ongoing and future clinical trials.
VX-970 was detected by a triple quadrupole mass spectrometer in MRM mode after separation from matrix components by reverse-phase chromatography. We developed a facile “dilute and shoot” method of sample preparation that included an additional post-protein precipitation dilution step with water that resolved an observed aqueous-to-organic ratio incompatibility between initial mobile phase conditions and final sample extract.
Previous investigations have indicated that rodents produce a direct glucuronide metabolite but evidence of this biotransformation in humans has yet to be demonstrated. Incubations of VX-970 permeabilized human microsomes revealed no occurrence of metabolism. To guard against potential interference from this metabolite, we made efforts to ensure instability and in-source fragmentation would not affect our assay by analyzing bile samples from rats treated with VX-970. From our investigations of metabolite conversion back to VX-970, we determined that the metabolite is stable under our assay conditions and its presence does not affect VX-970 response. Complete chromatographic separation of the analytes nullifies potential effects from in-source fragmentation of metabolite into the VX-970 channel. Additionally, no metabolite was detected in the analysis of the patient accrued onto a VX-970 phase I trial, which further suggests any glucuronide metabolite will not interfere with VX-970 quantification.
This is the first quantitative assay for VX-970 to be published. Our assay range captured the entire time-concentration curve of the analyzed patient treated with VX-970, including a 72 h sample which was still above the LLOQ. While the observed Cmax was vastly below our ULQ, this assay will accommodate increasing VX-970 concentrations as dose is escalated.
In conclusion, we have successfully developed a rapid and facile LC-MS/MS assay for VX-970 quantitation in human plasma, which is expected to cover the entire clinical concentration range. This assay will prove to be a valuable tool in supporting the clinical development of VX-970.
Supplementary Material
Acknowledgments
Support: Grant UM1-CA186690 (NCI-CTEP), RO1 CA204173, and R50 CA211241. This project used the UPCI Cancer Pharmacokinetics and Pharmacodynamics Facility (CPPF) and was supported in part by award P30-CA47904. Andre Iffland is an employee of and holds stock in Vertex Pharmaceuticals.
We thank Martin Howard (Vertex) for help in obtaining VX-970 treated rat bile sample.
References
- 1.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Molecular cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C, Orntoft T, Lukas J, Bartek J. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434(7035):864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 3.Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268(5218):1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 4.Cimprich KA, Shin TB, Keith CT, Schreiber SL. cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(7):2850–5. doi: 10.1073/pnas.93.7.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Choi S, Bakkenist CJ. Brd4 shields chromatin from ATM kinase signaling storms. Science signaling. 2013;6(293):pe30. doi: 10.1126/scisignal.2004622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nature medicine. 2004;10(8):789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 7.Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nature chemical biology. 2011;7(7):428–30. doi: 10.1038/nchembio.573. [DOI] [PubMed] [Google Scholar]
- 8.Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, Bartek J, Yaffe MB, Hemann MT. The combined status of ATM and p53 link tumor development with therapeutic response. Genes & development. 2009;23(16):1895–909. doi: 10.1101/gad.1815309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vendetti FP, Lau A, Schamus S, Conrads TP, O’Connor MJ, Bakkenist CJ. The orally active and bioavailable ATR kinase inhibitor AZD6738 potentiates the anti-tumor effects of cisplatin to resolve ATM-deficient non-small cell lung cancer in vivo. Oncotarget. 2015;6(42):44289–305. doi: 10.18632/oncotarget.6247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foote KM, Lau A, Nissink JW. Drugging ATR: progress in the development of specific inhibitors for the treatment of cancer. Future medicinal chemistry. 2015;7(7):873–91. doi: 10.4155/fmc.15.33. [DOI] [PubMed] [Google Scholar]
- 11.Fokas E, Prevo R, Pollard JR, Reaper PM, Charlton PA, Cornelissen B, Vallis KA, Hammond EM, Olcina MM, Gillies McKenna W, Muschel RJ, Brunner TB. Targeting ATR in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell death & disease. 2012;3:e441. doi: 10.1038/cddis.2012.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prevo R, Fokas E, Reaper PM, Charlton PA, Pollard JR, McKenna WG, Muschel RJ, Brunner TB. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer biology & therapy. 2012;13(11):1072–81. doi: 10.4161/cbt.21093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, Hare B, Johnson MA, Milton S, Murphy CE, Takemoto D, Tolman C, Wood M, Charlton P, Charrier JD, Furey B, Golec J, Reaper PM, Pollard JR. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014;5(14):5674–85. doi: 10.18632/oncotarget.2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KP, Parise RA, Holleran JL, Lewis LD, Appleman L, van Erp N, Morris MJ, Beumer JH. Simultaneous quantitation of abiraterone, enzalutamide, N-desmethyl enzalutamide, and bicalutamide in human plasma by LC-MS/MS. Journal of pharmaceutical and biomedical analysis. 2017;138:197–205. doi: 10.1016/j.jpba.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Little JL, Wempe MF, Buchanan CM. Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2006;833(2):219–30. doi: 10.1016/j.jchromb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 16.Zhang G, Wujcik CE. Overcoming ionization effects through chromatography: a case study for the ESI-LC-MS/MS quantitation of a hydrophobic therapeutic agent in human serum using a stable-label internal standard. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2009;877(22):2003–10. doi: 10.1016/j.jchromb.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 17.Seo KA, Kim HJ, Jeong ES, Abdalla N, Choi CS, Kim DH, Shin JG. In vitro assay of six UDP-glucuronosyltransferase isoforms in human liver microsomes, using cocktails of probe substrates and liquid chromatography-tandem mass spectrometry. Drug metabolism and disposition: the biological fate of chemicals. 2014;42(11):1803–10. doi: 10.1124/dmd.114.058818. [DOI] [PubMed] [Google Scholar]
- 18.Corona G, Elia C, Casetta B, Toffoli G. Fast liquid chromatography-tandem mass spectrometry method for routine assessment of irinotecan metabolic phenotype. Therapeutic drug monitoring. 2010;32(5):638–46. doi: 10.1097/FTD.0b013e3181ec3bf5. [DOI] [PubMed] [Google Scholar]
- 19.U.S. Department of Health and Human Services Food and Drug Administration. Guidance for Industry-Bioanalytical Method Validation. U.S. Department of Health and Human Services; Food and Drug Administration; Center for Drug Evaluation; and Research Center for Veterinary Medicine; 2001. [Google Scholar]
- 20.Rosing H, Man WY, Doyle E, Bult A, Beijnen JH. Bioanalytical Liquid Chromatographic Method Validation. A Review of Current Practices and Procedures. Journal of Liquid Chromatography & Related Technologies. 2000;23(3):329–354. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.