

Abstract
Application of the proteasome inhibitor Bortezomib for the treatment of haematopoietic malignancies such as multiple myeloma significantly improves the average overall survival of patients. However, one of the most severe side effects is the development of peripheral neuropathies caused by neurotoxic effects of Bortezomib limiting its therapeutic efficacy. With ONX-0914 a specific inhibitor of the β5i (LMP7)-immunosubunit containing proteasomes was developed that targets exclusively the proteasome subtypes mainly expressed in immune cells including B lymphocytes as the origin of multiple myeloma. Furthermore, immunosubunitspecific inhibitors have been shown to be promising tools for the therapy of autoimmune disorders. In the presented study, we analysed the concentration-dependent impact of both inhibitors on primary neurons regarding survival rate, morphological changes, and overall viability. Our results clearly demonstrate that ONX-0914, compared to Bortezomib, is less neurotoxic suggesting its potential as a putative antineoplastic drug and as a candidate for the treatment of autoimmune disorders affecting the peripheral and/or central nervous system.
Keywords: Bortezomib, ONX-0914, immunoproteasome, proteasome, peripheral neuropathy, neuronal cell culture, β5i/LMP7
Introduction
In 2003 the U.S. FDA approved Bortezomib (BZ) as the first proteasome inhibitor for the therapy of multiple myeloma [1]. Since then, several second-generation proteasome inhibitors have been developed including Carfilzomib, Ixazomib, and Delanzomib, which are currently tested in pre-clinical trials [2–4] as promising therapeutics for the treatment of multiple myeloma and, possibly in the future, of other cancers as well [4–6].
The molecular function of BZ is based on the inhibition of the proteasome system, thereby interfering with cellular protein degradation [7, 8].
The barrel-shaped multi-catalytic proteasome complex contains three different catalytic subunits, namely β1, β2, and β5 with caspase-like, trypsin-like, and chymotrypsin-like proteolytic activity, respectively [9, 10]. BZ reversibly interferes with the chymotrypsin-like β5 subunit [2]. Consequences are disturbed protein homeostasis and an intracellular accumulation of damaged and misfolded proteins leading to cell stress and finally apoptosis [8]. Notably several studies demonstrated an augmented susceptibility of cancer cells to the cytotoxic effects produced by proteasome inhibition compared to healthy cells. Reasons are the accelerated metabolism and a higher demand of protein degradation caused by an induction of cell cycle arrest and inhibition of cellular growth in combination with an upregulation of pro-apoptotic factors due to proteasome inhibition [11–13]. In addition, prolonged activation of the inflammation-associated NFκB signalling pathway, which is observed in advanced stages of cancer and suppresses apoptosis, is diminished by proteasome inhibition via stabilization of the NFκB inhibitor IκB [2, 14, 15].
Application of drug combinations including proteasome inhibitors such as BZ increases the average overall survival of patients with multiple myeloma significantly by promoting chemosensitizing effects that restore sensitivity to conventional chemotherapeutical drugs [12, 16]. However, there are limitations for BZ therapy including an impaired efficacy on solid tumours, the development of resistances, and toxic side effects [17, 18]. Indeed, up to 50% of BZ-treated patients develop peripheral neuropathies including peripheral nerve changes and neuropathic pain and/or gastrointestinal side effects. In 15–20% of the patients a severity of grade 3 and 4 according to the National Cancer Institute common toxicity criteria score is observed [19, 20].
Therefore, the development of pharmacologically improved proteasome inhibitors is of pivotal therapeutic interest. Promising targets are immunoproteasomes (i-proteasome) and immunosubunit (i-subunit)-containing proteasomes, which are induced in most cells upon elevated levels of proinflammatory cytokines such as type I and type II interferons as well as oxidative stress. Thereby, the i-subunits β1i (LMP2), β2i (MECL-1), and β5i (LMP7) are incorporated into de novo synthesized proteasome complexes instead of the standard proteasome subunits b1 (delta), b2 (zeta) and b5 (MB1) [21–23]. In cells of hematopoietic origin i-proteasomes and i-subunit-containing proteasomes are constitutively expressed. I-proteasomes exhibit an altered proteolytic activity and display an enhanced protein substrate turnover compared to standard proteasomes. Thus, they generate a quantitatively and also qualitatively altered set of peptides that plays an important role in the MHC class I antigen processing and presentation pathway [24–28]. However, several studies point out that the functional repertoire of i-proteasomes goes far behind MHC class I antigen presentation including regulation of cytokine release, regulation of T-cell recruitment, activation, and differentiation especially under pathophysiological conditions [29–33].
Application of selective i-subunit inhibitors may allow for the targeted treatment of cells featuring high i-proteasome expression levels as in (malignant) haematopoietic cells. Cells of healthy and non-immune relevant tissue, which exhibit low or absent expression levels of i-subunits, would, if at all, only be marginally affected. Due to this, clinical application of i-proteasome inhibitors in the therapy of haematopoietic malignancies and autoimmune diseases could lower side effects of proteasome inhibition and thereby improve future therapy options [34]. The first i-proteasome inhibitor selectively inhibiting the β5i-subunit is ONX-0914 (formerly named PR-957) [31]. Several studies using animal models showed a beneficial effect of selective β5i-inhibition in different autoimmune pathologies including experimental arthritis, experimental autoimmune encephalomyelitis, and neuritis [35], diabetes, colitis, and colitis-associated cancer [34, 36–38]. Furthermore, selective inhibition of the β2i i-subunit resulted in significant inhibition of tumour-growth in a xenograft murine model of prostate cancer [39], and anti-proliferative activity in myeloma patient samples indicating that specifically targeting the i-subunits might be a successful strategy in the treatment of cancer [40].
In the present study we analysed the neurotoxic effect of ONX-0914 as a selective β5i i-subunit inhibitor in comparison to BZ using primary neuronal cultures as a potent and easy accessible pharmacological in vitro model system to gain detailed information about the potential of ONX-0914 as a new drug for therapy of cancer and autoimmune diseases.
Material and methods
Inhibitors and antibodies
Proteasome inhibitors BZ (Cat. No S1013) and ONX-0914 (Cat. No S7172) were purchased from Selleckchem Chemicals (USA) and stored as 10 mM stock solutions in DMSO (Sigma-Aldrich) at –20°C according to manufacturer’s instructions.
Primary antibodies used in immunocytochemistry included mouse monoclonal anti-MAP2 (1:1000, M1406, Sigma-Aldrich), guinea pig polyclonal anti-Synaptophysin 1 (1:2000, 101004, Synaptic Systems), and chicken polyclonal anti-GFAP (1:1000, ab4674, abcam). For immunofluorescence the secondary antibodies donkey anti-mouse Alexa Flour® 488-conjugated (1:2000, A21202, Invitrogen), donkey anti-chicken Alexa Flour® 647-conjugated (1:1500, 703-606-155, Jackson Immunoresearch), and donkey anti-guinea pig Cy3™-conjugated (1:2000, 706-165-148, Jackson Immunoresearch) were used.
Primary antibodies used for immunoblotting were rabbit polyclonal anti-cleaved Caspase 3 (1:1000, #9661, Cell Signaling), rabbit polyclonal anti-phospho-NF-κB p65 (Ser536) (1:5000, #3033, Cell Signaling), rabbit monoclonal anti-CREB (1:2000, #9197, Cell Signaling), rabbit monoclonal anti-phospho-CREB (1:2000, #9198, Cell Signaling), rabbit polyclonal anti-alpha 6 (1:750, PA5-22288, Thermo Scientific), mouse monoclonal anti-ß-actin (1:5000, #3700, Cell Signaling), chicken polyclonal anti-GFAP (1:500, ab4674, abcam), goat polyclonal anti-Iba 1 (1:500, ab5076, abcam), rabbit monoclonal anti-AMPA receptor (1:2000, #13185, Cell Signaling), and guinea pig polyclonal anti-Synaptophysin 1 (1:10000, 101004, Synaptic Systems). Horseradish peroxidase-conjugated secondary antibodies raised against mouse, rabbit, guinea pig, chicken, and goat (all from Jackson ImmunoResearch Laboratories) were applied 1:7500 for WB analyses.
Animals
Primary neuron-glia co-cultures were obtained from pregnant E18 Wistar rats. Pregnant rats were housed at a constant temperature of 22 °C, relative humidity of 50% and under a 12 h light-dark cycle with food and water ad libitum at the animal facilities of the Otto-von-Guericke University in Magdeburg.
Cell culture and drug treatment
Primary cortical neuron-glia co-cultures were prepared as described previously [41]. Briefly, pregnant rats were deeply anaesthetized with Isofluran Baxter (Baxter Deutschland GmbH) and decapitated. Embryos were rapidly removed and placed on ice. Embryo brains were dissected, meninges removed, and the cortices underwent enzymatic digestion using Hank’s balanced salt solution (HBSS, Invitrogen) with 0,25 % trypsin (life technologies). After tissue dissociation with cannulas (Sterican® G21 and G25, from B.Braun), cells were seeded on coverslips coated with poly-D-lysine at densities of 20 000 cell/well in 24-well plates, 500 000 cells/well in 6-well plates, and 5 million cells per 75 cm2 culture flask.
Cells were kept in Dulbecco’s modified eagle medium (DMEM, from Gibco) with 10% foetal calf serum, 2 mM glutamine and antibiotics (100 U/ml penicillin, 100 µg/ml streptomycin, all from Invitrogen) at 37 °C and 5% CO2. After 24 h media was replaced with Neurobasal® (NB, Gibco) medium supplemented with 1x B-27 (life technologies), 0.8 mM glutamine, and 50% (v/v) conditioned medium. Cells were fed once a week with fresh NB medium supplemented with 1× B-27 and 0.8 mM glutamine.
After 14 days in vitro (DIV14) cells were treated with the proteasome inhibitors BZ or the i-proteasome inhibitor ONX-0914 for either 24 or 48 h. Final inhibitor concentrations ranged from 0.001 to 0.1 µM for BZ and from 0.01 to 0.5 µM for ONX-0914 treatment.
Immunocytochemistry
Cultured and treated cells were rinsed with 1 mM MgCl2, 0.1 mM CaCl2 in 1× PBS, pH7.4 and fixed with PLP fixative [42] for 30 min at room temperature (RT) and washed with 1x PBS, pH 7.4 three times for 10 min. Subsequently, cells were incubated in blocking solution (10% horse serum, 5% sucrose, 2% bovine albumin, 0.2% Triton™ X-100 in 1× PBS, pH 7.4) for 1 h at RT. Fixed and permeabilised cells were incubated with primary antibodies over night at 4 °C, washed 3 times with with 1× PBS, pH7.4 for 10 min each, and afterwards were incubated for 1.5 h at RT with secondary antibodies followed by 3 washing steps for 10 min with 1x PBS, pH7.4. Coverslips were mounted in 10% mowiol 4-88 (Calbiochem), 25% glycerol and 2.5% DACO in Tris-HCl buffer.
Microscopy and quantification of cell and synapse numbers
Confocal images of immunocytochemically stained cells were acquired with an Axio observer.Z1 microscope equipped with a LSM 710 confocal unit (both from Carl-Zeiss® Jena).
For cell number assessment, 16 µm thick z-stacks at 4 µm-intervals were generated with a 20× objective and merged to maximum intensity projections for 40 visual fields/condition and experiment. The number of MAP2-positive neurons after inhibitor treatment was counted using Adobe Photoshop CS5 software and referred to the control condition (mock-treated cells).
For quantification of synapse numbers, 4.6 µm thick z-stacks at 0.77 µm-intervals of the main dendrite from individual neurons were generated with a 63× oil objective and merged to maximum intensity projections. Using FIJI software (version 1.49, NIH), pictures of the dendrites were marked and straightened. Synapse numbers were determined by measuring the numbers of signals in the Synaptophysin 1-positive channel of defined dendrite sections of 50 µm using the ImageJ plugin Punctae Analyzer (by Bary Wark). For each experiment, 15–16 confocal images per condition were processed and analysed.
Semi-quantitative immunoblot analysis
Cells were harvested in the presence of 10% protease inhibitor, 2% phosphatase inhibitor (both from Roche), and benzonase nuclease (Sigma-Aldrich) in 1× PBS, pH 7.4 followed by solubilisation in sample buffer with a final concentration of 0.25% SDS, 10% glycerol, 5% β-Mercaptoethanol, 62.5 mM Tris pH 6.8 and 0.001% Bromphenolblue at 95° C for 10 min.
Protein concentrations were determined using the amidoblack assay. 20 µg protein per sample was loaded per lane, separated by SDS PAGE (5–20% mini-Tris gradient gels, 12 mA/gel), and transferred on nitrocellulose membranes (0.22 µm, LI-COR® Biosciences GmbH) via Western Blot (200 mA for 1.5 h). After Ponceau-staining unspecific binding was blocked with 5% BSA in 1× TBS, pH 7.4 for 1 h at RT under gentle agitation.
Membranes were probed with primary antibodies diluted in 5% BSA, 0.1% Tween in 1× TBS, pH 7.4 over night at 4 °C followed by thorough rinsing of the membranes before application of the appropriate secondary antibodies diluted in 0.1% BSA, 0.1% Tween for 2 h at RT. Proteins of interest were detected with chemiluminescent enhancer solution (Pierce) in an Odyssey Fc scanner (LI-COR® Biosciences GmbH).
Three independent experiments with 2 to 3 technical replicates were performed. Quantification of signal intensities was accomplished using LI-COR® Images Studio Lite 5.0 software. To ensure equal loading samples were first normalized using identically loaded Coomassie stained sister gels; absolute values were subsequently normalized to control samples.
Statistical analysis
All statistical analyses were conducted with GraphPad Prism 6 software (GraphPad Software Inc., USA). A oneway ANOVA with Bonferroni post-hoc testing was used to determine statistical significant differences between neuron numbers after inhibitor treatment. For assessment of differences in synapse numbers and protein expression levels, a Student’s t-test was used for identical inhibitor concentrations. A one-sample t-test against the hypothetical value 1 was carried out to assess changes of Creb phosphorylation. All data were presented as mean ± standard error of mean (SEM) and p values <0.05 were considered as statistically significant.
Ethics statement
All animal experiments were approved and conducted under established standards of the German federal state of Sachsen-Anhalt.
Results
BZ and ONX-0914 differentially affect the survival of primary neurons in vitro
To determine the neurotoxic effects of proteasome inhibition, we analysed the effects of the proteasome inhibitor BZ and the β5i i-subunit inhibitor ONX-0914 (ONX) first regarding neuronal cell death in primary rat cortical cultures. For this, cultures were supplemented for 24 and 48 h with inhibitor concentrations ranging from 0.001 to 0.5 µM.
Within the first 24 h of treatment the number of surviving neurons with an intact morphology, i.e. no neurite blebbing or formation of varicosities, remained constant for both inhibitors at all used concentrations, except for 0.05 µM ONX-0914 (Fig. 1B). The cells retained an intact morphology with a ramified network of dendrites (Fig. 1A).
Fig. 1.
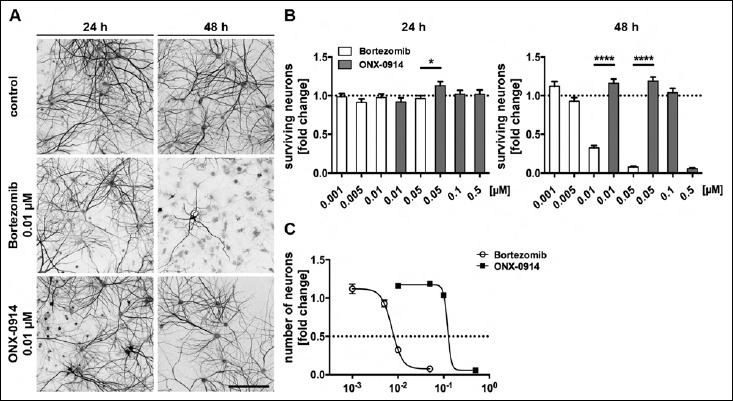
Bortezomib and ONX-0914 differentially affect the number of surviving neurons in a dose and time dependent manner. Neurotoxic effects of inhibitor treatment on the number of surviving neurons were determined in relation to the control condition. At DIV 14, primary rat cortical co-cultures were supplemented for 24 and 48 h either with the proteasome inhibitor Bortezomib (0.001 to 0.05 µM) or with the i-proteasome inhibitor ONX-0914 (0.01 to 0.5 µM), respectively. (A) Representative images of MAP2-immunostained neurons supplemented with either Bortezomib or ONX-0914, respectively. (B) Quantification of MAP2-positive cells revealed differences in the number of surviving neurons especially after 48 h of treatment. Application of Bortezomib led to a massive decrease of the number of neurons already at lower doses after 48 h compared to ONX-0914, whereas only a small difference was seen after 24 h at a concentration of 0.05 µM. (C) Correlation between inhibitor concentration and neuron number after 48 h exposure is shown, concentrations of 0.01 and 0.05 µM led to massive toxic effects of Bortezomib but not yet for ONX-0914. Calculated TD50 were 0.007 µM for Bortezomib and 0.121 µM for ONX-0914. Scale bar: 150 µm; presented data are mean ±SEM of 2–3 independent experiments with 40 images/condition each; One-way ANOVA, *: p = 0.0223, ****: p < 0.0001
In contrast, a strong effect on neuronal survival was observed after 48 h treatment especially for the β5/β5 i-subunit proteasome inhibitor BZ. Here, the number of unaffected neurons already started to decline at a concentration of 0.005 µM. Exposure to 0.05 µM BZ reduced the number of neurons further down to 7.5% compared to controls (Fig. 1B). In addition, we observed shortened processes and, thus, spatially separated neurons (Fig. 1A).
In comparison to BZ, ONX had fewer effects on the number of neurons after 48 h. The number of neurons remained unaffected up to a concentration of 0.1 µM ONX. However, at 0.5 µM ONX the number of neurons was massively diminished to approximately 5.7% of the controls (Fig. 1B). Hence, for concentrations of 0.01 and 0.05 µM we detected significant differences between both inhibitors.
Using a pharmacological dose-response analysis, the half-maximal toxic dose values (TD50) after 48 h treatment, here defined as the concentration at which the number of neurons is reduced for about 50%, were calculated. The TD50 for BZ (~0.0073 µM) was found to be much lower than for ONX treatment with ~0,1212µM (Fig. 1C).
In the next set of experiments, we focused on 48 h inhibitor treatment and determined the effects and toxic doses of ONX and BZ in more detail. Single cells, immunocytochemically stained for MAP2, Synaptophysin and GFAP, were imaged and analysed using different inhibitor concentrations (Fig. 2A). In BZ-treated neurons first changes of altered cell morphology were observed at 0.01 µM. The cells developed varicosities in dendrites and showed membrane blistering and blebbing. Furthermore, somata lost their structural integrity. After 48 h of BZ treatment with concentrations of 0.1 µM and higher, cell bodies were completely disintegrated.
Fig. 2.
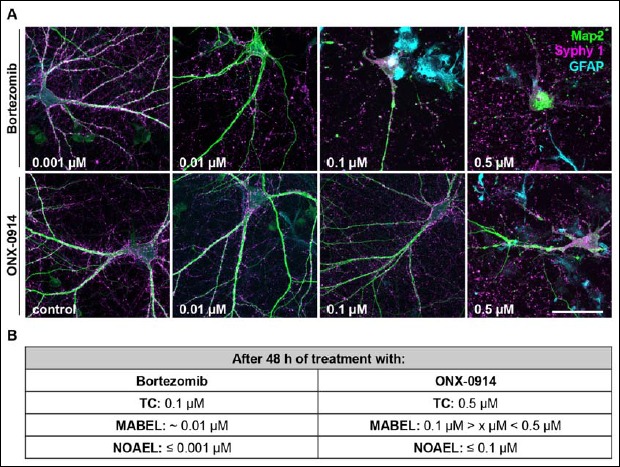
ONX-0914 is less neurotoxic than Bortezomib in in vitro neural cultures. (A) Representative images of primary rat cortical co-cultures (DIV14) after 48 h inhibitor treatment with indicated concentrations. For the immunocytochemical staining antibodies raised against MAP2 (green), Synaptophysin 1 (Syphy 1, magenta) and GFAP (cyan) were used. Cell viability was assessed using confocal microscopy. Note, in Bortezomib-treated cultures cellular disintegration and cell death appear already at much lower concentrations compared to cultures supplemented with ONX-0914. (B) Pharmacological threshold values for apparent toxicity after 48 h of application were specified based on morphological analysis including structure disintegration and membrane blebbing. Values determined for ONX-0914 were much higher than for Bortezomib. Scale bar: 50 µm, representative images of 3 independent experiments, TC: toxic concentration, MABEL: minimal anticipated biological effect level, NOAEL: no observed effect level
In contrast, only high concentrations of ONX (0.5 µM) caused neuronal cell death after 48 h treatment (Fig. 2A). At lower doses none of the mentioned alterations of cell morphology, as observed for BZ treatment, appeared.
Notably, neurons analysed after 24 h inhibitor treatment required higher inhibitor concentrations in order to exhibit morphological alterations. For BZ first changes were observed at 0.1 µM and for ONX membrane blistering/blebbing as well as dendrite varicosities became visible with a concentration of 2 µM (data not shown).
In summary, for primary cortical cultures at DIV14 we assessed a BZ concentration of ≤0.001 µM as the dosage with no observed effects (NOAEL). For ONX the NOAEL concentration was two decimal powers higher and was calculated at ≤0.1 µM. The minimal anticipated biological effect level (MABEL) was ~0.01µM for BZ and between 0.1 and 0.5 µM for ONX (Fig. 2B). The toxic dose for the i-proteasome inhibitor ONX was calculated to be 0.5 µM and, thus, much higher in comparison to the proteasome inhibitor BZ with its respective toxic concentration of 0.1 µM.
Bortezomib diminishes the number of neuronal synapses
Brain function and neuronal signalling rely on synapse functionality and ultimately on synapse number. To assess any adverse side effects of the two proteasome inhibitors, primary cortical cultures were treated for 48 h with BZ and ONX and afterwards immunostained for Synaptophysin 1 and MAP2 to count synapse numbers. To ensure, that only viable cells were taken into account, experiments were conducted using a concentration of 0.01 µM for both inhibitors, as this dose was identified as the one showing first signs of neuronal cell death for BZ in the previous experiment.
First, main dendrites of BZ-treated neurons showed morphological changes including shrinkage and neurite degeneration (Fig. 3A). In contrast, treatment with ONX had no effect on dendrite morphology and was comparable to the control. Particularly striking was the massive decline of synapses at the BZ treated neurons, where the Synaptophysin 1 signal was significantly reduced compared to the ONX treated neurons and the control (Fig. 3A, B), pointing towards a rapid loss of neuronal functionality.
Fig. 3.
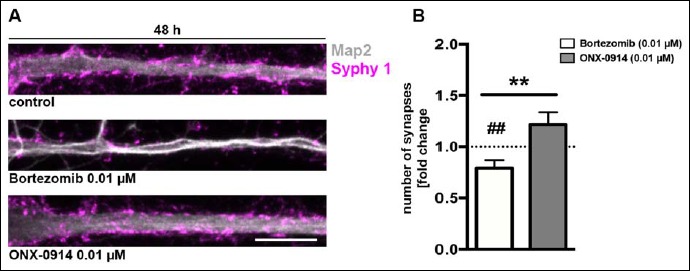
Bortezomib diminishes synapse numbers at lower concentrations as ONX-0914. Synapse numbers in primary rat cortical cultures (DIV14) were analysed after 48 h treatment with a dose of 0.01 µM for both inhibitors. (A) Representative confocal images of a representative main neuronal dendrite immunostained for MAP2 (grey) and Synaptophysin 1 (Syphy 1, magenta). (B) Synapse numbers were quantified in straightened dendrite sections of 50 µm using the Image J plugin punctae analyser. Notably, cells treated with Bortezomib exhibit morphological changes in their dendrite structure including a reduction of signal intensity for Synaptophysin 1. Synapses are significantly decreased after Bortezomib treatment compared to ONX-0914-treated cultures and the control condition. Scale bar: 10 µm, presented data are mean ±SEM of 3 independent experiments with 16 analysed images/condition each, Student’s t-test, **: p = 0.003, ##: p = 0.0062
Bortezomib and ONX-0914 alter protein expression levels in primary cortical co-cultures
The observed morphological changes in neurons after proteasome inhibitor treatment, point towards massive changes of the cellular protein homeostasis. Therefore, we quantified the expression of selected proteins using semiquantitative immunoblot analysis. We focused on proteins known to play an important role in cell signalling, viability and cytoskeleton architecture such as NFκB, CREB, phosphorylated CREB (pCREB), cleaved caspase 3, the proteasomal alpha6-subunit, β actin, AMPA Receptor and Synaptophysin. To assess glial function Iba1 (microglia) and GFAP (astrocytes) signals were quantified.
The transcription factor NFκB is known to play an important role during apoptosis and inflammation, where induction of NFκB signalling results in enhanced inflammation and reduced apoptosis. Induction of NFκB depends on the activation of IKK, which phosphorylates IκB inhibitory proteins and targets them for proteasomal degradation. Further, phosphorylation of NFκB-p65, a subunit of the NF-kappa-B transcription complex, is needed for an optimal activation of NFκB. Therefore, we analysed the expression level of phospho-NFκB-p65 after 24 and 48 h of BZ and ONX treatment in primary cortical cultures.
After 24 h we observed mainly a reduction of phospho-p65 in the presence of 0.01 and 0.1 µM BZ and 0.5 µM ONX, indicating an impaired activation of NFκB at these concentrations. Lower concentrations did not seem to influence the level NFκB activation and were comparable to the control. However, compared to ONX, BZ had, already at lower concentrations a stronger effect on NFκB phosphorylation (Fig. 4A, B).
Fig. 4.
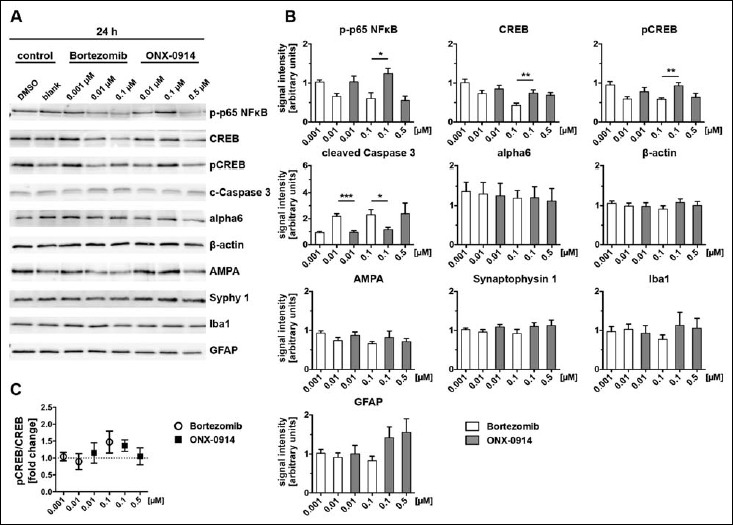
Bortezomib and ONX-0914 differentially affect neural protein expression after 24 h. Cell lysates of primary cortical cultures (DIV14), treated for 24 h with indicated inhibitor concentrations, were analysed via immunoblot for selected proteins (A) and signal intensities were subsequently determined (B). Normalization was ensured via loading same protein amounts and quantitative analysis of the respective Coomassie stained sister gel. Data were expressed in relation to the control condition (100%). Note, protein levels for NFκB, CREB and pCREB showed a tendency towards decreased expression with increasing inhibitor concentrations, whereas this effect was evident earlier and stronger on Bortezomib treatment compared to ONX-0914. Conversely, levels of the apoptosis marker Caspase 3 were significantly increased after Bortezomib treatment at doses of 0.01 and 0.1 µM after 24 h. No significant differences were detected for the protein level of alpha6, β-actin, AMPA and Synaptophysin 1 as well as the glial marker Iba1 and GFAP after 24 h inhibitor treatment (4B). Activation of CREB signalling was assessed by calculation of the pCREB/CREB ratio (4C). A slight tendency for CREB activation indicated by phosphorylation was observed after 24 h at doses of 0.1 µM for both inhibitors. Presented data are mean ±SEM of 2–3 technical replicates each from 3 independent experiments, B: Student’s t-test, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, C: one-sample t-test against hypothetical value 1
In contrast, phospho-p65 increased after 48 h of 0.001 and 0.01 µM BZ treatment and was clearly decreased at 0.1 µM BZ. ONX treatment increased phospho-p65 levels at 0.01 and 0.1 µM too, but led to decreased levels at 0.5 µM after 48 h (Fig. 5A, B). Thus, proteasome inhibitor treatment results in reduced NFκB activation in primary cortical co-cultures, remarkably at lower concentrations of BZ compared to ONX.
Fig. 5.
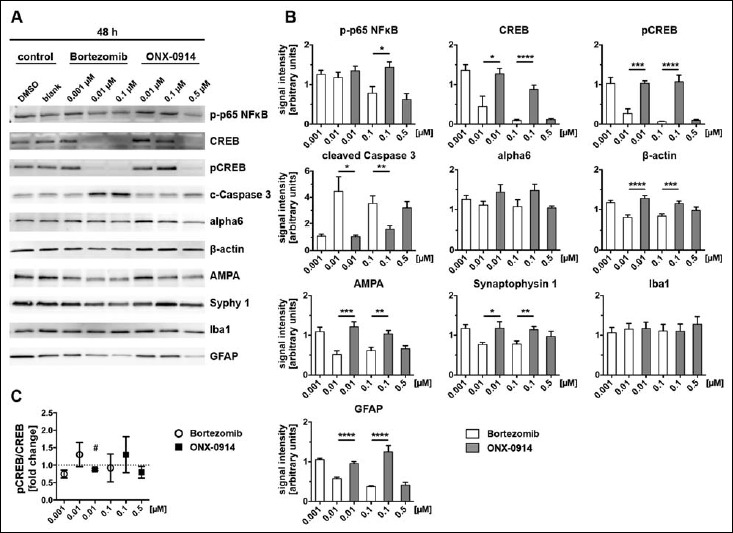
Altered neural protein expression after 48 h of inhibitor treatment. Alterations of protein levels after proteasome inhibition were detected by immunoblot analysis (A) and the signal intensities subsequently quantified (B). Cell lysates were prepared from primary cortical co-cultures after 48 h inhibitor treatment. Identical protein concentrations were loaded on gels and signals normalized to Coomassie stained sister gels. Signal intensities are expressed in relation to the control (100%). After 48 h, effects on protein expression after inhibitor treatment became even more obvious for NFκB and Caspase 3. The prolonged inhibitor incubation at a dose of 0.01 µM additionally effected massively decreased levels for CREB and pCREB after 48 h Bortezomib application compared to ONX-0914. Further, treatment with 0.01 and 0.1 µM Bortezomib over 48 h lead to decreased protein levels for β-actin, AMPA, Synaptophysin 1 and GFAP in comparison to ONX-0914-treated cultures. Expression levels for alpha6 subunit and Iba1 were unaffected by inhibitor treatment over 48 h (5B). CREB activation indicated by phosphorylation was, compared to 24 h (4C), nearly unaffected after 48 h (5C). Presented data are mean ±SEM of 2–3 technical replicates each from 3 independent experiments, B: Student’s t-test, *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001, C: one-sample t-test against hypothetical value 1, #: p < 0.05
Because cAMP/CREB-dependent gene expression is one of the major pathways mediating synaptic plasticity, we examined the status of CREB and pCREB after 24 and 48 h of BZ and ONX treatment.
After 24 h expression levels of CREB and pCREB were decreased in a dose-dependent manner to approximately 50 and 60% of the control for BZ treated primary cultures (Fig. 4A, B). Even though to a lower extent, the same effect was observed for ONX treatment at the indicated time point (Fig. 4A, B). Prolonged inhibitor treatment of 48 h revealed the same tendency, whereas concentrations of 0.01 and 0.1 µM BZ caused a significantly larger decrease in protein expression compared to ONX treatment. At the highest concentrations used for BZ and ONX treatment, CREB and pCREB protein levels were diminished to a minimum (Figs 4 and 5B) possibly suggesting that proteasome inhibition affects synaptic plasticity. Importantly, ONX treatment resulted in significant reduction of CREB/pCREB only after a long incubation period (48 h) using a high ONX-concentration (0.5 µM).
Based on the calculation of the pCREB/CREB ratio, it is possible to predict whether signalling cascades of CREB are regulated after inhibitor treatment. Incubation periods of 24 h induced elevated phosphorylation of CREB and, thus, its activation at a dose of 0.1 µM for both inhibitors (Fig. 4C). Treatment over 48 h did not clearly effect the ratio of CREB phosphorylation (Fig. 5C).
Cleaved Caspase 3, a typical apoptosis marker, increased after inhibitor treatment before cell death became obvious (compare to Fig. 1B). We detected 2-fold higher expression levels for cleaved Caspase 3 after 24 h treatment with BZ starting from 0.01 µM compared to the control (Fig. 4A, B). Also ONX treatment caused an increased level of cleaved Caspase 3 after 24 h but in comparison to BZ only at a higher concentration of 0.5 µM (Fig. 4A, B). This effect was enhanced with a longer incubation period and matched to the observed cell death after 48 h (compare Fig. 1B).
Cleaved caspase 3 levels rose further to approximately 4 to 5-fold levels after 48 h BZ treatment at 0.01 µM and higher concentrations as well as ONX treatment at 0.5 µM (Fig. 5A, B). Comparing both inhibitors, effects of BZ and ONX clearly differed at 0.01 µM and 0.1 µM, at which ONX, related to the control, did not yet have any obvious influence and BZ already produced elevated levels of active Caspase 3.
The alpha6 subunit is part of the outer ring of the 20S core particle of the proteasome complex and is present in the standard proteasome as well as in the i-proteasome. To exclude the possibility that the expression of the whole proteasome complex is non-specifically affected by proteasome inhibitor treatment, we determined alpha6 expression levels, which reflect the total amount of 20S core proteasomes. Treatment with proteasome inhibitors resulted in a minor trend towards decreasing expression levels of alpha6 after 24 h with increasing concentrations of BZ and ONX. Nevertheless, all means were above the level of the control after 24 h and after 48 h. Treatment for 48 h showed the same trend as observed for 24 h incubation and revealed that alpha6 was more increased in ONX than in BZ treatment at comparable concentrations of 0.01 and 0.1 µM, although the enhanced alpha6 expression was not significant for both inhibitors (Figs 4 and 5A, B).
One of the major components of the cytoskeleton is beta-actin. Application of proteasome inhibitors did not seem to have much influence on the beta-actin level of primary cortical cultures at least after 24 h (Fig. 4A, B). But after 48 h of BZ and ONX treatment, we observed a decrease in signal intensity in BZ-treated cells, indicating a reduction of beta-actin at concentrations of 0.01 and 0.1 µM with significant differences to ONX treatment, in which beta-actin expression levels were not below the control level (Fig. 5A, B).
In order to analyse synaptic structural integrity, we quantified the protein expression levels of Synaptophysin 1, a presynaptic vesicle protein and AMPAR, a postsynaptic receptor, after 24 and 48 h of proteasome inhibitor treatment. After 24 h of BZ and ONX treatment, we observed no effect on Synaptophysin 1 expression levels and a slight decrease for AMPAR signals in a dose-dependent manner (Fig. 4A, B). BZ treatment over 48 h resulted for both synaptic proteins in a decrease of the expression levels to roughly 50% for AMPAR and 75% for Synaptophysin compared to the control. Similar, ONX treatment reduced expression levels but to a lower extent than observed for BZ application. Comparison of the 0.01 and 0.1 µM concentrations at 48 h incubation time showed a higher expression level for AMPAR and Synaptophysin 1 in ONX-treated cells than after BZ treatment (Fig. 5A, B). In sum, synaptic structural integrity was affected by both proteasome inhibitors, although ONX displayed fewer effects than BZ.
To analyse whether glia cell growth and number are impaired in the presence of BZ or ONX, we finally determined the expression levels of glial marker proteins such as Iba1 for microglia cells and GFAP for astrocytes after proteasome inhibition. There was no obvious influence of BZ and ONX treatment on Iba1 protein levels detectable, neither for 24 h nor 48 h incubation periods (Figs 4 and 5A, B). In contrast, the application of these inhibitors had an effect on astrocytes. After 24 h we noticed an opposite trend of astrocyte activation for BZ and ONX treatment in primary cortical cultures (Fig. 4A, B). BZ resulted in a minor decrease of GFAP protein levels whereas ONX produced an 1.5-fold increase of GFAP compared to controls. However, GFAP expression levels were decreased below 50% of the control after 48 h at higher concentrations for both, BZ and ONX treatment (Fig. 5A, B). In sum, at comparable concentrations of 0.01 and 0.1 µM the GFAP level remained unchanged compared to the control for ONX treated cultures, whereby GFAP was strongly decreased after BZ treatment.
Discussion
The development of proteasome inhibitors such as BZ has significantly improved therapy options for patients suffering from multiple myeloma, although various side effects including the development of peripheral neuropathy complicate the therapy with this substance. Therefore, we focused in our study on neurotoxic effects of BZ and compared them with effects of the more selective β5i-specific inhibitor ONX-0914. As a model system, we opted for primary cortical co-cultures from rat as this system allows for a rapid and detailed analysis of cell survival, morphological integrity on cell and synapse level and facilitates to monitor the influence of drugs on cellular protein homeostasis.
Application of the β5/β5i proteasome inhibitor BZ induced pronounced neuronal cell death in a dose-dependent manner after 48 h of treatment (see Fig. 1). In contrast, ONX-0914, a selective inhibitor of the β5i i-subunit, had no influence on neuronal cell survival up to a concentration of 0.1 µM. Especially, when comparing the doses of 0.01 and 0.05 µM, the neurotoxic effect of BZ is pronounced whereas ONX-0914 clearly seems to be non-toxic for healthy neurons. A common side effect of BZ treatment of patients with multiple myeloma is the development of neuropathies [19]. Even though, the mechanism is not well understood, the reduced neurotoxicity of ONX-0914 in our results, strengthen the hypothesis to exploit specific proteasome subunit inhibition as a therapeutically promising target with the advantage of lower toxicity.
In line with this we observed cellular disintegration and membrane blebbing, a significant loss of synapses in BZ-treated cultures compared to ONX-0914 (see Figs 2 and 3). Changes in the axonal cytoskeleton and deformations at the sensory afferent terminals of nerve fibers are proposed to be involved in the development of peripheral neuropathy after BZ usage in multiple myeloma treatment [43, 44]. Although, we used primary cortical neurons and not a peripheral neuron model for our experiments, ONX-0914 application did not compromise the structural integrity and synapse density in comparison to BZ. These results indicate that specific i-proteasome inhibition causes less neurotoxicity and, thus, could possibly enable improved treatment possibilities not only in order to prevent dose limiting toxicity but also to enable prolonged treatment periods.
Further, we performed immunoblot analysis and determined expression levels of selected proteins after BZ and ONX-0914 treatment to examine effects of proteasome and i-proteasome inhibition on neuronal protein homeostasis (see Figs 4 and 5). Overall, comprehensive differences between ONX-0914-treated cells and the control were induced only at the highest dose of 0.5 µM. Conversely, application of BZ influences protein expression levels already at lower doses and to a more extensive manner. Thus, the neurotoxic effects of constitutive proteasome compared to selective i-proteasome inhibition in physiological neuronal cell cultures became even more obvious.
As expected, we observed an impaired NFκB activation in BZ-treated cells. Inhibition of NFκB activation indirectly inhibits also the expression of stress response enzymes, growth factors and apoptosis inhibitors which forces cells to die [12]. In a physiological state, neurons express mainly the standard proteasome and are more susceptible for proteasome inhibitor-induced apoptosis by BZ. In contrast, ONX-0914, which selectively targets β5i (LMP7)-i-subunit containing proteasomes, has no effect, illustrating the neuroprotective potential of ONX-0914 application as it would preferentially target cells with high β5i i-subunit expression and would not affect tissue expressing mainly standard proteasomes.
An important transcription factor for neuronal cells is CREB or rather the phosphorylated and active form pCREB. Especially after 48 h at the dose of 0.01 and 0.1 µM, BZ-treated cultures exhibit massively decreased levels of CREB and pCREB indicating either commencing decay of the total proteasome as a consequence of cell death or a stop of constitutive CREB expression based on cellular stress after inhibitor treatment. The incapacity of ONX-0914 to alter expression levels of CREB and pCREB up to a dose of 0.1 µM compared to the control, demonstrates both, i-proteasome inhibition by ONX-0914 seems to remain cellular protein homeostasis stable and does not seem to interfere with CREB signalling pathways in neural cells. This thesis is supported by the calculation of the pCREB/CREP ratio, which revealed no regulation of the CREB signalling way by proteasome inhibition.
According to the decreased cell numbers after 48 h treatment, we observed decreased levels of beta-actin and GFAP in BZ-treated cultures indicating the cell death and the degradation of these structural marker proteins. In addition, active Caspase 3 levels were elevated after BZ application application but none of these effects were detected for ONX-0914 treatment. Consequently, ONX-0914 features a lower potential to induce apoptosis via i-proteasome inhibition in healthy neurons, which could contribute to the protection against neurotoxicity.
However, Iba1 expression remained stable, which could be either due to a missing effect of BZ and ONX-0914 treatment or rather due to the fact that we were not able to analyse the impact of proteasome inhibition on microglia in the used cell culture system. Our primary cell cultures contain mainly neurons and astrocytes and only to a small proportion microglia. But since microglia are the main immune cells of the physiological brain and express i-proteasomes also constitutively, future research about the effect of proteasome and i-proteasome inhibition on these cells is highly warranted.
Moreover, we could confirm the immunocytochemically observed morphological changes in cell integrity and synapse condition and detected significant differences between BZ and ONX-0914-treated cells in the expression levels of AMPA and Synaptophysin 1 after 48 h. These results demonstrate the substantial influence of proteasome inhibition by BZ on synapse morphology, affecting possibly also the functional level. As the mechanisms leading to neuropathy in multiple myeloma treatment are not fully understood, these synaptic alterations should be taken into account for the clarification and analysis of BZ-induced side effects concerning peripheral neuropathies. Notably, ONX-0914 did not alter the levels of these synaptic markers and was comparable to the control condition. Hence, i-proteasome inhibition under the stated conditions had no effect on the synaptic morphology and prevents a loss and deformation of synapses.
In conclusion, we report here that selective i-proteasome inhibition by ONX-0914 has less neurotoxic effects concerning cell survival, morphological integrity and cellular protein homeostasis than the application of BZ in primary neuronal cells in vitro. Therefore, the development of i-proteasome inhibitors for the clinical use could be beneficial for the reduction of neuropathy side effects in multiple myeloma treatment as well as in autoimmune diseases [40].
In sum, our data underline the hypothesis of i-proteasomes as a promising clinical target that could be combined with established therapies for hematopoietic malignancies such as the multiple myeloma and autoimmune disease affecting the peripheral and/or central nervous system. Thereby, drug toxicity and the risk to develop resistances and/or side effects could be reduced with a high degree of specificity and efficiency.
Acknowledgements
The authors thank Karina Schäfer and Evelyn Dankert for expert technical assistance and the provision of cell cultures. Thanks goes to Johanna Rehfeld for the kind support in image analysis.
Footnotes
Funding sources
This work was supported by the DFG (Deutsche Forschungsgemeinschaft) within the CRC854 (to DCD and US).
Authors’ contributions
PS, PL, and DCD planned experiments; PS and LvB performed experiments; PS, LvB, and PL analysed data; US and CC provided reagents; PL and DCD supervised experiments; PS, LvB, PL, and DCD wrote the manuscript.
Conflict of interest
The authors report no financial or other relationship relevant to the subject of this article.
References
- 1.Kane RC, Bross PF, Farrell AT, Pazdur R: Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. The Oncologist 8, 508–513 (2003) [DOI] [PubMed] [Google Scholar]
- 2.Crawford LJ, Walker B, Irvine AE: Proteasome inhibitors in cancer therapy. J Cell Commun Signal 5, 101–110 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dou QP, Zonder JA: Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets 14, 517–536 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richardson PG, Hideshima T, Mitsiades C, Anderson K: Proteasome inhibition in hematologic malignancies. Ann Med 36, 304–314 (2004) [DOI] [PubMed] [Google Scholar]
- 5.Adams J: The proteasome: a suitable antineoplastic target. Nat Rev Cancer 4, 349–360 (2004) [DOI] [PubMed] [Google Scholar]
- 6.Landgren O, Iskander K: Modern multiple myeloma therapy: deep, sustained treatment response and good clinical outcomes. J Intern Med [Epub ahead of print], doi: 10.1111/joim.12590 (2017) [DOI] [PubMed] [Google Scholar]
- 7.Dou QP, Zonder JA: Overview of proteasome inhibitor-based anti-cancer therapies: perspective on bortezomib and second generation proteasome inhibitors versus future generation inhibitors of ubiquitin-proteasome system. Curr Cancer Drug Targets 14, 517–536 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Almond JB, Cohen GM: The proteasome: a novel target for cancer chemotherapy. Leukemia 16, 433–443 (2002) [DOI] [PubMed] [Google Scholar]
- 9.Glickman MH, Ciechanover A: The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 82, 373–428 (2002) [DOI] [PubMed] [Google Scholar]
- 10.Goldberg AL, Akopian TN, Kisselev AF, Lee DH, Rohrwild M: New insights into the mechanisms and importance of the proteasome in intracellular protein degradation. Biol Chem 378, 131–140 (1997) [PubMed] [Google Scholar]
- 11.Adams J: Preclinical and clinical evaluation of proteasome inhibitor PS-341 for the treatment of cancer. Curr Opin Chem Biol 6, 493–500 (2002) [DOI] [PubMed] [Google Scholar]
- 12.Adams J: The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421 (2004) [DOI] [PubMed] [Google Scholar]
- 13.Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC: The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res 61, 3071–3076 (2001) [PubMed] [Google Scholar]
- 14.Mateos MV, San Miguel JF: Bortezomib in multiple myeloma. Best Pract Res Clin Haematol 20, 701–715 (2007) [DOI] [PubMed] [Google Scholar]
- 15.Viatour P, Merville M-P, Bours V, Chariot A: Phosphorylation of NF-κB and IκB proteins: implications in cancer and inflammation. Trends Biochem Sci 30, 43–52 (2005) [DOI] [PubMed] [Google Scholar]
- 16.Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau J-L, Ben-Yehuda D, Lonial S, Goldschmidt H, Reece D, San-Miguel JF, Bladé J, Boccadoro M, Cavenagh J, Dalton WS, Boral AL, Esseltine DL, Porter JB, Schenkein D, Anderson KC: Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 352, 2487–2498 (2005) [DOI] [PubMed] [Google Scholar]
- 17.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BAC, Scheper RJ, Jansen G: Molecular basis of bortezomib resistance: proteasome sub-unit beta5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 112, 2489–2499 (2008) [DOI] [PubMed] [Google Scholar]
- 18.Verbrugge SE, Al M, Assaraf YG, Niewerth D, Meerloo J, van Cloos J, Veer M, van der Scheffer GL, Peters GJ, Chan ET, Anderl JL, Kirk CJ, Zweegman S, Dijkmans BA, Lems WF, Scheper RJ, Gruijl TD, de Jansen G: Overcoming bortezomib resistance in human B cells by anti-CD20/rituximab-mediated complement-dependent cytotoxicity and epoxyketone-based irreversible proteasome inhibitors. Exp Hematol Oncol 2, 2 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banach M, Juranek JK, Zygulska AL: Chemotherapy-induced neuropathies-a growing problem for patients and health care providers. Brain Behav 7, e00558 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bruna J, Udina E, Alé A, Vilches JJ, Vynckier A, Monbaliu J, Silverman L, Navarro X: Neurophysiological, histological and immunohistochemical characterization of bortezomib-induced neuropathy in mice. Exp Neurol 223, 599–608 (2010) [DOI] [PubMed] [Google Scholar]
- 21.Brown MG, Driscoll J, Monaco JJ: Structural and serological similarity of MHC-linked LMP and proteasome (multi-catalytic proteinase) complexes. Nature 353, 355–357 (1991) [DOI] [PubMed] [Google Scholar]
- 22.Monaco JJ, McDevitt HO: The LMP antigens: A stable MHC-controlled multisubunit protein complex. Hum Immunol 15, 416–426 (1986) [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Waters JB, Früh K, Peterson PA: Proteasomes are regulated by interferon gamma: implications for antigen processing. Proc Natl Acad Sci U S A 89, 4928–4932 (1992) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrington DA, Gregerson DS: Immunoproteasomes: Structure, function, and antigen presentation. Prog Mol Biol Transl Sci 109, 75–112 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seifert U, Bialy LP, Ebstein F, Bech-Otschir D, Voigt A, Schröter F, Prozorovski T, Lange N, Steffen J, Rieger M, Kuckelkorn U, Aktas O, Kloetzel P-M, Krüger E: Immunoproteasomes preserve protein homeostasis upon interferoninduced oxidative stress. Cell 142, 613–624 (2010) [DOI] [PubMed] [Google Scholar]
- 26.Yun YS, Kim KH, Tschida B, Sachs Z, Noble-Orcutt KE, Moriarity BS, Ai T, Ding R, Williams J, Chen L, Largaespada D, Kim D-H: mTORC1 coordinates protein synthesis and immunoproteasome formation via PRAS40 to prevent accumulation of protein stress. Mol Cell 61, 625–639 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishto M, Liepe J, Textoris-Taube K, Keller C, Henklein P, Weberruß M, Dahlmann B, Enenkel C, Voigt A, Kuckelkorn U, Stumpf MPH, Kloetzel PM: Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol 44, 3508–3521 (2014) [DOI] [PubMed] [Google Scholar]
- 28.Toes REM, Nussbaum AK, Degermann S, Schirle M, Emmerich NPN, Kraft M, Laplace C, Zwinderman A, Dick TP, Müller J, Schönfisch B, Schmid C, Fehling H-J, Stevanovic S, Rammensee HG, Schild H: Discrete cleavage motifs of constitutive and immunoproteasomes revealed by quantitative analysis of cleavage products. J Exp Med 194, 1–12 (2001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Basler M, Mundt S, Muchamuel T, Moll C, Jiang J, Groettrup M, Kirk CJ: Inhibition of the immunoproteasome ameliorates experimental autoimmune encephalomyelitis. EMBO Mol Med 6, 226–38 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ebstein F, Kloetzel P-M, Krüger E, Seifert U: Emerging roles of immunoproteasomes beyond MHC class I antigen processing. Cell Mol Life Sci CMLS 69, 2543–2558 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muchamuel T, Basler M, Aujay MA, Suzuki E, Kalim KW, Lauer C, Sylvain C, Ring ER, Shields J, Jiang J, J, Shwonek P, Parlati F, Demo SD, Bennett MK, Kirk CJ, Groettrup M: A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med 15, 781–787 (2009) [DOI] [PubMed] [Google Scholar]
- 32.Kalim KW, Basler M, Kirk CJ, Groettrup M: Immunoproteasome subunit LMP7 deficiency and inhibition suppresses Th1 and Th17 but enhances regulatory T cell differentiation. J Immunol 189, 4182–4193 (2012) [DOI] [PubMed] [Google Scholar]
- 33.Hensley SE, Zanker D, Dolan BP, David A, Hickman HD, Embry AC, Skon CN, Grebe KM, Griffin TA, Chen W, Bennink JR, Yewdell JW: Unexpected role for the immunoproteasome subunit LMP2 in antiviral humoral and innate immune responses. J Immunol 184, 4115–4122 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M: The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol 33, S74-9 (2015) [PubMed] [Google Scholar]
- 35.Liu H, Wan C, Ding Y, Han R, He Y, Xiao J, Hao J: PR-957, a selective inhibitor of immunoproteasome subunit low-MW polypeptide 7, attenuates experimental autoimmune neuritis by suppressing Th 17-cell differentiation and regulating cytokine production. FASEB J 31, 1756–1766 (2017) [DOI] [PubMed] [Google Scholar]
- 36.Basler M, Dajee M, Moll C, Groettrup M, Kirk CJ: Prevention of experimental colitis by a selective inhibitor of the immunoproteasome. J Immunol Baltim Md 1950 185, 634–641 (2010) [DOI] [PubMed] [Google Scholar]
- 37.Vachharajani N, Joeris T, Luu M, Hartmann S, Pautz S, Jenike E, Pantazis G, Prinz I, Hofer MJ, Steinhoff U, Visekruna A: Prevention of colitis-associated cancer by selective targeting of immunoproteasome subunit LMP7. Oncotarget [Epub ahead of print], doi: 10.18632/oncotarget.14579 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zaiss DMW, Bekker CPJ, Grone A, Lie BA, Sijts AJAM: Proteasome immunosubunits protect against the development of CD8 T cell-mediated autoimmune diseases. J Immunol 187, 2302–2309 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wehenkel M, Ban J-O, Ho Y-K, Carmony KC, Hong JT, Kim KB: A selective inhibitor of the immunoproteasome subunit LMP2 induces apoptosis in PC-3 cells and suppresses tumour growth in nude mice. Br J Cancer 107, 53–62 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuhn DJ, Hunsucker SA, Chen Q, Voorhees PM, Orlowski M, Orlowski RZ: Targeted inhibition of the immunoproteasome is a potent strategy against models of multiple myeloma that overcomes resistance to conventional drugs and nonspecific proteasome inhibitors. Blood 113, 4667 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Banker G, Goslin K: Developments in neuronal cell culture. Nature 336, 185–186 (1988) [DOI] [PubMed] [Google Scholar]
- 42.McLean IW, Nakane PK: Periodate-lysine-paraformaldehyde fixative. A new fixation for immunoelectron microscopy. J Histochem Cytochem 22, 1077–83 (1974) [DOI] [PubMed] [Google Scholar]
- 43.Staff NP, Podratz JL, Grassner L, Bader M, Paz J, Knight AM, Loprinzi CL, Trushina E, Windebank AJ: Bortezomib alters microtubule polymerization and axonal transport in rat dorsal root ganglion neurons. NeuroToxicology 39, 124–131 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng H, Xiao WH, Bennett GJ: Mitotoxicity and bortezomib-induced chronic painful peripheral neuropathy. Exp Neurol 238, 225–234 (2012) [DOI] [PubMed] [Google Scholar]