

Abstract
The development of a general catalytic system for the palladium-catalyzed carbocyclization of unactivated alkyl bromides with alkenes is described. This approach uses a commercially available bisphosphine ligand, and avoids the use of carbon monoxide atmosphere present in prior studies involving alkyl iodides. Detailed mechanistic studies of the transformation are performed, which are consistent with auto-tandem catalysis involving atom-transfer radical cyclization followed by catalytic dehydrohalogenation. These studies also suggest that reactions involving alkyl iodides may proceed through a metal-initiated, rather than metal-catalyzed, radical chain process.
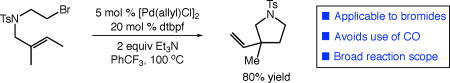
Graphical Abstract
INTRODUCTION
The Mizoroki-Heck reaction is a premier method for catalytic C–C bond construction across a wide range of synthetic contexts.1 The reactions commonly use aryl or vinyl halides (or sulfonates) as substrates in inter- or intramolecular cross couplings with alkenes. Extension to widely available alkyl electrophiles containing β-hydrogens has been a longstanding challenge in catalysis.2 Two issues to be addressed include the relative reluctance of sp3-hybridized versus sp2-hybridized electrophiles to participate in oxidative addition3 and the susceptibility of alkylmetals to β-hydride elimination.4 Despite early studies demonstrating potential cross couplings with limited scope (e.g., styrenes),5 the need for a general solution to this problem remained.
In recent years, several studies have demonstrated the ability of a variety of metals to catalyze formal alkyl-Mizoroki-Heck reactions, including Ti, Co, Ni, Au, and Pd. The Ti- and Co-catalyzed processes significantly increased the substrate scope of these carbocyclizations, but required highly reducing alkyl-magnesium reagents for catalyst turnover, limiting their broader use in the synthesis of complex targets.6 Recent studies involving Ni, Au, and Co catalysts provided useful Mizoroki-Heck-type reactions of unactivated alkyl halides that proceed under mild conditions.7 Pioneering work from the Fu group detailed the first examples of Pd-catalyzed alkyl-Mizoroki-Heck-type reactions to construct cyclopentanes with primary alkyl halides and terminal alkenes (Figure 1).8
Figure 1.

Carbocyclizations of unactivated alkyl halides with alkenes involving palladium.
Our prior studies towards a general, Pd-catalyzed carbocyclization encompassed both primary and secondary alkyl halides and a diverse range of alkenes.9 However, a major limitation was the requirement of alkyl iodides as substrates. Furthermore, reactions involving primary alkyl iodides required a CO atmosphere (10 atm) to maximize reaction yield (Figure 1). Thus, while the substrate scope was attractive, these system limitations served to hinder potential applications.
Another major question that remained was the role of the metal catalyst in these carbocyclizations. Fu demonstrated the presence of alkylpalladium(II) intermediates formed from SN2-type oxidative addition.8 While our initial mechanistic studies provided evidence for radical intermediates in reactions involving alkyl iodides, it was unclear whether these reactions were palladium-initiated, innate chain reactions or palladium-catalyzed transformations.10 These questions have important consequences regarding the potential for catalyst control in reaction development.
Herein, we report a second-generation catalytic system for the palladium-catalyzed carbocyclization of unactivated alkyl halides with alkenes that significantly increases the reaction scope and practicality of the transformation. This new catalytic process constitutes the first general carbocyclization of alkenes involving unactivated alkyl bromides. Importantly, this system also removes the requirement for CO atmosphere present in our previous studies. The mechanistic analysis of the hybrid organometallic-radical reactivity involved uncovered some intriguing insights into these reactions, consistent with an auto-tandem catalysis pathway for the present reaction, while suggesting that reactions involving alkyl iodides may proceed via metal-initiated, innate chain processes.
RESULTS AND DISCUSSION
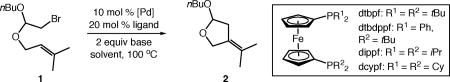
Our studies commenced with the carbocyclization of unactivated alkyl bromide 1 (Table 1). The use of catalytic systems previously developed in our laboratory for the carbonylation of alkyl bromides (entry 1)11 or aromatic C–H alkylation using alkyl bromides (entry 2)12 did not lead to successful carbocyclization. The ligand screen was then expanded to bidentate ferrocenyl ligand systems, as this class of ligands has proven useful in previous carbocyclizations involving alkyl electrophiles. The dtbdppf ligand recently used by Gevorgyan in an endo silyl-methyl Mizoroki-Heck-type process13 provided carbocyclization product 2 in poor yield (entry 3). The use of 1,1’-bis(dialkylphosphino)ferrocene ligands dcypf or dippf failed to improve reactivity (entries 4–5). Switching the ligand to 1,1'-bis(di-tert-butylphosphino)ferrocene (dtbpf) provided a promising result, with a significantly increased reaction yield (33%, entry 6). Substituting [Pd(allyl)Cl]2 as precatalyst (entry 7) and Et3N as base (entry 8) further increased the yield and selectivity of the reaction. The use of trifluorotoluene as solvent further increased the reaction efficiency by limiting reductive dehalogenation (entry 9). Interestingly, the use of the prepared Pd(dtbpf)Cl2 as catalyst was less successful (entry 10).
Table 1.
Identification of a catalytic system for the carbocyclization of unactivated alkyl bromides.
| |||||
|---|---|---|---|---|---|
|
| |||||
| entry | catalyst | ligand | base | solvent | yield (%)a |
| 1 | [Pd(allyl)Cl]2 | IMes | iPr2NEt | PhMe | <2 |
| 2b | Pd(PPh3)4 | – | PMP | PhtBu | <2 |
| 3 | Pd(OAc)2 | dtbdppf | iPr2NEt | PhMe | 5 |
| 4 | Pd(OAc)2 | dcypf | iPr2NEt | PhMe | <2 |
| 5 | Pd(OAc)2 | dippf | iPr2NEt | PhMe | <2 |
| 6 | Pd(OAc)2 | dtbpf | iPr2NEt | PhMe | 30(3) |
| 7 | [Pd(allyl)Cl]2 | dtbpf | iPr2NEt | PhMe | 65(12) |
| 8 | [Pd(allyl)Cl]2 | dtbpf | Et3N | PhMe | 73(5) |
| 9 | [Pd(allyl)Cl]2 | dtbpf | Et3N | PhCF3 | 85(5) |
| 10 | Pd(dtbpf)Cl2 | – | Et3N | PhCF3 | 14(2) |
Yield determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. Yield in parenthesis is that of a minor regioisomer, see Supporting Information for details.
Reaction temperature 130 °C.
With optimized conditions in hand, a comparison of the previously reported carbocyclizations of alkyl iodides to the current system using alkyl bromides was performed (Table 2). Each carbocyclization proceeded with similar or increased yields across a range of primary and secondary alkyl halides with diverse alkene substitution, as compared to our previous report.9 Importantly, the carbocyclizations of primary alkyl bromides using the second-generation catalytic system avoids the 10 atm CO previously required in cyclizations of alkyl iodides, increasing the practicality of the current system.14
Table 2.
Comparison of previously reported first-generation catalytic systems (alkyl iodides)9 with current protocol (alkyl bromides).

|
Reactions of alkyl iodides as reported in ref. 9. All reactions of alkyl bromides were performed with [substrate]0 = 0.25 M in PhCF3 at 100 °C with 5 mol % [Pd(allyl)Cl]2, 20 mol % dtbpf, and 2 equiv Et3N as base.
Isolated yield.
Isolated with minor regioisomers, see Supporting Information for details.
We next studied the carbocyclization of a range of substrates to assess the full reaction scope (Table 3). The 5-exo cyclizations of primary alkyl bromides were successful with di- and trisubstituted alkenes (entries 1–3), styrenes (entry 4), and terminal alkenes (entry 5). Simple substitution of the tether unit provides access to important heterocycles (e.g., pyrrolidines, tetrahydrofurans) or carbocycles, as desired. Importantly, this approach is not limited to five-membered ring synthesis, as demonstrated by the 6-exo cyclizations of entries 6–8. Secondary alkyl bromides also undergo efficient cyclization, as demonstrated by the 5-exo and 6-exo cyclizations of substrates 38 and 40, respectively (entries 9–10).
Table 3.
Catalytic carbocyclization of diverse unactivated alkyl bromides.
| entry | substrate | product | yield (%)a |
|---|---|---|---|
| 1 |
|
|
86b |
| 2 |
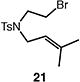
|
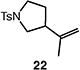
|
81b |
| 3 |
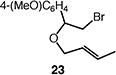
|
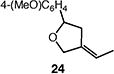
|
71 (1:1 E:Z) |
| 4 |
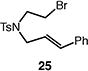
|
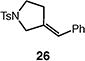
|
65 (1:1 E:Z) |
| 5c |
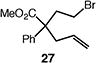
|
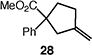
|
65 |
| 6d |
|
|
92 (30:31 = 1.9:1) |
| 7d |
|
|
70 (33:34 = 3.3:1) |
| 8d |
|
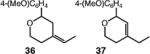
|
40 (36:37 = 7.0:1) |
| 9 |
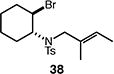
|
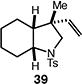
|
63 (>95:5 dr) |
| 10 |
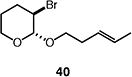
|
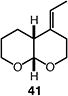
|
75 (>95:5 dr) (1.5:1 E:Z) |
Reactions were performed with [substrate]0 = 0.25 M in PhCF3 at 100 °C with 5 mol % [Pd(allyl)Cl]2, 20 mol % dtbpf, and 2 equiv Et3N as base.
Isolated yield.
Isolated with minor regioisomers, see Supporting Information for details.
DBU used as base instead of Et3N.
Reactions were performed at 120 °C with 5 mol % [Pd(allyl)Cl]2, 20 mol % dtbpf, and 2 equiv Cy2NMe as base.
In addition to carbocyclization, the present system is also effective in promoting intermolecular couplings. As an initial demonstration, we have studied the intermolecular cross coupling of secondary alkyl bromide 42 with styrene (eq 1), which delivers product 43 in moderate yield (40% by 1H NMR analysis). Previous reports of such palladium-catalyzed, intermolecular couplings required the use of alkyl iodides as substrates, used directly or formed in situ via Finkelstein reactions with iodide salts.15
 |
(1) |
To glean insight into the reaction mechanism of the carbocyclization, we began by attempting to isolate intermediates at short reaction times and low conversion. Surprisingly, the reaction of primary alkyl bromide 8 proceeded to near full conversion after only 10 minutes, and the major product was alkyl bromide 44–the product of atom-transfer radical cyclization (ATRC, eq 2). In addition, the expected carbocyclization product 7 was isolated in 34% yield.
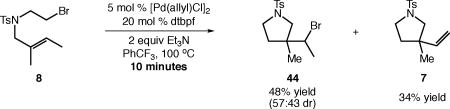 |
(2) |
A number of other representative substrates were studied to determine the role of ATRC in these carbocyclizations (Table 4). In order to maximize the potential for observing ATRC products, these reactions were performed at a slightly lower temperature (80 °C) with a decreased amount of base added (0.5 equiv). In each case, the formation of the ATRC product was significant. The low yield observed in the production of tertiary ATRC product 47 is attributed to the high propensity towards elimination to the alkene product. Interestingly, these results represent a rare example of ATRC processes involving unactivated alkyl halides,16 and the first involving unactivated alkyl bromides.
Table 4.
Palladium-catalyzed atom-transfer radical cyclizations of unactivated alkyl bromides.
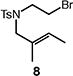
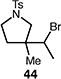
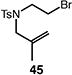
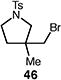
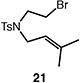
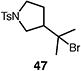
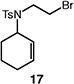
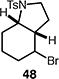
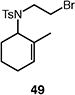
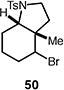
Reactions performed with [substrate]0 = 0.25 M in PhCF3 at 80 °C with 5 mol % [Pd(allyl)Cl]2, 20 mol % dtbpf, and 0.5 equiv Et3N as base.
Isolated yield.
Yield determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard.
Preliminary mechanistic investigations of the carbocyclizations of alkyl iodides were consistent with the presence of free radical intermediates in the reaction pathway.9 Two major mechanistic questions remained: 1) whether or not the carbocyclizations of alkyl iodides and alkyl bromides proceeded via the same pathway, and 2) the role of the palladium as either an initiator or a catalyst.
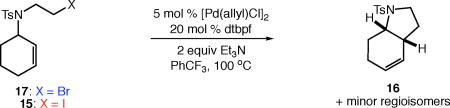
The reactions of alkyl bromide 17 and alkyl iodide 15 afforded bicyclic product 16 in 77% and 48% yield, respectively (Table 5, entries 1–2). Reaction of alkyl bromide 17 in the presence of single electron transfer inhibitor 1,4-dinitrobenzene resulted in a complete return of starting material (<2% conversion, entry 3). On the other hand, the reaction of alkyl iodide 15 went to complete conversion, and 16 was actually formed in higher yield (78%, entry 4). Similar results were observed upon the addition of persistent radical TEMPO. The reaction of alkyl bromide 17 proceeded with low conversion, and neither the ATRC nor the Heck-type product were observed (entry 5). Conversely, the reaction of the corresponding iodide delivered 16 in moderate yield with complete consumption of starting material.
Table 5.
Comparing reactions of alkyl bromides and iodides in the presence of radical and electron-transfer inhibitors.
| |||
|---|---|---|---|
|
| |||
| entry | conditions | conversion (%)a | yield (%)a |
| 1 | X = Br, no additive | 100 | 77 |
| 2 | X = I, no additive | 100 | 48 |
| 3 | X = Br, with 10 mol % dinitrobenzene | <2 | <2 |
| 4 | X = I, with 10 mol % dinitrobenzene | 100 | 78 |
| 5 | X = Br, with 1 equiv TEMPO | 31 | <2 |
| 6 | X = I, with 1 equiv TEMPO | 100 | 55 |
Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. For details regarding the distribution of minor regioisomers, see Supporting Information.
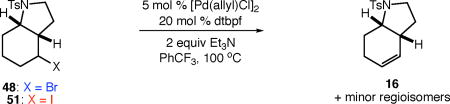
We next studied the dehydrohalogenation of ATRC intermediates 48 and 51 in a similar manner (Table 6). Both substrates underwent high yielding dehydrohalogenation under the reaction conditions (entries 1–2).17 In the presence of 1,4-dinitrobenzene, the reaction of alkyl bromide 48 was significantly inhibited, while the dehydrohalogenation of iodide 51 was unaffected (entries 3–4). In the absence of metal catalyst or ligand, no dehydrohalogenation of bromide 48 occurred, while the elimination of iodide 51 still proceeded to a significant extent (entries 5–6).
Table 6.
Comparing the dehydrohalogenation of alkyl bromide and iodide ATRC intermediates.
| |||
|---|---|---|---|
|
| |||
| entry | conditions | conversion (%)a | yield (%)a |
| 1 | X = Br, no additive | 100 | 100 |
| 2 | X = I, no additive | 100 | 97 |
| 3 | X = Br, with 10 mol % dinitrobenzene | 22 | 9 |
| 4 | X = I, with 10 mol % dinitrobenzene | 100 | 100 |
| 5 | X = Br, no metal or ligand | <2 | <2 |
| 6 | X = I, no metal or ligand | 56 | 37 |
Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. For details regarding the distribution of minor regioisomers, see Supporting Information.
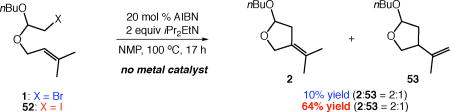
ATRC reactions may be initiated by transition metals or other simple radical initiators. We hypothesized that particularly in the case of alkyl iodides, palladium could be functioning as an initiator for an innate chain reaction, rather than as a catalyst. Alkyl iodides are known to participate in ATRC with alkenes in chain fashion.18 The carbocyclizations of alkyl bromide 1 and iodide 52 were studied in the absence of palladium using 20 mol % AIBN as a representative radical initiator (eq 3). While the reaction of alkyl bromide 1 proceeded with low conversion, a good yield of the Heck-type products 2 and 53 was achieved using alkyl iodide 52.
 |
(3) |
A plausible mechanism consistent with our observations is depicted in Scheme 1. We hypothesize that palladium is serving as a true catalyst, rather than a simple initiator, in the carbocyclization of unactivated alkyl bromides. The catalytic cycle commences with a bromine atom abstraction by the palladium catalyst, producing a carbon-centered radical and a putative palladium(I) intermediate. Facile radical cyclization forms the desired C–C bond and generates a new radical intermediate. At this stage, the ATRC product 48 is formed by bromine atom transfer from the catalyst.
Scheme 1.

Plausible mechanism for the palladium-catalyzed carbocyclization of unactivated alkyl bromides with alkenes involving auto-tandem catalysis.
Following the formation of the ATRC product, the palladium catalyst reengages bromide 48 to form the secondary alkyl radical once again. This is followed by formation of an alkylpalladium(II) intermediate, which upon β-hydride elimination delivers the product 16. As a single catalyst promotes two different transformations (ATRC and dehydrohalogenation), we hypothesize that this is an example of auto-tandem catalysis.19 While our results are consistent with ATRC as the major pathway for most substrates, it is possible that the direct formation of an alkylpalladium(II) species could also occur preferentially to bromine atom transfer, leading directly to product.20
Our results involving the reactions of alkyl iodides in the presence of inhibitors, and in the absence of palladium, suggest that an alternative mechanistic scenario may be involved. While palladium is required to abstract an iodine atom to begin the reaction, upon radical cyclization chain transfer with another equivalent of substrate is viable (Scheme 2). Furthermore, palladium is not necessarily required for the dehydrohalogenation of ATRC iodide products. While other pathways may contribute overall, in reactions of alkyl iodides involving palladium the role of the metal as an initiator, rather than catalyst, must be considered.
Scheme 2.

Plausible mechanism for the palladium-initiated carbocyclization of alkyl iodides involving an innate chain reaction.
CONCLUSIONS
In conclusion, we have developed an efficient second-generation system for the carbocyclization of unactivated alkyl halides with alkenes. This catalytic system promotes a general carbocyclization of unactivated alkyl bromides for the first time, and avoids the undesired use of the CO atmosphere previously required. This reaction efficiently transforms an array of primary and secondary alkyl bromides in cyclizations with diverse alkenes to form valuable 5- and 6-membered carbo- and heterocycles. We propose the reactions of alkyl bromides involve auto-tandem catalysis, involving the first examples of ATRC with unactivated alkyl bromides. We anticipate this work will significantly expand the use of these catalytic cyclizations in chemical synthesis and provide important insights into the reactivity of palladium catalysts with unactivated alkyl halides in new reaction development.
Supplementary Material
Acknowledgments
This work was supported by Award No. R01 GM107204 from the National Institute of General Medical Sciences.
Footnotes
ASSOCIATED CONTENT
Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Oestrich M, editor. The Mizoroki-Heck Reaction. John Wiley & Sons; West Sussex, U.K.: 2009. [Google Scholar]
- 2.For reviews on metal-catalyzed cross-coupling reactions employing unactivated alkyl halides, see: Luh T-Y, Leung M-K, Wong K-T. Chem. Rev. 2000;100:3187. doi: 10.1021/cr990272o.Frisch AC, Beller M. Angew. Chem. Int. Ed. 2005;44:674. doi: 10.1002/anie.200461432.Kambe N, Iwasaki T, Terao J. Chem. Soc. Rev. 2011;40:4937. doi: 10.1039/c1cs15129k.
- 3.(a) Collman JP. Acc. Chem. Res. 1975;8:342. [Google Scholar]; (b) Pearson RG, Figdore PE. J. Am. Chem. Soc. 1980;102:1541. [Google Scholar]
- 4.(a) Ozawa F, Ito T, Yamamoto A. J. Am. Chem. Soc. 1980;102:6457. [Google Scholar]; (b) Hartwig J. Organotransition Metal Chemistry: From Bonding to Catalysis. Chapter 10. University Science Books; Sausalito, CA: 2009. pp. 398–402. [Google Scholar]
- 5.Alkyl Mizoroki-Heck-type reactions with styrene: Lebedev SA, Lopatina VS, Petrov ES, Beletskaya IP. J. Organomet. Chem. 1988;344:253.Alkyl Mizoroki-Heck-type reactions with alkyl halides not prone to β-hydride elimination: Bräse S, Waegell B, de Meijere A. Synthesis. 1997:148.
- 6.Alkyl-Mizoroki-Heck-type reactions with superstoichiometric alkylmagnesium reagents: (Titanocene catalysis) Terao J, Watabe H, Miyamoto M, Kambe N. Bull. Chem. Soc. Jpn. 2003;76:2209.Terao J, Kambe N. Bull. Chem. Soc. Jpn. 2006;79:663.(Cobalt catalysis) Affo W, Ohmiya H, Fujioka T, Ikeda Y, Nakamura T, Yorimitsu H, Oshima K, Imamura Y, Mizuta T, Miyoshi K. J. Am. Chem. Soc. 2006;128:8068. doi: 10.1021/ja061417t.Wakabayashi K, Yorimitsu H, Oshima K. J. Am. Chem. Soc. 2001;123:5374. doi: 10.1021/ja0100423.
- 7.Nickel catalysis: Millán A, Álvarez de Cienfuegos L, Miguel D, Campaña AG, Cuerva JM. Org. Lett. 2012;14:5984. doi: 10.1021/ol3028913.Gold catalysis: Xie J, Li J, Weingand V, Rudolph M, Hashmi ASK. Chem. Eur. J. 2016;22:12646. doi: 10.1002/chem.201602939.Cobalt catalysis: Weiss ME, Kreis LM, Lauber A, Carreira EM. Angew. Chem. Int. Ed. 2011;50:11125. doi: 10.1002/anie.201105235.Busato S, Tinembart O, Zhang Z, Scheffold R. Tetrahedron. 1990;46:3155.Bhandal H, Pattenden G, Russell JJ. Tetrahedron Lett. 1986;27:2299.
- 8.Firmansjah L, Fu GC. J. Am. Chem. Soc. 2007;129:11340. doi: 10.1021/ja075245r. [DOI] [PubMed] [Google Scholar]
- 9.Bloome KS, McMahen RL, Alexanian EJ. J. Am. Chem. Soc. 2011;133:20146. doi: 10.1021/ja2091883. [DOI] [PubMed] [Google Scholar]
- 10.For a discussion of the role of metal reagents as catalysts or initiators in radical-mediated reactions, see: Studer A, Curran DP. Angew. Chem. Int. Ed. 2016;55:58. doi: 10.1002/anie.201505090.
- 11.Sargent BT, Alexanian EJ. J. Am. Chem. Soc. 2016;138:7520. doi: 10.1021/jacs.6b04610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venning ARO, Bohan PT, Alexanian EJ. J. Am. Chem. Soc. 2015;137:3731. doi: 10.1021/jacs.5b01365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parasram M, Iaroshenko VO, Gevorgyan V. J. Am. Chem. Soc. 2014;136:17926. doi: 10.1021/ja5104525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.For comparison, reactions involving the alkyl chloride or tosylate of substrate 8 proceeded with significantly lower yields than the bromide providing 6% and <2% 1H NMR yield respectively, of cyclization products after 16 h with significant amounts of starting material remaining. Additionally, reactions of the alkyl iodides in Table 2 using the current protocol provided carbocyclization products in generally poor yields. See the Supporting Information for details.
- 15.(a) McMahon CM, Alexanian EJ. Angew. Chem. Int. Ed. 2014;53:5974. doi: 10.1002/anie.201311323. [DOI] [PubMed] [Google Scholar]; (b) Zou Y, Zhou J. Chem. Commun. 2014;50:3725. doi: 10.1039/c4cc00297k. [DOI] [PubMed] [Google Scholar]
- 16.(a) Liu H, Qiao Z, Jiang X. Org. Biomol. Chem. 2012;10:7274. doi: 10.1039/c2ob25990g. [DOI] [PubMed] [Google Scholar]; (b) Shen Y, Cornella J, Juliá-Hernández F, Martin R. ACS Catal. 2017;7:409. [Google Scholar]; (c) Monks BM, Cook SP. Angew. Chem. Int. Ed. 2013;52:14214. doi: 10.1002/anie.201308534. [DOI] [PubMed] [Google Scholar]
- 17.Bissember AC, Levina A, Fu GC. J. Am. Chem. Soc. 2012;134:14232. doi: 10.1021/ja306323x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Curran DP, Chen M-H, Kim D. J. Am. Chem. Soc. 1986;108:2489. doi: 10.1021/ja00269a082. [DOI] [PubMed] [Google Scholar]; (b) Curran DP, Chen M-H, Spletzer E, Seong CM, Chang C-T. J. Am. Chem. Soc. 1989;111:8872. [Google Scholar]
- 19.For reviews on auto-tandem catalysis featuring transition metal catalysts, see: Shindoh N, Takemoto Y, Takasu K. Chem. Eur. J. 2009;15:12168. doi: 10.1002/chem.200901486.Camp JE. Eur. J. Org. Chem. 2017:425.
- 20.Reductive elimination of an alkylpalladium(II) species could also potentially deliver bromide 48. For example, see: Hao W, Wei J, Geng W, Zhang W-X, Xi Z. Angew. Chem Int. Ed. 2014;53:14533. doi: 10.1002/anie.201408341.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.